

Abstract
Host-induced antibodies and their contributions to cancer inflammation are largely unexplored. IgG4 subclass antibodies are present in IL-10–driven Th2 immune responses in some inflammatory conditions. Since Th2-biased inflammation is a hallmark of tumor microenvironments, we investigated the presence and functional implications of IgG4 in malignant melanoma. Consistent with Th2 inflammation, CD22+ B cells and IgG4+-infiltrating cells accumulated in tumors, and IL-10, IL-4, and tumor-reactive IgG4 were expressed in situ. When compared with B cells from patient lymph nodes and blood, tumor-associated B cells were polarized to produce IgG4. Secreted B cells increased VEGF and IgG4, and tumor cells enhanced IL-10 secretion in cocultures. Unlike IgG1, an engineered tumor antigen-specific IgG4 was ineffective in triggering effector cell–mediated tumor killing in vitro. Antigen-specific and nonspecific IgG4 inhibited IgG1-mediated tumoricidal functions. IgG4 blockade was mediated through reduction of FcγRI activation. Additionally, IgG4 significantly impaired the potency of tumoricidal IgG1 in a human melanoma xenograft mouse model. Furthermore, serum IgG4 was inversely correlated with patient survival. These findings suggest that IgG4 promoted by tumor-induced Th2-biased inflammation may restrict effector cell functions against tumors, providing a previously unexplored aspect of tumor-induced immune escape and a basis for biomarker development and patient-specific therapeutic approaches.
Introduction
Despite numerous reports investigating the clinical significance of immune cells in the circulation and in tumor lesions, the nature of local B cell responses and functional contributions of antibodies produced in cancer are largely unexplored (1–4). Recent studies have mainly focused on the immunoregulatory roles of B cells in mouse models of cancer through mechanisms such as effector cell engagement of Fcγ receptors and production of cytokines such as TNF-α and IL-10 (5, 6).
B cells respond to a variety of local stimuli to differentiate, undergo class switching, and produce antibodies of specific classes and subclasses. Human B cells are known to produce 4 subclasses of IgG (IgG1, IgG2, IgG3, IgG4), with each subclass having different biological functions (7, 8). These antibody types vary in their ability to activate immune system components, including the formation of the complement complex or the engagement of Fc receptors on the surface of effector cells (9). However, whether IgG subclasses and their effector functions are of significance in cancer inflammation is relatively unknown.
IgG4 is considered a “weak” subclass due to its poor ability to bind complement and Fc receptors and to activate effector cells. IgG4 production is normally associated with prolonged exposure to antigens and has been reported to interact with antibodies of the IgG and IgE classes through their Fc domains, potentially influencing antibody-mediated functions (10, 11). In healthy adult serum, IgG1, IgG2, IgG3, and IgG4 represent 65%, 25%, 6%, and 4% of the total IgG pool, respectively, but these proportions may be altered in certain disease contexts (8, 12). Associations of IgG4 antibodies are reported in a range of chronic inflammatory and autoimmune conditions that feature infiltration of target organs by IgG4-expressing cells (13, 14). Despite association with inflammatory pathologies, in allergy, elevated serum IgG4 antibody titers correlate with a reduction of allergic symptoms and successful allergen immunotherapy (15, 16). In this context, IgG4 antibodies are thought to interfere with IgE-mediated effector cell activation. This indirectly implies a functional significance of IgG4 in modulating antigen-specific antibody-mediated effector mechanisms and in inducing clinical tolerance (17, 18).
The relationship between IgG4 and malignancy is largely unexplored. Infiltrating IgG4+ cells in lesions of patients with extrahepatic cholangiocarcinomas and pancreatic cancers were recently reported (19, 20), and early studies have indicated abnormalities in serum titers of IgG4 in patients with melanoma (21). Both the presence and potential biological role of IgG4 subclass antibodies in melanoma tumor lesions remain largely unknown. Th2-mediated immune responses represent the classical hallmarks of local inflammation in solid tumors such as melanomas (22). The immunoregulatory cytokine IL-10 has been shown to trigger “a modified Th2 response” by inducing differentiation of IgG4+ B cells and, in the presence of IL-4, to direct antibody class switching of B cells to secrete IgG4 (23, 24).
The association between induction of IL-10 and production of IgG4 antibodies has been shown in IgG4-related diseases and also in allergic individuals undergoing allergen immunotherapy (25). Th2-type inflammation in tumor tissues is dominated by IL-10–producing cells, such as Tregs and M2-type macrophages (26, 27). We therefore reasoned that these Th2-type tumor inflammatory microenvironments may favor “alternatively activated” humoral immunity and local expression of IgG4 antibodies.
In this study, we show mature B cells and IgG4 antibodies in melanoma lesions in the presence of key Th2-type cytokines that may trigger IgG4 production. Using engineered IgG1 and IgG4 antibodies of the same specificity against a tumor-associated antigen, we demonstrate the capacity of IgG4 to counteract antitumor immunity in vivo.
Results
CD22+ B cells infiltrate melanoma lesions and IgG antibodies are expressed in situ.
The presence of mature CD22+ B cells was observed in melanoma lesions by immunohistochemistry (Figure 1A). CD22+ cell infiltrates were found both within tumor lesions and in surrounding stroma areas rich in CD45+ cells, indicating that this population is part of the immune inflammatory infiltrate in tumors. Furthermore, CD22+ cells were also seen in close proximity to FoxP3+ cells, which may imply associations between regulatory elements and B cells in tumors, as reported in the context of cholangiocarcinoma (19). CD22+ cell infiltrates were found in 8 out of 9 melanoma lesions examined, and 6 of these CD22+ tumors were also positive for FoxP3 (Table 1). Quantitative immunohistochemical evaluations also confirmed infiltration of CD22+ B cells, associated with 6 out of 6 primary and 5 out of 7 metastatic tumor lesions, while we detected sporadic CD22+ infiltrates in 2 out of 8 healthy skin specimens (Figure 1B and Supplemental Figure 1, left panel; supplemental material available online with this article; doi: 10.1172/JCI65579DS1). Primary and metastatic melanoma tumors had ≥7 CD22+ B cells per high-powered microscope field, compared with very low or absent CD22+ B cell infiltration in healthy skin (P < 0.05 and P < 0.001, respectively; Figure 1B).
Figure 1. B cells (CD22+) infiltrate melanoma lesions and produce IgG.

(A) Immunohistochemistry showing the presence of lymphocytes (CD45+), mature B cells (CD22+), activated lymphocytes (FoxP3+) (alkaline phosphatase [red], hematoxylin [blue]), and colocalization of all 3 within cutaneous metastases (scale bar: 100 μm; original magnification, ×10). CD22+ cells in melanoma are shown at higher magnification (original magnification, ×40). (B) Significantly increased CD22+ B cell infiltration was measured in primary (n = 6) and metastatic (n = 7) melanoma lesions compared with healthy skin (n = 8). HPF, high-powered microscope field. (C) Comparative real-time PCR showed significantly elevated CD22 expression in primary (n = 10) and metastatic (n = 10) melanomas compared with healthy skin (n = 9). (D) Increased expression of mature IgG mRNA in metastatic melanoma lesions (n = 10) compared with primary melanomas (n = 10) and healthy skin (n = 9) measured by comparative real-time PCR analysis. (E) IgG expression (by comparative real-time RT-PCR) is elevated in melanoma lesions of stage IV patients compared with lesions of stage I–III patients. (F) Immunofluorescent evaluations of IgG+ B cells in human metastatic melanoma lesions (CD22+ B cells in red; left) (IgG+ cells in green; middle) and CD22+IgG+ B cell infiltrates (right). Scale bar: 10 μm; original magnification, ×63. (B and E) *P < 0.01, **P < 0.01, ***P < 0.001, Mann-Whitney U test. (C and D) *P < 0.05, **P < 0.01, ***P < 0.001, Kruskal-Wallis 1-way ANOVA with Dunn’s post-hoc test. Horizontal lines in box plots represent the mean, and whiskers indicate minimum and maximum values
Table 1.
Pathological evaluations of IgG4, CD22, and FoxP3 of melanoma lesions, with corresponding clinical parameters
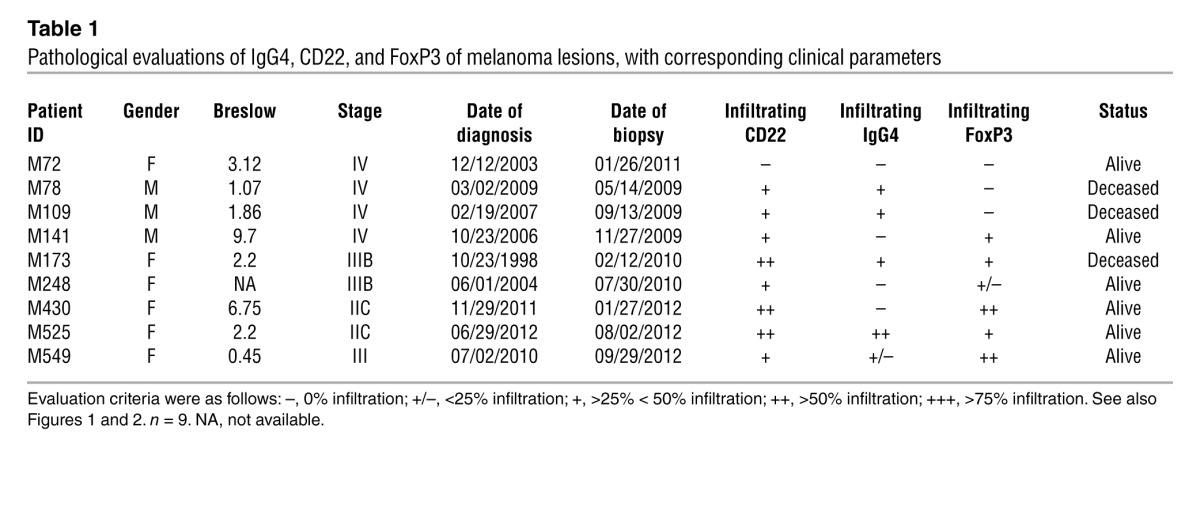
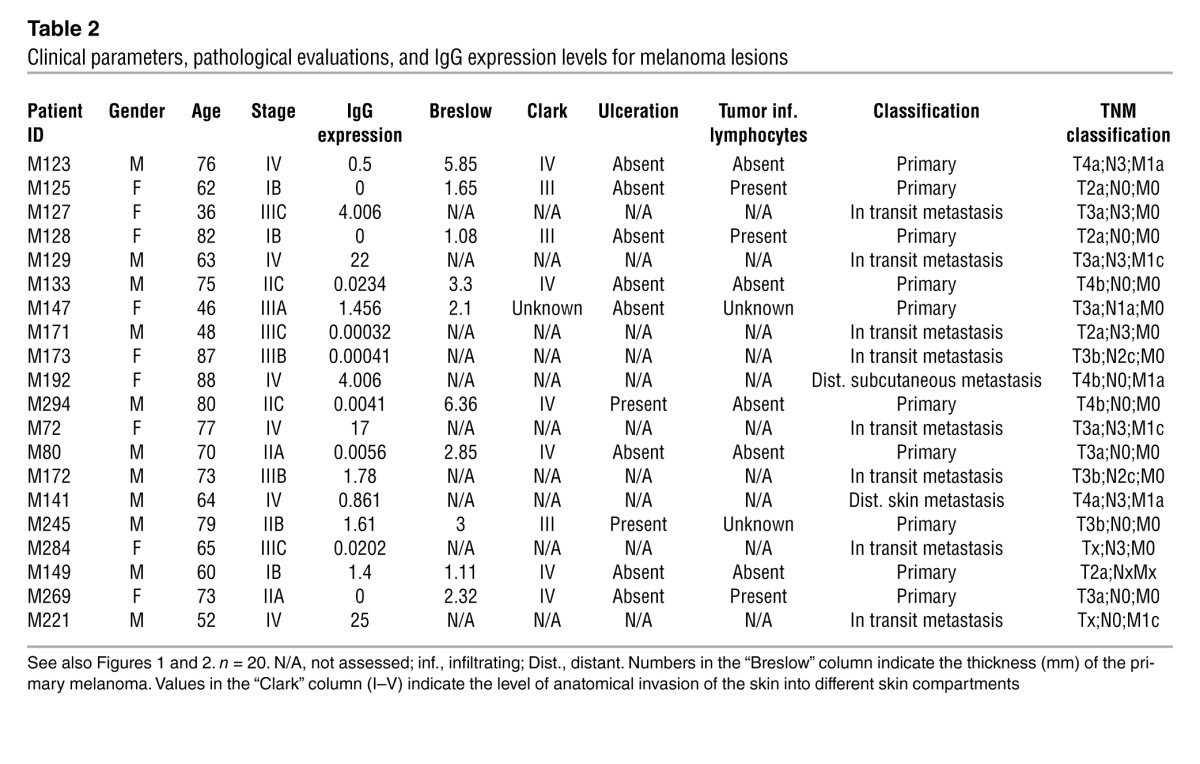
The presence of CD22+ B cells in melanoma lesions was also confirmed by quantitative RT-PCR analyses. CD22 mRNA expression was determined in healthy skin (n = 9), primary melanoma lesions (n = 10), and melanoma skin metastases (n = 10) (Figure 1C). We detected low to no expression of CD22 mRNA in healthy skin samples and significantly elevated levels in primary and metastatic melanoma skin lesions (P < 0.01 and P < 0.001, respectively), suggesting recruitment of mature B cells to tumors (Figure 1C). Levels of mature IgG mRNA were elevated in metastatic melanoma lesions (n = 10) compared with those in both primary melanomas (n = 10) (P < 0.05) and healthy skin samples (n = 9) (P < 0.001), in which there was little to no expression (Figure 1D and Table 2). When we analyzed IgG4 expression by disease stage, we found significantly higher expression of IgG mRNA in lesions from patients with stage IV disease (n = 6) compared with expression in lesions from patients at stages I–III (n = 14) (Figure 1E). Local expression of CD22+IgG+ B cell infiltrates was detected in melanoma lesions, as determined by dual-color immunofluorescence (Figure 1F). These data confirm that mature antibody-expressing B cells infiltrate melanoma lesions and that IgG antibodies are expressed locally in metastatic melanoma.
Table 2.
Clinical parameters, pathological evaluations, and IgG expression levels for melanoma lesions
Tumor-associated B cells are polarized to produce IgG4 antibodies in Th2-biased inflammatory melanoma microenvironments.
To examine the proportions of IgG subclasses produced in melanoma, B cells from melanoma skin lesions (n = 2, stages III and IV), patient lymph nodes (n = 3, stage III), patient blood (n = 6, stages III and IV), and healthy volunteer blood (n = 4) were cultured ex vivo (Tables 2 and 3 and Supplemental Table 1). IgG subclass titers (IgG1, IgG2, IgG3, IgG4) in culture supernatants were then measured by ELISA. Proportional IgG subclass expression and IgG4/IgGtotal ratios from healthy volunteer peripheral blood B cells (IgG1: 79.5% ± 8.5%; IgG2: 14.3% ± 7.9%; IgG3: 4.3% ± 1.5%; IgG4: 1.5% ± 1%), patient peripheral blood B cells (IgG1: 76.4% ± 14.3%; IgG2: 15.8% ± 10.2%; IgG3: 4.1% ± 3.2%; IgG4: 3.9% ± 2.2%), and B cells from patient lymph nodes (IgG1: 83.2% ± 3.6%; IgG2: 6.5% ± 0.5%; IgG3: 3.3% ± 1.0%; IgG4: 7.0% ± 2.1%) were consistent with those commonly detected in human sera (IgG4/IgGtotal ratio range: 0.01–0.083, each condition tested in triplicate) (Figure 2A and ref. 12). However, B cells derived from metastatic melanoma lesions exhibited higher proportional IgG4 subclass production (IgG4/IgGtotal ratio range: 0.209–0.334) (IgG1: 43.2% ± 1.7%; IgG2: 4.1% ± 1.4%; IgG3: 24.8% ± 3.5%; IgG4: 27.5% ± 5.3%) (Figure 2A).
Table 3.
Clinical parameters and pathological evaluations for lymph node specimens

Figure 2. B cells in melanoma lesions are polarized to produce IgG4 antibodies with reactivity against tumor cells.

(A) Polarization of IgG subclasses in B cells derived from metastatic melanoma skin tumor lesions (n = 2), patient lymph nodes (n = 3), peripheral blood from patients with melanoma (n = 6), and from healthy volunteers (n = 4). Cells were cultured ex vivo, and IgG subclass expression profiles were analyzed from culture supernatants by ELISA (mean ± SD; each sample condition tested in triplicate). (B) Immunohistological evaluations confirm significantly increased IgG4+ cell infiltration in melanoma lesions (n = 10) but not healthy skin (n = 10) (***P < 0.001), and representative metastatic melanoma depicts IgG4+ cells (red; hematoxylin [blue]) (scale bar: 20 μm; original magnification, ×10). (C) Reactivity of patient B cell–derived IgG1 and IgG4 antibodies to A375 tumor cells evaluated using a cell-based ELISA. Dashed black and gray lines indicate cut-off points for IgG1 and IgG4 tumor-reactive antibodies, respectively (lines defined as 2 SDs above isotype control antibodies). (D) Immunofluorescent staining of IgG4+ (red) colocalized with S100+ (green) cells in metastatic melanoma (scale bar: 50 μm; original magnification, ×40). (E) Increased expression of IL4, IL10 and IFNG in metastatic melanomas (n = 10) compared with primary melanomas (n = 10) and healthy skin (n = 9) by comparative real-time PCR. Horizontal lines in box plots represent mean, and whiskers indicate minimum and maximum values. *P < 0.05, **P < 0.01, Kruskal-Wallis 1-way ANOVA with Dunn’s post-hoc test. (F) Cytokines IL-4, IL-10 and IFN-γ are secreted in metastatic melanoma lesion ex vivo cultures.
Using immunohistochemical evaluations, we also detected substantial levels of IgG4-immunopositive cells infiltrating tumor areas in primary and metastatic melanoma lesions, while IgG4+ cells were rarely observed in healthy skin (Figure 2B, left, and Supplemental Figure 1, right panels). These observations were confirmed with quantitative assessments of sections, which showed IgG4 positivity in 9 out of 12 melanomas and sporadic expression in 2 out of 10 healthy skin specimens (P < 0.001; Figure 2B, right). Furthermore, immunohistochemical evaluations of IgG4+ infiltrates were examined in relation to clinical parameters for a cohort of 9 patients. Although these observations are limited by the small number of patients, it is noteworthy that 3 patients who are deceased demonstrated IgG4 positivity in the lesions tested (Table 1). Expression of IgG4 was also confirmed by RT-PCR sequence alignments of patient specimens (representative clone in Supplemental Figure 2). These data suggest that IgG4 production occurs in situ in the melanoma microenvironment.
We then asked whether IgG4 antibodies in the tumor microenvironment and in patient circulation may recognize tumor cells. For this, we examined the reactivities against tumor cells of IgG1 and IgG4 antibodies produced by B cells derived from patient blood (n = 2, patients in stage III and IV), cutaneous metastases (n = 3, 2 patients in stage III and 1 patient in stage IV), and a lymph node metastasis (n = 1, a patient in stage III) and cultured ex vivo for 5 days. Tumor cell reactivity evaluations were conducted using a previously described cell-based ELISA (28). We found detectable levels of IgG4 reactivity against A375 metastatic melanoma cells, above background set by human IgG4 antibody controls, in 1 blood sample and 1 cutaneous metastasis (Figure 2C). In the same specimens, we found no equivalent detectable reactivity of IgG1 antibodies against these tumor cells. In contrast, we detected IgG1 antibody reactivity to melanoma cells in the lymph node sample without detectable equivalent IgG4 reactivity. Additionally, using 2-color immunofluorescence on a cutaneous melanoma lesion, we demonstrated colocalization of IgG4 with the melanoma-associated antigen S100, further supporting the presence of tumor reactive IgG4 antibodies in situ (Figure 2D). These experiments, together with detected IgG4 expression in melanoma lesions by RT-PCR analysis (Supplemental Figure 2), suggest that tumor-reactive antibodies of the IgG4 subclass are expressed by patient B cells situated in both melanoma lesions and the circulation. We reasoned that the polarization of IgG4 in situ may be influenced by local factors, such as production of IL-10 in combination with IL-4, which is known to induce differentiation of IgG4+ B cells, drive class switching, and to maintain polarization of humoral immune responses in favor of IgG4 (29). Accordingly, we found significantly elevated mRNA expression of the Th2 cytokines IL4 and IL10 and of the inflammatory cytokine IFNG, reported to be involved in tumorigenesis, by quantitative RT-PCR in both primary (n = 10) and metastatic (n = 10) melanoma skin specimens when compared with healthy skin samples (n = 9) (P < 0.05; Figure 2E and ref. 30). Secretion of these cytokines was also confirmed in ex vivo cultures of tumor lesions (Figure 2F). These findings indicate a potential association of Th2-biased local expression of IL-10 and IL-4 with IgG4 production at inflammatory tumor sites.
Melanoma tumor cells trigger Th2-driven polarization, inducing B cells to secrete IgG4.
To elucidate whether melanoma cells can influence polarized expression of IgG4 by B cells, we attempted to recreate the proximity between B cells and tumor cells in an ex vivo stimulation assay. We cocultured melanoma patient peripheral blood B cells with autologous irradiated PBMCs in the presence or absence of A375 metastatic melanoma cells or human primary melanocytes. Similarly to that in B cells isolated from melanoma lesions, we detected increased IgG4/IgGtotal ratios from blood B cells cultured with PBMCs in the presence of melanoma cells (IgG1: 35.0% ± 5.6%; IgG2: 23.1% ± 0.9%; IgG3: 30.3% ± 4.6%; IgG4: 20.0% ± 1.1%), while IgG4/IgGtotal ratios remained low when B cells were cultured with PBMCs in the absence of melanoma cells (IgG1: 54.8% ± 3.5%; IgG2: 29.3% ± 8.6%; IgG3: 9.2% ± 1.3%; IgG4: 6.7% ± 1.2%) (n = 9, Figure 3A, left). When we compared IgG4 polarization of B cells plus PBMCs in the absence or presence of melanocytes (IgG1: 51.9% ± 2.9%; IgG2: 21.5% ± 4.2%; IgG3: 14.0% ± 1.5%; IgG4: 12.6% ± 2.8%), we observed a small, but not statistically significant, difference in IgG4/IgGtotal ratios with the addition of melanocytes, indicating that IgG4 polarization may be partly due to an allogeneic HLA mismatch effect in these assays. We demonstrated significant changes in IgG4 polarization (higher IgG4/IgGtotal ratios) when comparing B cells, PBMCs, and melanocytes with samples of B cells, PBMCs, and melanoma cells (P < 0.05). The overall titers of IgG antibodies produced in the presence or absence of melanoma cells in these assays were comparable (Figure 3A, right).
Figure 3. Ex vivo stimulation assays demonstrate that tumor cells polarize B cells to produce IgG4.

(A) IgG subclass production by patient peripheral blood B cells. Cells were cocultured with irradiated PBMCs with or without metastatic melanoma cells or primary melanocytes for 5 days. Culture supernatants were harvested, and IgG subclass expression profiles were analyzed by IgG subclass ELISA. IgG subclass fractions and total IgG concentrations were illustrated for each sample as mean ± SD of 3 independent experiments (all conditions performed in triplicates). *P < 0.01. Secretion of Th2 cytokines IL-10 and IL-4 from B cell cultures treated with or without (B) tumor cells or (C) primary melanocytes were analyzed by cytokine multiplex assay analysis. Both cytokines were significantly increased in cultures containing tumor cells but not in cocultures with melanocytes (*P < 0.01; **P < 0.01, Mann-Whitney U test; n = 9 replicates per coculture condition). Horizontal lines in box plots represent mean, and whiskers indicate minimum and maximum values. Comparative real-time PCR analysis of IL-4, IL-10, and VEGF expression by A375 melanoma cells isolated by flow cytometric sorting before (–) and after (+) cocultures with B cells and PBMCs shows (D) increased IL-10 expression by cocultured melanoma cells and (E) confirmation that A375 cells secrete IL-10 but not IL-4 in culture. ND, not detected. (F) Comparative real-time PCR analysis of IL-4, IL-10, and VEGF expression by B lymphocytes isolated by flow cytometric sorting from monocultures or from cultures with tumor cells, showing increased expression of VEGF expression by B cells when cocultured with tumor cells.
We next asked whether ex vivo production of IgG4 in the presence of melanoma cells is associated with Th2-biased inflammatory cytokines. Consistent with our findings in metastatic melanoma lesions, ex vivo stimulation of B cells with metastatic melanoma cells triggered enhanced IL-4 and IL-10 levels, as compared with B cell cultures in the absence of melanoma cells (P < 0.01 and P < 0.05, respectively) (Figure 3B). We also detected increased titers of VEGF, IL-6, and MCP-1, but not IFN-γ, in cocultures with melanoma cells (Supplemental Figure 3A). In contrast, when we compared IL-4 and IL-10 titers in cultures of B cells and PBMCs in the presence or absence of melanocytes, we did not observe any significant increases (Figure 3C). These findings indicate that melanoma cells drive Th2-biased inflammatory environments favoring increased IgG4/IgGtotal production ratios.
To further investigate the origin of Th2 cytokine production, we harvested A375 melanoma cells before and after coculture assays by flow cytometric cell sorting (strategy for flow cytometric sorting shown in Supplemental Figure 3B). We demonstrated increased expression of IL-10 by these cells following coculture with B cells and PBMCs. Message for IL-4 was not detected, and VEGF mRNA was present but was not upregulated following coculture (Figure 3D). When cultured alone under normal culture conditions, melanoma cells did not express or secrete IL-4, but the cells secreted detectable levels of IL-10 (Figure 3E). These assays point at enhanced expression of IL-10 by melanoma cells in these coculture assays. Neither B cells nor tumor cells expressed IL-4 message under these culture conditions, and therefore, IL-4 may be contributed by another cell type in PBMCs (Supplemental Figure 3C) or possibly stromal cells in the tumor microenvironment.
Having shown that tumor cells produce IL-10 in culture, next we investigated the cytokines produced by sorted B cells in these culture conditions (strategy for flow cytometric sorting shown in Supplemental Figure 3D). B cells in coculture experiments upregulated VEGF expression, while expression of IL-10 by these cells was detected but not upregulated following coculture with tumor cells (Figure 3F). These experiments suggest that B cells may contribute to polarized humoral immunity by enhancing production of VEGF.
IgG4 subclass antibodies lack antitumoral effector functions and block IgG1-mediated tumor cell killing by patient monocytes.
To explore potential mechanisms by which IgG4 may impact on antitumoral responses, we engineered 2 antibodies, with IgG1 and IgG4 Fc regions, that share identical variable regions recognizing the melanoma-associated antigen CSPG4. With these, we attempted to mimic a clinically relevant situation, in which IgG1 and IgG4 were found alone or together in the presence of tumor antigen-expressing cancer cells and FcγR-expressing immune effector cells, such as patient monocytes/macrophages (5, 19). Both antibodies bound to FcγR-expressing U937 monocytes, to patient monocytes (Figure 4, A–C), and to a panel of melanoma cells (Figure 4D).
Figure 4. Biological properties of engineered IgG1 and IgG4 antibodies recognizing the melanoma-associated antigen CSPG4.

Anti-CSPG4 IgG1 and anti-CSPG4 IgG4 antibodies bind to receptors on the cell surface of (A) U937 monocytic cells, (B) primary monocytes, and (C) tumor cells. ImageStream and flow cytometric evaluations confirm IgG1 and IgG4 antibody binding to Fcγ receptors (top and middle panels) and also to the antigen CSPG4 (bottom panel) expressed on the surface of SK-MEL-28 melanoma cells. ImageStream images are shown on the left. IgG FITC, cell surface (area indicated by circle on top left of each panel) binding of antibodies on cells; BF, bright-field image of cells; IgG FITC/BF, composite image of antibody and bright-field images, confirming anti-CSPG4 antibody binding to the cell surface. Quantitative analyses are based on the acquisition of 20,000 cells. Control samples for U937 and monocytes were incubated with goat anti-human IgG (Fab′)2-FITC antibody. Control samples for antibody binding to tumor cells were human IgG1 and IgG4 isotype antibodies of irrelevant specificity. (D) Flow cytometric analysis of CSPG4 IgG1 or CSPG4 IgG4 antibody binding to a panel of tumor cell lines or primary human melanocytes. Antibody binding was detected with an anti-human IgG (Fab′)2-FITC, and specific binding was depicted by MFI values above isotype control antibody binding.
First, we examined the ability of each antibody to activate patient-derived monocytes to kill tumor cells in vitro. Patient monocytes incubated with anti-CSPG4 IgG4 and A375 melanoma cells mediated nonsignificant tumor cell killing compared with a nonspecific IgG4 (antibody clone SP211-IgG4) after 3 hours (n = 6). In contrast, the corresponding anti-CSPG4 IgG1 of the same specificity triggered statistically significant increases in phagocytosis (antibody-dependent cellular phagocytosis [ADCP]) (21.1% ± 2.5%) of melanoma tumor cells by monocytes compared with anti-CSPG4 IgG4 (3.2% ± 0.3%) and controls (2.6% ± 0.1%) (P < 0.001; Figure 5, A and B). Consistent with reduced IgG4 antitumor activity, we detected lower levels of IFN-γ (P < 0.01), IL-1β (P < 0.01), and TNF-α (P < 0.05) in supernatants from cultures treated with anti-CSPG4 IgG4 compared with those treated with anti-CSPG4 IgG1 (n = 6) (Figure 5C).
Figure 5. IgG4 irrespective of tumor specificity is ineffective in triggering anti-melanoma effector functions in vitro and prevents IgG1 from mediating tumor cell killing.

(A) Dot plots depicting CFSE+ tumor cells and CD89-PE+ monocytes (effector cells) were used to quantify CFSE+ tumor cells present within PE+ effector cells, indicating phagocytosis (ADCP) (CFSE+/PE+ cells) (top panel). CFSE+/DAPI+ double-positive tumor cell events indicate tumor targets killed by cytotoxicity (ADCC, bottom panel) by IgG1 but not IgG4. (B) Quantification of tumor antigen-specific IgG1 and IgG4 antibody induced ADCC/ADCP by monocytes. Tumor antigen-specific IgG1 mediated tumor cell killing compared with an unspecific control antibody or cells alone. The corresponding anti-CSPG4 IgG4 antibody induced no tumor cell killing above isotype control levels. (C) Titers of secreted IFN-γ, IL-1b, and TNF-a, but not of IL-10, were higher in culture supernatants of monocytes and tumor cells stimulated with anti-CSPG4 IgG1 antibodies compared with those stimulated with anti-CSPG4 IgG4 or controls. All samples were tested in duplicates; mean ± SD (n = 6). (D) Tumor antigen-specific IgG1 against the melanoma antigen CSPG4 can induce monocytes to kill tumor cells, but addition of anti-CSPG4 IgG4 blocks the antitumor functions of the same concentrations of IgG1. (E) Similar blocking of IgG1-mediated tumor cell killing can be observed when CSPG4 IgG1 is coincubated with a nonspecific human IgG4 antibody. (B, D, and E) All assays were performed in triplicates. Data represent percentage tumor cell killing (mean ± SD) (n = 6). *P < 0.05, **P < 0.01; ***P < 0.001.
When anti-CSPG4 IgG1 and anti-CSPG4 IgG4 were combined with patient-derived monocytes and melanoma cells, the tumoricidal capacity of anti-CSPG4 IgG1, previously observed in the antibody-dependent cellular cytotoxicity/ADCP (ADCC/ADCP) assays, was significantly reduced in the presence of anti-CSPG4 IgG4 (P < 0.001, Figure 5D). This suggested that anti-CSPG4 IgG4 is not only incapable of triggering effector cell–mediated tumor killing but may additionally suppress tumor cell killing capacity of tumor-specific IgG1. To evaluate whether specificity for the tumor antigen was necessary for an IgG4 antibody to block IgG1 effector cell functions against tumor cells, we performed ADCC/ADCP (cytotoxicity/phagocytosis) assays, incubating tumor cells and patient-derived monocytes with anti-CSPG4 IgG1 in combination with a human IgG4 antibody of irrelevant specificity. Nonspecific IgG4 partly hindered tumor-specific IgG1 tumor cell killing (P < 0.001; Figure 5E), suggesting that tumor cell specificity may only be partly responsible for the antibody modulatory properties of IgG4. These data are in line with known properties of IgG4 interactions with Fc regions of immunoglobulins, and they are also consistent with reports that only a small fraction of IgG4 antibodies are allergen specific in allergic subjects following immunotherapy (31). In summary, IgG4 antibodies lack antitumoral effector functions and impair IgG1-mediated tumor cell killing by patient monocytes.
IgG4 blocks IgG1 tumor cell killing by inhibiting IgG1 binding and activation through FcγRI.
In order to elucidate whether IgG4 antibodies could hinder binding of IgG1 on the surface of effector cells, we performed a competition binding assay using monocytic cells. This showed decreased IgG1 binding on the surface of human monocytes with increasing concentrations of IgG4 (Figure 6A), suggesting that IgG4 competes with IgG1 for FcγR binding on the surface of effector cells.
Figure 6. IgG4 blocks IgG1 antibody-dependent tumor cell killing by inhibiting IgG1 binding and activation through FcγRI.

(A) Competition assay of IgG1 binding on the surface of monocytic cells displaced by addition of increasing concentrations of IgG4 antibody. Proportion of cells binding IgG1 is decreased with increasing concentrations of IgG4, demonstrated by flow cytometric evaluations and representative confocal images (yellow, by ImageStream). (B) Anti-CSPG-4 IgG1-mediated tumor cell killing (by flow cytometry) is inhibited by addition of an antibody known to block IgG Fc binding to FcγRI but not with addition of blocking antibodies to FcγRII or to FcγRIII. (C) Inhibitory functions of anti-CSPG-4 IgG4 are not lost by blocking FcγRII or FcγRIII with previously described specific FcγR blocking antibodies in flow cytometric antibody-dependent tumor cell killing assays. (D) Protein extracts of primary human monocytes isolated by flow cytometric sorting at different times during the antibody-mediated tumor cell killing assay were examined for phosphorylated products of the FcγR signaling pathway. Western blots of phospho-proteins and band density quantifications relative to freshly isolated monocytes demonstrate that IgG4 inhibits the activatory signaling cascades of FcγR (Src, AKT, MEK), while lack of pSHIP implies that FcγRII signaling is not involved in the IgG4 blockade. (B and C) Data are representative figures of 3 independent experiments.
In order to investigate which FcγR is predominantly responsible for the induction of tumor cell killing via IgG1, we conducted tumor killing assays during which individual FcγR families were blocked with antibodies described in the literature to have an inhibitory capacity for these specific receptor families. We demonstrate that, in this experimental system, blockade of FcγRI, but not of the other 2 receptor families, was responsible for the IgG1-mediated tumor cell killing (Figure 6B). We confirmed the widespread distribution of FcγRI along with the other 2 FcγRs (FcγRII and FcγRIII) in melanoma lesions (1 representative of 4 patient specimens shown in Supplemental Figure 4). Since FcγRs such as FcγRIIb are known to play immunomodulatory roles, we investigated whether specific inhibition of IgG binding to each of these receptor families had any bearing on the killing inhibition mediated by IgG4. In these experiments, we found that inhibition of CD32 is not involved in IgG1 blocking by IgG4 (Figure 6C).
Next, at different time points during in vitro tumor cell killing assays, we harvested monocytes by flow cytometric sorting and assessed phosphorylation of downstream signaling events triggered through engagement of FcγR engagement and activation (32). We observed increased phosphorylation of known members of the FcγR activatory signaling cascades (Src, MEK, and AKT) in the presence of IgG1 compared with the presence of both IgG1 and IgG4 antibodies (33). On the contrary, there were no differences in the levels of phosphorylated SHIP1 in relation to CSPG4 IgG1 or in relation to coincubation with IgG1 and IgG4 antibodies together at any of the time points tested (Figure 6D). These data suggest that inhibition of IgG1-mediated tumor cell killing by IgG4 is attributed to lack of effector cell activation rather than induction of inhibitory signals via FcγRs such as FcγRIIb. We conclude that in this experimental system IgG4 functions by competing with IgG1 for FcγRI binding and therefore blocking the activatory cascades through reduced phosphorylation of Src, MEK, and AKT.
IgG4 impairs antitumor immunity in a human melanoma xenograft model.
Next, we compared the efficacies of anti-CSPG4 IgG1 and IgG4 in vivo in a subcutaneous human melanoma xenograft mouse model engrafted with human immune effector cells (Supplemental Figure 5 and ref. 34). Intravenous treatment with anti-CSPG4 IgG1 (3 × 10 mg/kg weekly doses) significantly reduced tumor size compared with treatments with nonspecific mAb or vehicle controls (n = 6, P < 0.05; Figure 7A). However, tumor size in anti-CSPG4 IgG4–treated mice increased in line with that in controls, suggesting that IgG4 was significantly less effective than IgG1 at restricting tumor growth in vivo (P < 0.001). Furthermore, when anti-CSPG4 IgG1 and IgG4 were administered together, tumors grew similarly to vehicle-treated controls (n = 6, P < 0.001 for IgG1/IgG4 combinations compared with IgG1 treatment alone; Figure 7A), indicating that anti-CSPG4 IgG4 might counteract IgG1 antitumor functions in vivo.
Figure 7. IgG4 has no antitumoral effector functions in vivo and blocks tumor-specific IgG1 function.

(A) Anti-CSPG4 IgG1 antibody is capable of restricting the growth of subcutaneous melanoma lesions in NSG mice engrafted with human immune effector cells, while tumors in mice treated with an anti-CSPG-4 IgG4 antibody or coadministered with anti-CSPG4 IgG1 and IgG4 antibodies grow similarly to those from mice treated with nonspecific antibody or vehicle alone (n = 7 mice per group; mean ± SEM tumor volume in mm3). *P < 0.05, ***P < 0.001, 2-way ANOVA with Bonferroni post-hoc test. Data are representative of 2 experiments. (B) Representative NanoSPECT/CT images of Indium-111–labeled anti-CSPG4 IgG4 antibody (red), demonstrating accumulation of antibody in subcutaneous melanoma lesions at 20 minutes, 8 hours, and 24 hours following intravenous administration (n = 3). (C) Representative immunohistological images and (D) quantitative analyses of sections from human melanoma tumors grown in NSG mice treated with vehicle alone, anti-CSPG4 IgG1, anti-CSPG4 IgG4, or a combination of anti-CSPG4 IgG1 plus anti-CSPG4 IgG4 antibody, demonstrating elevated levels of human CD68+ (brown) immune cell infiltration in human xenograft tumors in animals treated with anti-CSPG4 IgG1 antibody. Horizontal bar indicate the mean, and individual symbols indicate individual tumors. Scale bar: 50 μm; original magnification ×20. ***P < 0.001.
To ascertain that impaired antitumoral functions of anti-CSPG4 IgG4 were not due to inability of IgG4 to localize in CSPG4-expressing tumors in the mice, 111In radio-labeled anti-CSPG4 IgG4 biodistribution was monitored by PET/SPECT imaging of human xenografts (n = 3). Accumulation in tumors was observed within 24 hours following antibody treatment (Figure 7B). Furthermore, we observed that subcutaneous human melanoma xenografts from mice treated with anti-CSPG4 IgG1 attracted higher levels of human CD68+ cells (macrophages) compared with mice treated with IgG4 or combinations of IgG1 and IgG4 antibodies (Figure 7, C and D). These findings indicate that macrophages may be recruited and potentially play a role in restricting tumor growth when engaged by CSPG4 IgG1 in situ.
Serum IgG4 antibodies may be predictive of clinical outcomes in melanoma.
Having investigated the immunomodulatory potential of IgG4 in melanoma in vitro and in vivo, we then evaluated whether the presence of IgG4 in patient circulation could be used as a possible biomarker. We performed IgG1–IgG4 subclass Luminex bead array assays using sera from 33 patients with stage III and IV melanoma (Figure 8 and Table 4). Spearman correlation analyses were performed to examine any significance among IgG subclass titers in sera in relation to patient survival. We demonstrated a negative correlation only between IgG4 serum titers and patient survival (r = –0.31, P < 0.05), while no significant correlations were found in relation to the other IgG subclasses (Figure 8A).
Figure 8. Elevated levels of IgG4 antibodies in patient sera correlate with patient survival.

(A) IgG subclass ELISA evaluations of sera from 33 patients with stage III and IV melanoma were evaluated using Spearman correlation analyses for IgGsubclass/IgGtotal, fractions in relation to patient survival (months), showing a statistically significant negative correlation between IgG4 fraction only (r = –0.32; P = 0.00353). (B) Kaplan-Meier cumulative survival analysis of overall patient survival was compared with respect to IgG4/IgGtotal fractions (stratified according to fractions above (blue) or below (black) the 75 percentile of the IgG4/IgGtotal fractions measured from the 33 metastatic melanoma patient cohort), indicating that high IgG4/IgGtotal ratios in sera are associated with a significantly lower overall survival (P = 0.003; hazard ratio 0.19; 95% CI 0.0635–0.5685).
Table 4.
Clinical parameters and disease staging at the time of sampling for peripheral blood donor cohort used for serum detection of IgG subclass and correlations
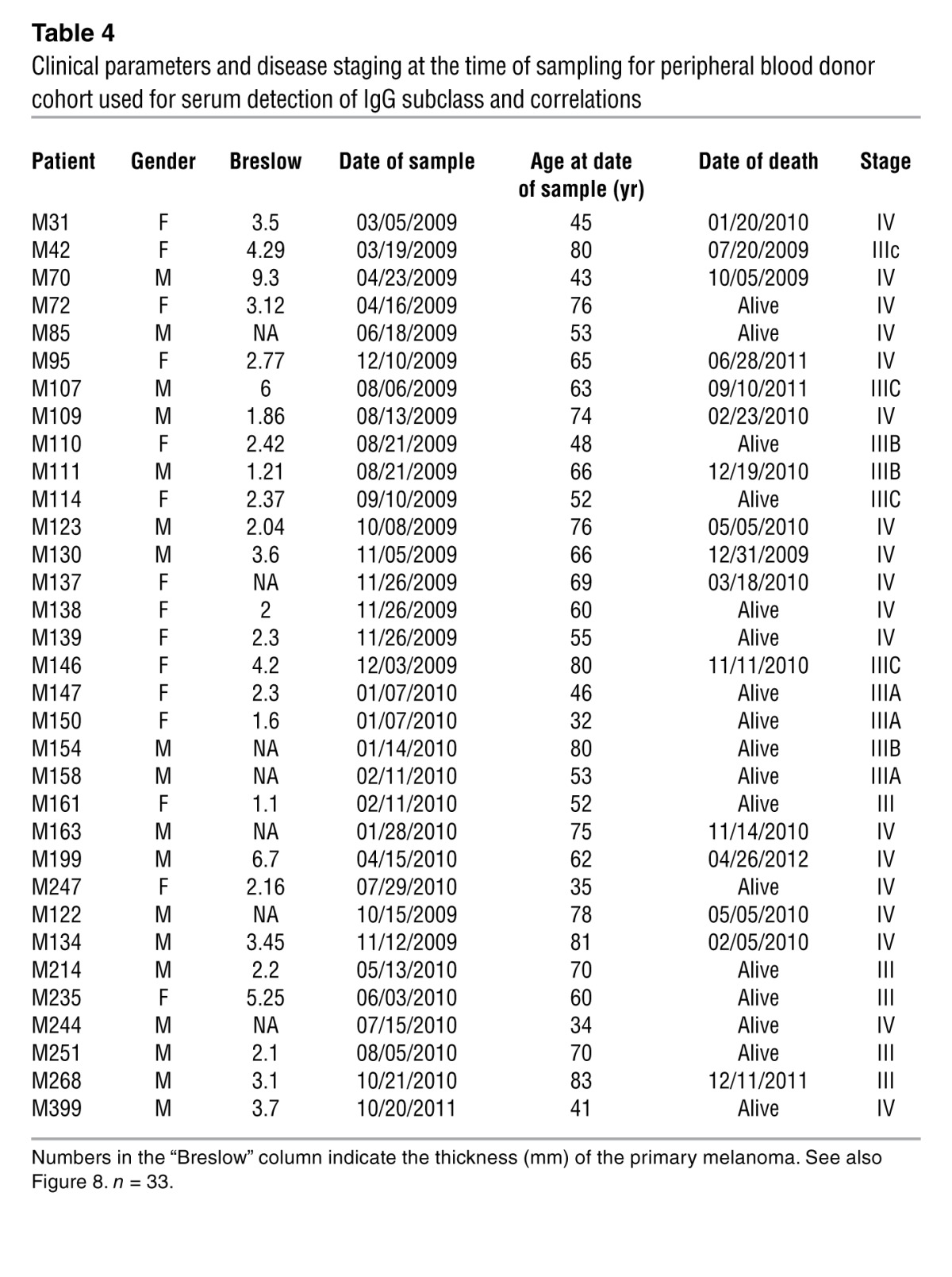
Normal IgG4/IgGtotal ratios in human sera have been reported around 0.025 (35). In this study, we confirmed this (Figure 2A) and additionally found that IgG4/IgGtotal ratios in patient sera had a median of 0.034 (95% CI of mean, 0.024–0.044). We therefore reasoned that serum IgG4/IgGtotal ratios above the 75% percentile (IgG4/IgGtotal = 0.04) may be considered high in patient sera (Supplemental Figure 6). To gain an insight into the significance of IgG4/IgGtotal ratios in patient sera, we analyzed cumulative survival on a Kaplan-Meier survival plot in our 33 patient cohort. We found that patients with higher serum IgG4/IgGtotal ratios (>0.04, n = 13) had significantly lower survival rates (median survival 8 months; hazard ratio, 0.19; 95% CI, 0.0635–0.5685) compared with patients whose IgG4/IgGtotal serum ratios were <0.04 (n = 20) (P < 0.01) (Figure 8B).
Overall, we have shown that IgG4 subclass antibodies are present in tumor lesions and that they are inefficient in restricting melanoma tumor cell growth. In addition, IgG4 suppressed IgG1-mediated melanoma cell killing in vitro and in a clinically relevant humanized mouse engrafted with human melanoma. When examined in relation to clinical outcomes, data presented here suggest that IgG4 is associated with reduced survival and may be evaluated as a prognostic biomarker in melanoma.
Discussion
Despite the fact that B lymphocytes constitute important sentinels of adaptive immunity and the antibody-mediated immune response, the mechanisms by which they participate in tumor immunity remain only partially understood (2–4, 36–39). We described a hitherto undiscovered aspect of local humoral immunity in melanoma, characterized by B cell infiltration and local expression of IgG4 antibodies, in the context of prominent expression of IgG4 polarizing cytokines IL-10, IL-4, and VEGF. We demonstrated that, in the presence of melanoma cells, production of these cytokines is enhanced and B cells are polarized to produce IgG4 ex vivo. Importantly, we showed that IgG4 antagonizes IgG1-mediated human anti-melanoma immunity in vitro and in vivo and that IgG4 blockade is mediated through competition with IgG1 for FcγR binding and therefore inhibits downstream signaling activation of effector cells (i.e., monocytes).
Previous studies have reported the presence of CD20+ cells in melanoma lesions (2, 3), but this pan–B cell marker is also expressed by melanoma stem cells (40, 41). We selected the B cell marker CD22 to differentiate B cells from melanoma stem cells and to study mature B cells that may express class-switched antibodies. We detected CD22 at the mRNA and protein levels in melanoma lesions but at very low levels in normal skin (Figure 1). These cells were observed within tumor lesions and also surrounding stroma areas rich in CD45+ cells, indicating that B cells form part of the immune inflammatory infiltrate; however, the significance of CD22+ B cell infiltrates in cancer remains unclear. B cell and plasma cell (CD138) infiltration in melanoma have been associated with favorable clinical outcomes, and lymphoid structures containing B cells (CD20) and also plasma cells (CD138) were recently reported (4, 42). Other reports highlight immunomodulatory roles for B cells in cancer, suggesting that their contribution may reflect a balance between effective immunity and tumor-induced inflammation (20, 43).
The presence of mature IgG mRNA in metastatic melanoma lesions and CD22+IgG+ and IgG4+ cells infiltrating tumor cell areas suggest that B cells may participate in local tumor surveillance through production of antibodies. This study demonstrates the expression of IgG antibodies in situ rather than being sequestered or diffused into tumors from the circulation. This was shown by the detection of mature IgG mRNA, IgG4 clone expression, and IgG4+ infiltrates in melanoma lesions as well as by the production of tumor-reactive IgG4 antibodies by tumor-resident B cells (Figure 2). Recent findings implicate increased IgG4 serum titers and IgG4+ tissue infiltrates in a range of inflammatory conditions triggered by chronic exposure to nonmicrobial antigens (14, 18, 31). Our findings demonstrating increased expression of IgG, the presence of IgG4+ infiltrates in tumors, and tumor-reactive B cells in lesions are consistent with exposure to tumor antigens, as this might allow for antibody class switch recombination (44).
We found a striking polarization in IgG subclass distribution produced by tumor-derived B cells in favor of IgG4 (Figure 2). We directly showed a role for tumor cells in influencing IgG4 production by B cells (Figure 3). Furthermore, ex vivo stimulation of human B cells with melanoma cells also resulted in a similar proportional increase in IgG4/IgGtotal ratios compared with unstimulated B cells, and we demonstrated that melanoma tumor cells, but not normal human melanocytes, are capable of polarizing B cell responses in favor of IgG4 production. In melanoma tumor lesions, these increased IgG4/IgGtotal ratios were accompanied by expression of Th2-biased inflammatory cytokines known to drive production of IgG4 by B cells. Tumors circumvent immune-mediated clearance by triggering inflammatory cells with immunoregulatory properties and immunosuppressive mediators (e.g., IL-10, VEGF, TGF-β) in situ (34, 45, 46). The immunoregulatory cytokine, IL-10, can direct “a modified Th2 response” favoring B cell production of IgG4 (23, 25, 47). In support of this, we have found induction of increased IL-10 production by tumor cells cultured with B cells ex vivo. IL-10 has been implicated in modified Th2 responses in the context of IgG4-related diseases (48, 49). Although IgE and IgG4 are triggered by similar Th2-biased signals, IL-10 is thought to divert a classical Th2-type immunity, bypassing IL-4–induced IgE responses in favor of IgG4 (23, 29). Classically, high IgG4/IgGtotal ratios are considered a natural effort by the immune system to contain immune activation. These alterations, however, have also been documented in individuals chronically exposed to occupational or environmental antigens and are not normally accompanied by development of IgE (50, 51). Consistent with this, we detected IgE mRNA in only 2 out of 10 patient lesions that we examined (data not shown), which also points to the involvement of altered Th2 responses in tumor microenvironments (52).
As reported by others in the context of IgG4-related diseases, we detected local expression of tissue-resident IgG4 together with enhanced expression of IL-10 and IL-4 cytokines in tumors (Figure 3 and ref. 49). We also demonstrated that tumor cells are capable of stimulating such modified Th2 responses, featuring production of IL-10 and IL-4 and secretion of IgG4 by B cells. Also consistent with a tumor-associated inflammatory environment, VEGF, known to polarize Th2-biased cytokine production by PBMCs through induction of IL-10–secreting cells such as Tregs (53), along with well-described tumor-induced inflammatory mediators IL-6 and MCP-1 (53–56), was found at significantly increased levels (P < 0.001; Supplemental Figure 3A), upregulated by B cells in cultures of B cells with melanoma cells compared with cultures without tumor cells. While tumor cells contributed enhanced expression of IL-10 in coculture assays, neither B cells nor tumor cells expressed IL-4 under these coculture conditions, and it is possible that IL-4 production may be contributed by other stromal or immune cells in tumor microenvironments. Our findings therefore identify melanoma tumor cells as potential inducers of IgG4 in tumor microenvironments via B cell–produced VEGF, triggering enhanced IL-10 production by tumor cells.
Insights into how IgG4 antibodies contribute to immune homeostasis and to different disease pathologies are starting to emerge. IgG4 is considered a “nonactivating” subclass associated with chronic inflammation and limited immune activating functions (57). Links between IgG4 and tissue inflammation (14), in the context of well-characterized inflammatory mechanisms at play in tumors, raise the possibility of a key role for this antibody subclass in tumor inflammation. In support of this concept, we have shown the local presence of IgG4 antibodies in melanoma and their contributions to tumor inflammatory responses. In addition, we found that tumor antigen-specific IgG1, but not the corresponding IgG4, could activate patient-derived monocytes to kill tumor cells in vitro (Figure 5). These findings were confirmed by in vivo experiments, since tumor antigen-specific IgG4 was unable to restrict tumor growth, despite accumulating in tumor lesions (Figure 7). Polarizing humoral responses in favor of IgG subclasses with low or no potency in activating effector cells against tumors may therefore represent a mechanism of tumor escape.
Another potential mechanism relates to IgG4 interactions with other IgG antibodies and its capacity to impede IgG functions that would otherwise activate effector cells to eradicate tumors. We observed that a tumor antigen-specific IgG4 antibody hindered the ability of the corresponding IgG1 to induce ADCP of tumor cells by patient monocytes. This may be the outcome of one or more potential scenarios. First, IgG4 may compete with IgG1 for tumor antigen recognition, and, in support of this, we found tumor-reactive IgG4 antibodies associated with melanoma lesions. Second, IgG4 could bind to Fcγ receptors, preventing IgG1 interactions on the surface of effector cells. The implications of the latter mechanism would be that IgG4 of any specificity could neutralize IgG1-FcR–mediated effector functions against tumors through engagement of the main Fcγ receptor families (58). In agreement with this possibility, a nonspecific IgG4 also blocked IgG1-mediated tumor cell ADCP by patient monocytes almost as effectively as the tumor antigen-specific IgG4 (Figure 5). This suggested that antigen specificity may only partly account for the IgG4 antibody-neutralizing properties (31, 59, 60). Furthermore, here we demonstrated that tumor antigen-specific IgG4 inhibited IgG1-dependent tumor cell killing by blocking activatory signal cascades associated with FcγRI effector mechanisms, which would normally lead to functions such as ADCP (Figure 6). One can therefore envisage that if IgG4 antibodies are abundant in tumor microenvironments, their proximity to tumor cells may not only inhibit the effector functions of naturally expressed antibodies, but IgG4 could also impair therapeutic antibody effector functions in situ by one or more mechanisms, as has already been reported following antibody treatments for rheumatoid arthritis (61, 62). Our findings are also consistent with similar reported functional roles of IgG4 in blocking immune responses in atopic individuals receiving allergen immunotherapy (18, 31). While these effects signal the relief of symptoms and successful allergen immunotherapy, the presence of IgG4 in tumor microenvironments and its antibody-neutralizing functions may contribute to evasion of the humoral immune response to cancer.
Further supporting the potential significance of IgG4 in melanoma growth are our findings that elevated levels of IgG4 in patient sera are associated with poorer survival (Figure 8). Data presented here mandate a closer examination of IgG4 antibodies but also of IgG3 antibodies, which are also proportionally elevated in tumors and possess lower effector cell activatory properties compared with IgG1 antibodies (63), as potentially useful biomarkers in tumor lesions and/or in the circulation. Additionally, data demonstrating VEGF and IgG4 production by B cells in the presence of tumor cells imply that B cells may be “reeducated” through cross-talk with tumor cells and highlight the need to further dissect the mechanisms by which tumors escape humoral immune responses.
In conclusion, we present the first evidence of local IgG4 expression in melanoma, associated with the IgG4-polarizing cytokines IL-10, VEGF, and IL-4. Our data suggest that IgG4 antibodies triggered in the presence of tumor cells participate in local inflammation and may represent one regulatory mechanism by which tumors evade immune attack. Studies in larger cohorts of patients are required to elucidate associations of IgG4 with immunological, molecular, and clinical parameters and patient responses to treatments, paving the way for biomarker development and design of personalized medicine approaches. Future strategies to counteract IgG4 immunoregulatory pathways or to design antibody treatments and derivatives less prone to IgG4 blockade will be of potential therapeutic value.
Methods
Patient data and specimen collection.
Melanoma tumor skin lesions, lymph nodes, and blood were collected from a total of 57 patients diagnosed with stage I–IV malignant melanoma (Tables 1–3 and Supplemental Table 1). Patients were staged and classified according to the American Joint Committee on Cancer Melanoma Staging and Classification criteria (64). Samples were used fresh, or placed in OCT for cryosectioning or in RNAlater solution (Life Technologies) and stored at –70°C for subsequent RNA extraction, or fixed in formal saline and embedded in paraffin. Discarded human skin samples were obtained from 18 volunteers undergoing routine plastic surgery. Human peripheral blood lymphocytes (PBLs) and B cells were isolated from a cohort of 8 healthy volunteers. Human samples were collected with informed written consent, in accordance with the Helsinki Declaration, and study design was approved by the Guy’s Research Ethics Committee, Guy’s and St. Thomas’ NHS Foundation Trust.
Human cell isolation and ex vivo stimulation assays.
Peripheral blood B cells from patients with melanoma and healthy volunteers were isolated using the B cell enrichment cocktail (Stemcell Technologies) and cultured at a density of 500 cells per well (in 96-well plates) in combination with (3,010 cGy) irradiated autologous PBMCs, Epstein-Barr virus, and the TLR9 agonist CpG 2006, as previously described (28). Single cell suspensions were derived from patient tissues (lymph nodes or melanoma skin lesions) using a Gentle MACs Tissue Dissociator (Miltenyi) and filtered through a 100-μm strainer. In ex vivo stimulation experiments, human peripheral blood B cells were cocultured for 5 days together with irradiated PBMCs and tumor cells at 1:5:10 ratios in RPMI 1640 medium, 10% FCS, 2 mM l-glutamine, penicillin (5,000 U/ml), and streptomycin (100 μg/ml) (all Life Technologies) (65). Primary human monocytes from patients with melanoma were obtained using the monocyte enrichment cocktail (Stemcell Technologies) according to the manufacturer’s instructions (66).
Cell culture and reagents.
Cell lines were obtained from ATCC. Media for maintaining the melanoma cell lines were as follows: DMEM, 10% FCS for A375 (CRL-1619) and A-2058 (CRL-11147); McCoy’s medium, 10% FCS for G-361 (CRL-1424); MEM, 10% FCS for WM-115 (CRL-1675) and SK-MEL-28 (HTB-72); and Iscove’s modified Dulbecco’s medium, 20% FCS for Malme-3M (HTB-64). Primary human melanocytes (PCS-2000-012) were grown in Dermal Cell Basal Medium (ATCC) and supplemented with the Melanocyte Growth Kit (ATCC). The monocytic cell line U937 (CRL-1593.2) was grown in DMEM, 10% FCS. Cells were grown in 2 mM l-glutamine, penicillin (5,000 U/ml), and streptomycin (100 μg/ml). The B95-8 marmoset blood leukocyte cell line secreting Epstein-Barr virus was grown in RPMI 1640 medium, 10% FCS, 2 mM l-glutamine, penicillin (5,000 U/ml), and streptomycin (100 μg/ml). All cells were maintained in a 5% CO2 humidified incubator at 37°C.
RNA isolation from patient samples.
Specimens placed in RNAlater solution (Life Technologies) were stored at –70°C prior to RNA extraction. RNA isolation was performed using a Qiagen Homogenizer II and an RNeasy Kit (Qiagen). Reagents were from Qiagen unless otherwise indicated. Tissue was removed from RNAlater, placed in a 2-ml microfuge tube with lysis buffer, and homogenized. Total RNA was prepared using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s protocol. A total 500 ng of RNA was reverse transcribed into complementary DNA, using SuperScript II Reverse Transcriptase and OLIGO(dT) primer mix (Life Technologies) according to the manufacturers’ instructions.
RNA and protein isolation from in vitro assays.
Cells used in coculture experiments were labeled either with CSPG4-IgG-APC (Miltenyi) and CD45-PE-Cy7 (BD Biosciences) or with CSPG4-IgG-APC, CD45-PE-Cy7, and B cell lineage (CD19, CD22)-FITC and sorted using an Aria II (BD Biosciences). Cells from the ADCC/ADCP assay were labeled with anti-CD14-PE-Cy7 or CD89-PE. Sorted cells were resuspended either in RLT buffer for RNA isolation following the RNeasy Kit (Qiagen) or in Cell Lysis buffer (Cell Signaling) supplemented with 5 mM PMSF for protein isolation.
Western blot.
Protein lysates were separated by SDS-PAGE using 4%–15% polyacrylamide minigels (Bio-Rad) and transferred onto PVDF membranes using a Bio-Rad Trans-Blot Turbo Transfer system (Bio-Rad). Membranes were blocked with 5% skimmed milk in PBS-Tween 20 and incubated with rabbit antibodies specific for the unphosphorylated and phosphorylated forms of Src, Akt, MEK(1/2), and SHIP-1 (all from Cell Signaling) in 1% skimmed milk-PBS-Tween 20 according to manufacturer recommendations. Rabbit antibodies were detected with goat anti-rabbit-HRP (Cell Signaling) and visualized with the ECL Detection Kit (GE Healthcare) on a Hyperfilm (Amersham).
Cell-based ELISA.
Tumor cell–reactive antibodies were detected using cell-based ELISA, as previously described (28). Briefly, A375 tumor cells were plated at 3 × 105 to 6 × 105 cells on 96-well flat-bottom tissue culture plates, fixed in 0.5% formaldehyde, and stored at –80°C. On the day of the assay, plates were thawed, blocked with a 5% skimmed milk/PBS solution for 2 hours, and incubated with 100 μl of ex vivo culture supernatants, nonspecific IgG (125 μg/100 μl), or tumor-specific IgG1 or IgG4 antibodies (0.05 μg/100 μl) for 90 minutes at room temperature. Bound tumor cell–reactive antibodies were detected using a mouse anti-human IgG1 (AbD Serotec, 1:400) or mouse anti-human IgG4 (BD Biosciences, 1:250) antibody. This was followed by a 45-minute incubation with a goat anti-mouse IgG PE-labeled F(ab)′2 Fc-specific antibody (Jackson ImmunoResearch, 1:350). Fluorescence intensity depicting tumor cell reactivity was measured on an ELISA reader (BMG Labtech; excitation 488 nm, emission 540 nm). All samples were in duplicates, and MFIs were corrected against background and normalized against the MFI of the isotype control antibody. Antibodies were defined as reactive against tumor cells when the measured MFI was 2 SDs above the MFI of the specific isotype control antibody.
Quantitative real-time RT-PCR analysis.
CD22, mature IgG, IL4, IL10, IFNG, and VEGF mRNA expression were assessed by multiplex real-time quantitative RT-PCR using TaqMan Gene Expression Assays (primers and probes, Life Technologies) according to the manufacturers’ instructions. Primers and probes for the quantification of mature IgG mRNA were designed in-house using Primer Express (Life Technologies) as follows: probe, 5′ FAM-CATCGGTCTTCCCC-MGB 3′; J segment primer, 5′ ACCCTGGTCACCGTCTCCTCA 3′; mature IgG reverse primer, TGCAGCAGCGGGTCAAG). For each sample, mRNA abundance was normalized against the amount of human GAPDH mRNA (VIC) for the cytokine TaqMan Gene Expression Assays or against β2-microglobulin mRNA (VIC) for CD22 and mature IgG mRNA. Data analysis was performed using either a ΔCt method, comparing it to an arbitrary number, or a ΔΔCt method, comparing it to freshly isolated cells from blood. Results were expressed as relative mRNA expression units or ΔΔCt.
Immunohistochemical evaluations of frozen tissue specimens.
Freshly frozen tissue sections embedded in OCT were cut at 4-μm to 8-μm thickness using a cryostat (Leica), and sections were air dried and stored at –80°C until further use. Prior to staining, slides were equilibrated at room temperature for 10 minutes, followed by fixation in acetone for 20 minutes. Sections were then air dried and stained using standard operation procedures for immunohistochemistry and immunofluorescence using the following commercial antibodies: mouse anti-human CD22 mAb (eBioscience); goat anti-human IgG (DakoGlostrup); mouse anti-human IgG4 (BD Biosciences); mouse anti-human CD64 (Biolegend); mouse anti-human CD32 (Abcam); mouse anti-human CD16 (Abcam); goat anti-mouse IgG, biotinylated (DAKO); and rabbit anti-human S100. Subsequently, antibodies were detected with the VECTOR Red Alkaline Phosphatase Substrate Kit (Vector Labs) or with donkey anti-mouse Alexa Fluor 555 and donkey anti-goat Alexa Fluor 488 (both Life Technologies). Images were captured using a Zeiss Axio Observer.Z1/ Axiophot microscope using AxioVision (×10, ×20, and ×63 magnification lenses, Carl Zeiss).
Immunohistochemical evaluations of paraffin-embedded tissue specimens.
Paraffin sections were cut at 6-μm thickness on a microtome (Leica) and dried overnight at 60°C. Prior to staining, sections were deparaffinized with a 20-minute incubation in Xylene and then rehydrated by serial incubations in alcohol. Heat-induced antigen retrieval was performed in a 95°C water bath, using citric acid. Subsequently, sections were stained using standard operation procedures for immunohistochemistry with the following commercial antibodies: mouse anti-human CD22 (Abcam); rabbit anti-human FoxP3 (eBioscience); mouse anti-human CD45 (eBioscience); and mouse anti-human IgG4 (BD Biosciences). Antibodies were detected with goat/rabbit anti-mouse IgG-biotinylated (DAKO) and visualized using the VECTOR Red Alkaline Phosphatase Substrate Kit, including Levamisole (Vector Labs). All sections were mounted in DPX mounting solution and analyzed on a Zeiss Axiophot microscope using ×10 and ×20 magnification lenses (Carl Zeiss) and NIS-Elements imaging software (Nikon).
IgG subclass ELISA.
Immunoglobulin subclasses produced in cell culture supernatants were measured using an IgG1-, IgG2-, IgG3-, and IgG4-specific ELISA. Capture antibodies were from AbD Serotec (IgG1 and IgG3) and BD Biosciences (IgG2 and IgG4). Certified Reference Material 470 (67) (ERM-DA470, Institute for Reference Materials and Measurements) was used as the standard to quantify Ig subclass. Maxisorp 96-well plates were coated with mouse anti-human IgG1, IgG2, IgG3, or IgG4 antibodies in carbonate-bi-carbonate buffer (0.2 M, pH 9.4) overnight, and plates were blocked for 1 hour with 2% nonfat milk and PBS-Tween 20. Plates were incubated with culture supernatants diluted 1:1 in 50% RPMI media, 50% PBS, 0.025% Tween 20 for 3 hours, and subclass antibody binding was detected with goat anti-human IgG F(ab′)2-HRP incubated for 2 hours (Jackson ImmunoResearch), followed by a color reaction with 0.5 mg/ml o-phenylenediamine dihydrochloride substrate (Sigma-Aldrich) in peroxide substrate buffer (Pierce). Reactions were stopped with the addition of 1 M HCl. Optical density values were measured at 492 nm (reference wavelength: 650 nm). Standard curve fitting was performed using Graphpad Prism software (Graphpad) with a 4-parameter curve fit using a minimum of 6 points on the standard curve.
Luminex bead array analysis.
Luminex Bead Array Assay Kits, Milliplex for cytokines (Millipore) or Bioplex for IgG subclasses (Bio-Rad), were used according to the manufactures instructions, and data were acquired and analyzed using a FlexMap3 (Luminex Cooperation) or Bio-Plex200 (Luminex Cooperation), respectively.
Production of human anti-CSPG4 antibodies.
The cDNA of the heavy and light chain variable regions directed against the human CSPG4, a cell surface antigen expressed by >80% of malignant melanomas, were derived from previously published variable region sequences of the murine mAb 225.28S (68–70), and antibodies were subsequently cloned and produced in an method analogous to that previously described (71).
Flow cytometric and ImageStream evaluations of antibody binding.
For assessment of antibody binding to CSPG4 on a panel of human melanoma cell lines (A375, A-2058, G-361, WM-115, SK-MEL-28, Malme-3M), primary human melanocytes, U937 monocytes, or human primary monocytes, cells were incubated with 10 μg/ml mAbs for 30 minutes at 4°C, followed by 2 washes in PBS with 5% normal goat serum (FACS buffer). Cells were then treated with goat anti-human IgG (Fab′)2-FITC antibody or goat anti-human IgG1-PE (10 μg/ml) (Jackson ImmunoResearch) for 30 minutes at 4°C and washed in FACS buffer prior to acquisition and analysis on a FACSCanto flow cytometer (BD Biosciences) or ImageStreamX (Amnis Corporation). For ImageStreamX, quantitative analyses are based on the acquisition of 20,000 cell events, and MFIs were measured and calculated using an object mask to detect surface staining only.
Cytotoxicity/phagocytosis (ADCC/ADCP) assays.
Antibody-dependent cell-mediated killing of CSPG4-expressing cells by anti-CSPG4 antibodies was quantified by a 3-color flow cytometric ADCC/ADCP assay, as previously described (72). Briefly, A375 melanoma cells were stained 24 hours prior to assays with 5.0 μM CFSE (5-[and 6-] carboxyfluorescein diacetate succinimidyl ester, Life Technologies) in PBS for 10 minutes at 37°C; washed in DMEM medium, 10% FCS, 2 mM l-glutamine; and returned to standard culture conditions. The following day, CFSE-labeled tumor cells were washed and then mixed with human monocytes at an E/T cell ratio of 2:1, with or without (5 μg/ml) antibodies, followed by a 3 hours incubation at 37°C. Cells were then washed in PBS with 2% normal goat serum (FACS buffer), incubated for 30 minutes with a mouse anti-human CD89 PE (BD Biosciences), washed again in FACS buffer, and resuspended in PBS supplemented with DAPI (Life Technologies). All assay conditions were tested in triplicates and replicated in 6 independent experiments using peripheral blood monocytes derived from 6 patients with stage II melanoma.
Assessments of receptor function were conducted using blocking antibodies: mouse anti-human FcγRI (Biolegend); mouse anti-human FcγRII (Abcam); and mouse anti-human FcγRIII (Abcam), which have been previously described to block the antibody Fc-mediated functions of different FcγR family members (73–75). Blocking experiments with specific or nonspecific IgG4 antibodies were conducted using a 3:1 (IgG4/IgG1) ratio.
Samples were acquired immediately using a FACSCanto flow cytometer (BD Biosciences). For event acquisition and analysis, CFSE-labeled tumor cells were detected in FITC (530/30-nm band-pass filter and a 502-nm long-pass filter), CD89-PE-labeled primary monocytes were detected in PE (585/42-nm band-pass filter and a 556-nm long-pass filter), and DAPI+ dead cells were detected in Pacific Blue (345/20-nm band-pass filter) channels, while control samples were set for compensation adjustments between CFSE and PE. Two dual-color flow cytometric dot plots were generated to calculate ADCC and ADCP, as previously described (66, 72, 76).
Assessments of antibodies in a subcutaneous human melanoma model in NOD/SCID/Il2rg–/– mice engrafted with human immune cells.
Male and female NOD/SCID/Il2rg–/– mice (NOD.cg-Prkdc SCID Il2rg tm1Wjl /SzJ [NSG]; The Jackson Laboratory) were used at between 6 and 10 weeks of age. Mice were maintained under specific pathogen–free conditions and handled in accordance with the Institutional Committees on Animal Welfare of the UK Home Office (The Home Office Animals Scientific Procedures Act, 1986). NSG mice were injected subcutaneously with 5 × 105 A375 melanoma cells in 150 μl PBS. On day 5, mice received intravenous injections of 10 × 106 human PBLs (derived from whole human blood by lysis of red blood cells) and 10 mg/kg of antibody. Subsequent injections of antibody treatments were given 3 times on days 12, 18, and 25 at doses of 10 mg/kg each in 150 μl PBS. A control group was treated with 10 × 106 human PBLs on day 5 and injected with 150 μl of PBS on days 12, 18, and 25. Tumor growth was monitored and measured using calipers. Tumor size (mm3) was calculated using the following formula: mm3 = d2 × (D/2), where d stands for the small diameter of tumor and D stands for the large diameter of tumor.
Experiments were terminated once the first animal was measured with a subcutaneous tumor size no greater than 750 mm3. Spleen engraftment of 40%–70% human CD45+ cells was confirmed by flow cytometry for all experiments (Supplemental Figure 5).
NanoSPECT/CT imaging of anti-CSPG4 antibody in vivo.
NSG mice were injected subcutaneously with 5 × 105 A375 tumor cells on day 0. On day 5 mice were injected intravenously with 10 × 106 human PBLs. Spleen engraftment was analyzed as above (Supplemental Figure 5). Anti-CSPG4 IgG4 (7 mg) was conjugated with the bifunctional chelator CHX-A”-DTPA prior to experiments, and on day 24, the antibody was radiolabeled with 20 MBq of Indium-111 (St. Thomas’ and Guy’s Trust, KCL, London, Radiopharmacy) and administered intravenously at 10 mg/kg as above. Images were captured under anesthesia after 20 minutes, 8 hours, and 24 hours using a NanoSPECT/CT preclinical imager (Bioscan) equipped with a multipinhole (9 pinholes; aperture 1.0 mm) collimator and reconstructed using the Invivoscope software (Bioscan).
Statistics.
Comparative quantitative PCR analyses in human skin specimens were conducted using a Kruskal-Wallis analysis with a Dunn post-hoc test. Comparative analyses of immunohistochemical staining were performed using a Mann-Whitney U test. One-way ANOVA with Bonferroni post-hoc test was used to compare ADCC/ADCP of tumor cells by human monocytes, triggered by antibodies. Two-way ANOVA with Bonferroni post-hoc test was used to compare restriction of tumor cell growth in vivo. For correlation and patient survival analyses, Spearman and Mantel-Cox analyses were applied. All statistical analyses were performed using GraphPad Prism software (version 5.03, GraphPad). Error bars in all in vitro figures represent SD. Error bars in all in vivo figures and histological analyses represent SEM.
Supplementary Material
Acknowledgments
The authors thank Angela Clifford for recruitment of volunteers; Isabella Tosi, Katazryna Grys, Eduardo Calonje, and Tawatchai Suttikoon for help with sample processing and assessments; Rebecca Beavil and Andrew Beavil for expert assistance with antibody engineering and purification; Chung-Ching Chu for assistance with illustrations; and Hannah Gould and Pooja Takhar for advice and discussions. We acknowledge the Biomedical Research Centre Immune monitoring core Facility team at Guy’s and St. Thomas’ NHS Foundation Trust for assistance. We thank all patients and healthy volunteers who participated in this study. The authors are solely responsible for study design, data collection, analysis, decision to publish, and preparation of the manuscript. The research was supported by the NIHR Biomedical Research Centre based at Guy’s and St. Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health. The authors acknowledge support by CR UK/EPSRC/MRC/NIHR KCL/UCL Comprehensive Cancer Imaging Centre (C1519/A10331) (to D.H. Josephs, P.J. Blower, J.F. Spicer, and S.N. Karagiannis); Cancer Research UK (C30122/A11527) (to S.N. Karagiannis, J.F. Spicer, D.H. Josephs, L. Saul); Cancer Research UK (C16736/A8371) (to F.O. Nestle and S.N. Karagiannis); Mary Dunhill Trust (to F.O. Nestle); CR UK/NIHR in England/DoH for Scotland, Wales and Northern Ireland Experimental Cancer Medicine Centre (to F.O. Nestle and J.F. Spicer); BBSRC grant BB/H019634/1 (D.J. Fear); and the Overseas Research Students Award Scheme (to A.E. Gilbert).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(4):1457–1474. doi:10.1172/JCI65579.
References
- 1.Weide B, et al. Functional T cells targeting NY-ESO-1 or Melan-A are predictive for survival of patients with distant melanoma metastasis. J Clin Oncol. 2012;30(15):1835–1841. doi: 10.1200/JCO.2011.40.2271. [DOI] [PubMed] [Google Scholar]
- 2.Watson DB, Burns GF, Mackay IR. In vitro growth of B lymphocytes infiltrating human melanoma tissue by transformation with EBV: evidence for secretion of anti-melanoma antibodies by some transformed cells. J Immunol. 1983;130(5):2442–2447. [PubMed] [Google Scholar]
- 3.Kirkwood JM, Robinson JE. Human IgG and IgM monoclonal antibodies against autologous melanoma produced by Epstein-Barr-virus-transformed B lymphocytes. Cancer Immunol Immunother. 1990;32(4):228–234. doi: 10.1007/BF01741705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erdag G, et al. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012;72(5):1070–1080. doi: 10.1158/0008-5472.CAN-11-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andreu P, et al. FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell. 2010;17(2):121–134. doi: 10.1016/j.ccr.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schioppa T, et al. B regulatory cells and the tumor-promoting actions of TNF-alpha during squamous carcinogenesis. Proc Natl Acad Sci U S A. 2011;108(26):10662–10667. doi: 10.1073/pnas.1100994108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jefferis R. Isotype and glycoform selection for antibody therapeutics. Arch Biochem Biophys. 2012;526(2):159–166. doi: 10.1016/j.abb.2012.03.021. [DOI] [PubMed] [Google Scholar]
- 8.Papadea C, Check IJ. Human immunoglobulin G and immunoglobulin G subclasses: biochemical, genetic, and clinical aspects. Crit Rev Clin Lab Sci. 1989;27(1):27–58. doi: 10.3109/10408368909106589. [DOI] [PubMed] [Google Scholar]
- 9.Steplewski Z, Sun LK, Shearman CW, Ghrayeb J, Daddona P, Koprowski H. Biological activity of human-mouse IgG1, IgG2, IgG3, and IgG4 chimeric monoclonal antibodies with antitumor specificity. Proc Natl Acad Sci U S A. 1988;85(13):4852–4856. doi: 10.1073/pnas.85.13.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology. 2002;105(1):9–19. doi: 10.1046/j.0019-2805.2001.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aalberse RC, Stapel SO, Schuurman J, Rispens T. Immunoglobulin G4: an odd antibody. Clin Exp Allergy. 2009;39(4):469–477. doi: 10.1111/j.1365-2222.2009.03207.x. [DOI] [PubMed] [Google Scholar]
- 12.French M. Serum IgG subclasses in normal adults. Monogr Allergy. 1986;19:100–107. [PubMed] [Google Scholar]
- 13.Neild GH, Rodriguez-Justo M, Wall C, Connolly JO. Hyper-IgG4 disease: report and characterisation of a new disease. BMC Med. 2006;4:23. doi: 10.1186/1741-7015-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–551. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 15.Eifan AO, Shamji MH, Durham SR. Long-term clinical and immunological effects of allergen immunotherapy. Curr Opin Allergy Clin Immunol. 2011;11(6):586–593. doi: 10.1097/ACI.0b013e32834cb994. [DOI] [PubMed] [Google Scholar]
- 16.Frew AJ. Allergen immunotherapy. J Allergy Clin Immunol. 2010;125(2 suppl 2):S306–S313. doi: 10.1016/j.jaci.2009.10.064. [DOI] [PubMed] [Google Scholar]
- 17.Garcia BE, Sanz ML, Gato JJ, Fernandez J, Oehling A. IgG4 blocking effect on the release of antigen-specific histamine. J Investig Allergol Clin Immunol. 1993;3(1):26–33. [PubMed] [Google Scholar]
- 18.Shamji MH, et al. Functional rather than immunoreactive levels of IgG4 correlate closely with clinical response to grass pollen immunotherapy. Allergy. 2012;67(2):217–226. doi: 10.1111/j.1398-9995.2011.02745.x. [DOI] [PubMed] [Google Scholar]
- 19.Harada K, et al. Significance of IgG4-positive cells in extrahepatic cholangiocarcinoma: molecular mechanism of IgG4 reaction in cancer tissue. Hepatology. 2012;56(1):157–164. doi: 10.1002/hep.25627. [DOI] [PubMed] [Google Scholar]
- 20.Cipponi A, et al. Neogenesis of lymphoid structures and antibody responses occur in human melanoma metastases. Cancer Res. 2012;72(16):3997–4007. doi: 10.1158/0008-5472.CAN-12-1377. [DOI] [PubMed] [Google Scholar]
- 21.Daveau M, et al. IgG4 subclass in malignant melanoma. J Natl Cancer Inst. 1977;58(2):189–192. doi: 10.1093/jnci/58.2.189. [DOI] [PubMed] [Google Scholar]
- 22.Ellyard JI, Simson L, Parish CR. Th2-mediated anti-tumour immunity: friend or foe? Tissue Antigens. 2007;70(1):1–11. doi: 10.1111/j.1399-0039.2007.00869.x. [DOI] [PubMed] [Google Scholar]
- 23.Platts-Mills TA, Woodfolk JA, Erwin EA, Aalberse R. Mechanisms of tolerance to inhalant allergens: the relevance of a modified Th2 response to allergens from domestic animals. Springer Semin Immunopathol. 2004;25(3–4):271–279. doi: 10.1007/s00281-003-0149-8. [DOI] [PubMed] [Google Scholar]
- 24.Satoguina JS, Weyand E, Larbi J, Hoerauf A. T regulatory-1 cells induce IgG4 production by B cells: role of IL-10. J Immunol. 2005;174(8):4718–4726. doi: 10.4049/jimmunol.174.8.4718. [DOI] [PubMed] [Google Scholar]
- 25.Francis JN, et al. Grass pollen immunotherapy: IL-10 induction and suppression of late responses precedes IgG4 inhibitory antibody activity. J Allergy Clin Immunol. 2008;121(5):1120–1125. doi: 10.1016/j.jaci.2008.01.072. [DOI] [PubMed] [Google Scholar]
- 26.Robinson DS, Larche M, Durham SR. Tregs and allergic disease. J Clin Invest. 2004;114(10):1389–1397. doi: 10.1172/JCI23595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeannin P, Lecoanet S, Delneste Y, Gauchat JF, Bonnefoy JY. IgE versus IgG4 production can be differentially regulated by IL-10. J Immunol. 1998;160(7):3555–3561. [PubMed] [Google Scholar]
- 28.Gilbert AE, et al. Monitoring the systemic human memory B cell compartment of melanoma patients for anti-tumor IgG antibodies. PLoS One. 2011;6(4):e19330. doi: 10.1371/journal.pone.0019330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Punnonen J, de Waal Malefyt R, van Vlasselaer P, Gauchat JF, de Vries JE. IL-10 and viral IL-10 prevent IL-4-induced IgE synthesis by inhibiting the accessory cell function of monocytes. J Immunol. 1993;151(3):1280–1289. [PubMed] [Google Scholar]
- 30.Zaidi MR, et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature. 2011;469(7331):548–553. doi: 10.1038/nature09666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.James LK, et al. Allergen specificity of IgG(4)-expressing B cells in patients with grass pollen allergy undergoing immunotherapy. J Allergy Clin Immunol. 2012;130(3):663–670. doi: 10.1016/j.jaci.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura K, Malykhin A, Coggeshall KM. The Src homology 2 domain-containing inositol 5-phosphatase negatively regulates Fcgamma receptor-mediated phagocytosis through immunoreceptor tyrosine-based activation motif-bearing phagocytic receptors. Blood. 2002;100(9):3374–3382. doi: 10.1182/blood-2002-03-0787. [DOI] [PubMed] [Google Scholar]
- 33.van der Poel CE, Spaapen RM, van de Winkel JG, Leusen JH. Functional characteristics of the high affinity IgG receptor, FcgammaRI. J Immunol. 2011;186(5):2699–2704. doi: 10.4049/jimmunol.1003526. [DOI] [PubMed] [Google Scholar]
- 34.Chu CC, et al. Resident CD141 (BDCA3)+ dendritic cells in human skin produce IL-10 and induce regulatory T cells that suppress skin inflammation. J Exp Med. 2012;209(5):935–945. doi: 10.1084/jem.20112583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.French MA, Harrison G. Serum IgG subclass concentrations in healthy adults: a study using monoclonal antisera. Clin Exp Immunol. 1984;56(2):473–475. [PMC free article] [PubMed] [Google Scholar]
- 36.Anderson KS, et al. Serum antibodies to the HPV16 proteome as biomarkers for head and neck cancer. Br J Cancer. 2011;104(12):1896–1905. doi: 10.1038/bjc.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hussein MR, Elsers DA, Fadel SA, Omar AE. Immunohistological characterisation of tumour infiltrating lymphocytes in melanocytic skin lesions. J Clin Pathol. 2006;59(3):316–324. doi: 10.1136/jcp.2005.028860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeilding NM, Gerstner C, Kirkwood JM. Analysis of two human monoclonal antibodies against melanoma. Int J Cancer. 1992;52(6):967–973. doi: 10.1002/ijc.2910520623. [DOI] [PubMed] [Google Scholar]
- 39.Yamaguchi H, et al. Cell-surface antigens of melanoma recognized by human monoclonal antibodies. Proc Natl Acad Sci U S A. 1987;84(8):2416–2420. doi: 10.1073/pnas.84.8.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt P, Kopecky C, Hombach A, Zigrino P, Mauch C, Abken H. Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A. 2011;108(6):2474–2479. doi: 10.1073/pnas.1009069108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zabierowski SE, Herlyn M. Melanoma stem cells: the dark seed of melanoma. J Clin Oncol. 2008;26(17):2890–2894. doi: 10.1200/JCO.2007.15.5465. [DOI] [PubMed] [Google Scholar]
- 42.Ladanyi A, et al. Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol Immunother. 2011;60(12):1729–1738. doi: 10.1007/s00262-011-1071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zapata JM, Llobet D, Krajewska M, Lefebvre S, Kress CL, Reed JC. Lymphocyte-specific TRAF3 transgenic mice have enhanced humoral responses and develop plasmacytosis, autoimmunity, inflammation, and cancer. Blood. 2009;113(19):4595–4603. doi: 10.1182/blood-2008-07-165456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coker HA, Durham SR, Gould HJ. Local somatic hypermutation and class switch recombination in the nasal mucosa of allergic rhinitis patients. J Immunol. 2003;171(10):5602–5610. doi: 10.4049/jimmunol.171.10.5602. [DOI] [PubMed] [Google Scholar]
- 45.Whiteside TL. Immune suppression in cancer: effects on immune cells, mechanisms and future therapeutic intervention. Semin Cancer Biol. 2006;16(1):3–15. doi: 10.1016/j.semcancer.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 46.Gerlini G, Tun-Kyi A, Dudli C, Burg G, Pimpinelli N, Nestle FO. Metastatic melanoma secreted IL-10 down-regulates CD1 molecules on dendritic cells in metastatic tumor lesions. Am J Pathol. 2004;165(6):1853–1863. doi: 10.1016/S0002-9440(10)63238-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Platts-Mills T, Vaughan J, Squillace S, Woodfolk J, Sporik R. Sensitisation, asthma, and a modified Th2 response in children exposed to cat allergen: a population-based cross-sectional study. Lancet. 2001;357(9258):752–756. doi: 10.1016/S0140-6736(00)04168-4. [DOI] [PubMed] [Google Scholar]
- 48.Nakashima H, et al. An amplification of IL-10 and TGF-beta in patients with IgG4-related tubulointerstitial nephritis. Clin Nephrol. 2010;73(5):385–391. doi: 10.5414/cnp73385. [DOI] [PubMed] [Google Scholar]
- 49.Zen Y, Nakanuma Y. Pathogenesis of IgG4-related disease. Curr Opin Rheumatol. 2011;23(1):114–118. doi: 10.1097/BOR.0b013e3283412f4a. [DOI] [PubMed] [Google Scholar]
- 50.Krop EJ, Stapel SO, De Vrieze H, Van der Zee JS. Immunoglobulin E and G4 antibody responses in occupational airway exposure to bovine and porcine plasma proteins. Int Arch Allergy Immunol. 2006;139(3):237–244. doi: 10.1159/000091169. [DOI] [PubMed] [Google Scholar]
- 51.Matsui EC, et al. Mouse allergen-specific immunoglobulin G and immunoglobulin G4 and allergic symptoms in immunoglobulin E-sensitized laboratory animal workers. Clin Exp Allergy. 2005;35(10):1347–1353. doi: 10.1111/j.1365-2222.2005.02331.x. [DOI] [PubMed] [Google Scholar]
- 52.Strid J, Sobolev O, Zafirova B, Polic B, Hayday A. The intraepithelial T cell response to NKG2D-ligands links lymphoid stress surveillance to atopy. Science. 2011;334(6060):1293–1297. doi: 10.1126/science.1211250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nevala WK, Vachon CM, Leontovich AA, Scott CG, Thompson MA, Markovic SN. Evidence of systemic Th2-driven chronic inflammation in patients with metastatic melanoma. Clin Cancer Res. 2009;15(6):1931–1939. doi: 10.1158/1078-0432.CCR-08-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng W, et al. Down-modulation of TNFSF15 in ovarian cancer by VEGF and MCP-1 is a pre-requisite for tumor neovascularization. Angiogenesis. 2012;15(1):71–85. doi: 10.1007/s10456-011-9244-y. [DOI] [PubMed] [Google Scholar]
- 55.Maeda K, Mehta H, Drevets DA, Coggeshall KM. IL-6 increases B-cell IgG production in a feed-forward proinflammatory mechanism to skew hematopoiesis and elevate myeloid production. Blood. 2010;115(23):4699–4706. doi: 10.1182/blood-2009-07-230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hirano H, et al. TLR4, IL-6, IL-18, MyD88 and HMGB1 are highly expressed in intracranial inflammatory lesions and the IgG4/IgG ratio correlates with TLR4 and IL-6. Neuropathology. 2012;32(6):628–637. doi: 10.1111/j.1440-1789.2012.01310.x. [DOI] [PubMed] [Google Scholar]
- 57.Labrijn AF, Aalberse RC, Schuurman J. When binding is enough: nonactivating antibody formats. Curr Opin Immunol. 2008;20(4):479–485. doi: 10.1016/j.coi.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 58.Bruhns P, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113(16):3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 59.Rispens T, Ooievaar-De Heer P, Vermeulen E, Schuurman J, van der Neut Kolfschoten M, Aalberse RC. Human IgG4 binds to IgG4 and conformationally altered IgG1 via Fc-Fc interactions. J Immunol. 2009;182(7):4275–4281. doi: 10.4049/jimmunol.0804338. [DOI] [PubMed] [Google Scholar]
- 60.Leyendeckers H, et al. Correlation analysis between frequencies of circulating antigen-specific IgG-bearing memory B cells and serum titers of antigen-specific IgG. Eur J Immunol. 1999;29(4):1406–1417. doi: 10.1002/(SICI)1521-4141(199904)29:04<1406::AID-IMMU1406>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 61.van Schouwenburg PA, et al. IgG4 production against adalimumab during long term treatment of RA patients. J Clin Immunol. 2012;32(5):1000–1006. doi: 10.1007/s10875-012-9705-0. [DOI] [PubMed] [Google Scholar]
- 62.Svenson M, Geborek P, Saxne T, Bendtzen K. Monitoring patients treated with anti-TNF-alpha biopharmaceuticals: assessing serum infliximab and anti-infliximab antibodies. Rheumatology (Oxford). 2007;46(12):1828–1834. doi: 10.1093/rheumatology/kem261. [DOI] [PubMed] [Google Scholar]
- 63.Bruggemann M, et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. 1987;166(5):1351–1361. doi: 10.1084/jem.166.5.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balch CM, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lapointe R, Bellemare-Pelletier A, Housseau F, Thibodeau J, Hwu P. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003;63(11):2836–2843. [PubMed] [Google Scholar]
- 66.Karagiannis SN, et al. IgE-antibody-dependent immunotherapy of solid tumors: cytotoxic and phagocytic mechanisms of eradication of ovarian cancer cells. J Immunol. 2007;179(5):2832–2843. doi: 10.4049/jimmunol.179.5.2832. [DOI] [PubMed] [Google Scholar]
- 67.Schauer U, et al. IgG subclass concentrations in certified reference material 470 and reference values for children and adults determined with the binding site reagents. Clin Chem. 2003;49(11):1924–1929. doi: 10.1373/clinchem.2003.022350. [DOI] [PubMed] [Google Scholar]
- 68.Campoli M, Ferrone S, Wang X. Functional and clinical relevance of chondroitin sulfate proteoglycan 4. Adv Cancer Res. 2010;109:73–121. doi: 10.1016/B978-0-12-380890-5.00003-X. [DOI] [PubMed] [Google Scholar]
- 69.Price MA, et al. CSPG4, a potential therapeutic target, facilitates malignant progression of melanoma. Pigment Cell Melanoma Res. 2011;24(6):1148–1157. doi: 10.1111/j.1755-148X.2011.00929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Neri D, et al. Recombinant anti-human melanoma antibodies are versatile molecules. J Invest Dermatol. 1996;107(2):164–170. doi: 10.1111/1523-1747.ep12329566. [DOI] [PubMed] [Google Scholar]
- 71.Karagiannis P, et al. Characterisation of an engineered trastuzumab IgE antibody and effector cell mechanisms targeting HER2/neu-positive tumour cells. Cancer Immunol Immunother. 2009;58(6):915–930. doi: 10.1007/s00262-008-0607-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bracher M, Gould HJ, Sutton BJ, Dombrowicz D, Karagiannis SN. Three-colour flow cytometric method to measure antibody-dependent tumour cell killing by cytotoxicity and phagocytosis. J Immunol Methods. 2007;323(2):160–171. doi: 10.1016/j.jim.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 73.Bunk S, et al. Internalization and coreceptor expression are critical for TLR2-mediated recognition of lipoteichoic acid in human peripheral blood. J Immunol. 2010;185(6):3708–3717. doi: 10.4049/jimmunol.0901660. [DOI] [PubMed] [Google Scholar]
- 74.Tamm A, Schmidt RE. The binding epitopes of human CD16 (Fc gamma RIII) monoclonal antibodies. Implications for ligand binding. J Immunol. 1996;157(4):1576–1581. [PubMed] [Google Scholar]
- 75.Hober D, et al. Circulating and cell-bound antibodies increase coxsackievirus B4-induced production of IFN-alpha by peripheral blood mononuclear cells from patients with type 1 diabetes. J Gen Virol. 2002;83(pt 9):2169–2176. doi: 10.1099/0022-1317-83-9-2169. [DOI] [PubMed] [Google Scholar]
- 76.Karagiannis SN, et al. Role of IgE receptors in IgE antibody-dependent cytotoxicity and phagocytosis of ovarian tumor cells by human monocytic cells. Cancer Immunol Immunother. 2008;57(2):247–263. doi: 10.1007/s00262-007-0371-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.