

Abstract
The identification of a gain-of-function mutation in CACNA1C as the cause of Timothy Syndrome (TS), a rare disorder characterized by cardiac arrhythmias and syndactyly, highlighted unexpected roles for the L-type voltage-gated Ca2+ channel CaV1.2 in nonexcitable cells. How abnormal Ca2+ influx through CaV1.2 underlies phenotypes such as the accompanying syndactyly or craniofacial abnormalities in the majority of affected individuals is not readily explained by established CaV1.2 roles. Here, we show that CaV1.2 is expressed in the first and second pharyngeal arches within the subset of cells that give rise to jaw primordia. Gain-of-function and loss-of-function studies in mouse, in concert with knockdown/rescue and pharmacological approaches in zebrafish, demonstrated that Ca2+ influx through CaV1.2 regulates jaw development. Cranial neural crest migration was unaffected by CaV1.2 knockdown, suggesting a role for CaV1.2 later in development. Focusing on the mandible, we observed that cellular hypertrophy and hyperplasia depended upon Ca2+ signals through CaV1.2, including those that activated the calcineurin signaling pathway. Together, these results provide new insights into the role of voltage-gated Ca2+ channels in nonexcitable cells during development.
Introduction
Voltage-gated Ca2+ channels are the sine qua non of excitable cells — translating electrical activity into the cytoplasmic Ca2+ changes that regulate cellular responses such as neuronal activity, muscle contraction, and hormone release. Nonexcitable cells generally employ different means to increase cytoplasmic Ca2+, such as receptor-operated Ca2+ channels or release from intracellular stores. Although expression of certain voltage-gated Ca2+ channels, mainly the CaV1.2 L-type Ca2+ channel, has been documented in certain nonexcitable cells, the physiologic roles of voltage-gated Ca2+ channels in these cells have been enigmatic and largely unexplored.
The broad array of abnormalities within nonexcitable tissues in Timothy Syndrome (TS) patients (1), however, revealed that CaV1.2 controls critical, yet previously unknown, roles in multiple nonexcitable tissues. Identified as a novel cardiac arrhythmia syndrome associated with syndactyly and dysmorphic facial features (2), the TS defect was discovered to be a specific gain-of-function mutation (G406R) in CACNA1C, the gene encoding CaV1.2. The mutation greatly slows channel inactivation, and thereby prolongs cellular repolarization in cardiac myocytes, which provided a clear rationale for the cardiac arrhythmias. Yet how CaV1.2 affects nonexcitable cells, as indicated by additional phenotypes documented in TS, such as small teeth, baldness at birth, and dysmorphic facial features, is unclear. These phenotypes were not consistent with previously understood roles for CaV1.2. Here, we exploited both mouse and zebrafish models to define how Ca2+ influx through CaV1.2 affects craniofacial development.
Results
CaV1.2 is expressed in the developing jaw.
To determine how CaV1.2 might contribute to craniofacial development, we started by investigating whether CaV1.2 was present in jaw primordia during the appropriate developmental stages. We used a CaV1.2 reporter mouse (CaV1.2+/lacZ) in which lacZ disrupted one allele of Cacna1c, thereby marking CaV1.2-expressing cells. Although CaV1.2–/– mice die in utero, CaV1.2+/– mice are viable and fertile without any obvious differences in any aspect of morphology or development (3). Thus, analysis of β-gal expression in CaV1.2+/lacZ reporter embryos should approximate CaV1.2 temporal expression and accurately reflect CaV1.2 spatial expression. Examination of β-gal activity in CaV1.2+/lacZ E9.5 embryos revealed intense staining mainly in the developing heart (Figure 1A), as expected. By E11.5, however, we observed extensive activity in the first and second pharyngeal arches (Figure 1, B–D), as well as in the limb buds. Coronal sections through the first arch in E11.5 embryos showed that the β-gal activity was mainly in the periphery (Figure 1, E and F), where a subset of cranial neural crest–derived cartilage progenitors resides and from which jaw structures such as the mandible develop (4, 5); the identification of β-gal–positive cells in this region suggests that they are of neural crest lineage. Thus, the presence of CaV1.2-expressing cells in these structures provides a basis for the syndactyly and craniofacial abnormalities seen in TS patients.
Figure 1. CaV1.2 is expressed in developing jaw.
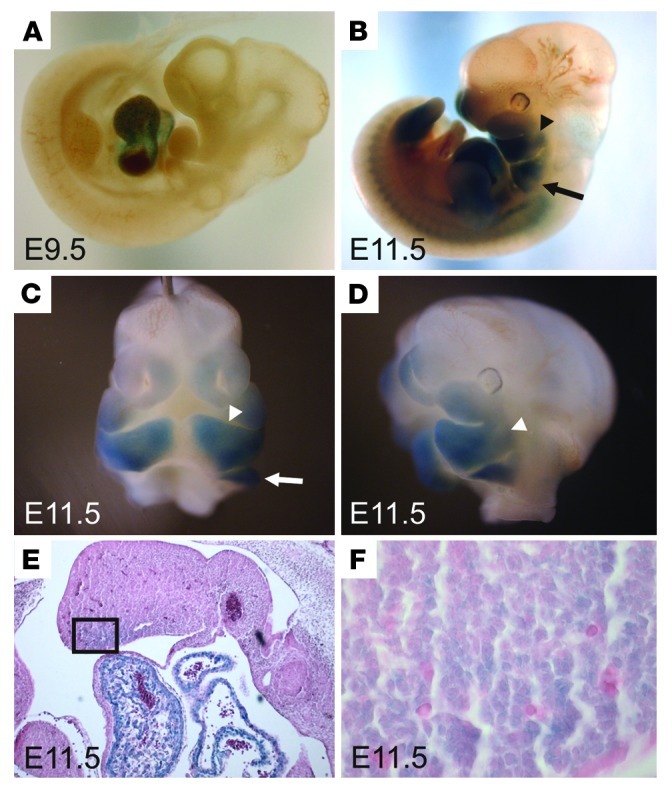
(A–D) Whole-mount β-gal staining of CaV1.2+/LacZ embryos. CaV1.2 is highly expressed in the developing heart around E9.5 (A). By E11.5 intense staining is present in the first (arrowhead) and second (arrow) pharyngeal arches (B–D). (E and F) β-gal staining of a coronal section through the first mandibular arch. F is a magnified section of the boxed area shown in E.
Altered CaV1.2 activity affects jaw development in mice.
To test whether abnormal signaling through CaV1.2 in the developing jaw causes craniofacial abnormalities, we exploited a mouse model in which a floxed STOP TS mutant CaV1.2 (CaV1.2TS) allele for the pore-forming α1C subunit of CaV1.2 (or CaV1.2WT as a control) had been knocked into the Rosa26 locus (6). To examine the effects of CaV1.2 in mandibular development uncomplicated by effects in other tissues, we expressed CaV1.2WT or CaV1.2TS in a subset of craniofacial mesenchyme by breeding these mice with a Prx1-Cre transgenic mouse. Within the branchial arches, Prx1 drives Cre recombinase expression starting around E11.5 in the ventral surfaces (7), where we observed endogenous CaV1.2 expression (Figure 1, C and D). Prx1 also drives expression in limb bud mesenchyme (7), in both chondrocytes and osteoblasts (8), which allowed us to obtain material to test whether expression levels of CaV1.2WT and CaV1.2TS were comparable in tissues in which Prx1 is active. We isolated articular chondrocytes from the femur and tibia of Prx1+-CaV1.2TS, Prx1+-CaV1.2WT, and Prx1–-CaV1.2TS mice and performed immunocytochemistry for α1C. An experimenter blinded to the genotypes used identical exposure settings for representative images from all three genotypes and observed that transgenic expression of α1C was elevated in Prx1+-CaV1.2TS compared with Prx1–-CaV1.2TS mice, but was equivalent to Prx1+-CaV1.2WT mice (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI66903DS1). Having established a similar expression of CaV1.2TS and CaV1.2WT driven by Prx1, we obtained radiographs of skulls to ascertain the effect of CaV1.2TS on jaw development. We observed that radiographs of skulls from mice expressing CaV1.2WT (Prx1+) were not different from those of their Prx1– littermate controls (Figure 2A) or wild-type animals (not shown). In contrast, the tip of the mandible (lower jaw) extended anteriorly as far as, or beyond, the tip of the maxilla (upper jaw) in 6 of 7 mice in which Prx1 drove CaV1.2TS expression (Figure 2B). In Prx1– littermates, the maxilla extended beyond the mandible (3 of 3 animals), as in the wild-type animals (Figure 2B). The relatively larger mandible in Prx1-CaV1.2TS mice recapitulated the phenotype reported in TS patients (Figure 2C and ref. 1). Together, these data demonstrate that expression of the TS mutant CaV1.2, and not just overexpression of CaV1.2WT, is necessary to recapitulate this TS phenotype.
Figure 2. CaV1.2 expression in pharyngeal arches affects craniofacial development.

(A) X-rays of a Prx-CaV1.2WT skull and a Prx1– littermate skull. Note that the upper jaw extends anteriorly to the mandible (arrows). (B) X-rays of a Prx1-CaV1.2TS skull and a Prx1– littermate skull. When CaV1.2TS is expressed (in the Prx1+ animal), the mandible extends more anteriorly, as is also seen in TS patients. (C) Image showing patient phenotype (arrow points to mandible). Image in C is reproduced with permission from the authors and publisher (1). (D) Mandibles from adult CaV1.2+/LacZ or wild-type (littermates). Note reduced width between coronoid processes in CaV1.2+/LacZ compared with wild-type mice. Scale bars: 5 mm.
Based on these results with the gain-of-function CaV1.2TS, we hypothesized that CaV1.2 haploinsufficiency might affect mandible size in the opposite manner. Although previous examination of CaV1.2+/– mice revealed no differences (compared with wild-type mice) in morphology or development (3) — and we also found that overall mouse size, weight, and tibial length were not different between the 2 genotypes — subtle extracardiac phenotypes might have been missed in that original study. Indeed, in a blinded analysis, we found that the normalized intracoronoid width was smaller in CaV1.2+/lacZ mice (0.40 ± 0.00, n = 6) compared with wild-type littermate controls (0.48 ± 0.01, n = 6, P < 0.001) (Figure 2D). Together, these data suggest that altered Ca2+ influx through CaV1.2 in a tissue-specific manner can affect craniofacial development.
Genetic and pharmacological manipulation shows that CaV1.2 regulates mandibular development in zebrafish.
We exploited zebrafish to begin dissection of the molecular mechanisms by which CaV1.2 regulates craniofacial development. Among the advantages of this model system is that, due to oxygen diffusion, embryos do not require heart function until approximately 5 days post fertilization, a developmental stage significantly beyond when a functioning heart is necessary in higher vertebrates and when key aspects of craniofacial development are complete. This is particularly relevant for CaV1.2, which is necessary for cardiac function in zebrafish (9) and mice (3). A previous study detected CaV1.2 expression in zebrafish embryos in the developing heart and pancreas at 26 hours post fertilization (hpf) by in situ hybridization (9), which we confirmed (Supplemental Figure 2). At 72 hpf, when jaw elements are visible by Alcian blue staining (Figure 3), we detected CaV1.2 expression in the developing mandible and the ceratohyal cartilage (Supplemental Figure 2). To determine the role of CaV1.2 in mandibular development, we designed a cocktail of 2 morpholinos targeting the donor and acceptor sites, respectively, for exon 4 of cacna1c (Supplemental Figure 3) and documented effective CaV1.2 knockdown by two different methods: we found >80% reduction in cacna1c mRNA at 72 hpf, and embryos at 48 hpf displayed the same abnormal cardiac phenotype (a non-beating, “silent” ventricle; see Supplemental Video 1) observed in cacna1c knockout embryos (9). In contrast, a control morpholino affected neither cacna1c mRNA levels nor cardiac function (Supplemental Video 2). Alcian blue staining for cartilage elements at 72 hpf showed that CaV1.2 morpholino knockdown significantly affected overall jaw development, reducing the size of all jaw components (Figure 3A). We quantified the reduction by measuring mandibular area (highlighted by dotted lines), with the observer blinded to the treatment. Compared with no morpholino, the control morpholino had minimal effects on mandibular area (Figure 3, A and B). Since overall embryo size and length were only minimally affected (Supplemental Figure 4) and mortality rates were unaffected by the control morpholino cocktail, we used the control morpholino as the comparator in subsequent experiments. The CaV1.2 knockdown morpholino cocktail reduced the mandibular area by 42 ± 3.8% after CaV1.2 knockdown (Figure 3, A and B). We confirmed that the observed jaw defects were due to CaV1.2 knockdown and not to a nonspecific effect of the morpholinos because coinjection of a morpholino-insensitive rabbit cRNA encoding a CaV1.2WT channel doubled the mandibular area compared with the CaV1.2 knockdown morpholino by itself, and restored mandible size almost to that observed in the control morpholino-treated embryos (43,536 ± 1,334 μm2 for control morpholino rescued with CaV1.2WT versus 44,530 ± 1,433 μm2 for control morpholino alone; P = 0.72) (Figure 3, A and C). As expected, coinjection of CaV1.2WT cRNA also rescued cardiac function (64.4% of embryos had a beating heart at 48 hpf compared with 10% of embryos treated with morpholinos only; n = 180 for each, χ2 = 554, P < 0.0001), providing additional evidence that the morpholinos were specific for cacna1c. To confirm that the reduced mandible size observed after morpholino treatment was independent of the effect upon cardiac function, we analyzed developing mandibles in the offspring of weak atrium (wea) and half-hearted (haf) double heterozygotes (wea;haf). The wea mutants lack atrial contractility and the haf mutants lack ventricular contractility (10). Homozygous double mutants were identified at 48 hpf by a complete lack of atrial and ventricular contractility. These were segregated from the remaining embryos (with normal cardiac function) and mandible size, analyzed by Alcian blue staining at 72 hpf, was measured in both groups. As shown in Supplemental Figure 5, the absence of cardiac activity (in the wea;haf homozygotes) did not reduce mandible size, suggesting that the specific knockdown of CaV1.2 by morpholinos affected mandible size independent of effects upon cardiac function.
Figure 3. Ca2+ influx through CaV1.2 regulates size of mandible in zebrafish.

(A) Alcian blue–stained embryos at 72 hpf. CON, uninjected control; CMO, control morpholino; MO, CaV1.2 morpholinos; MO + WT, CaV1.2 morpholinos coinjected with rabbit CaV1.2WT cRNA; MO + 4EQ, CaV1.2 morpholinos coinjected with rabbit CaV1.2 cRNA with mutations in the 4-pore glutamates (non-Ca2+ permeant); MO + TS, CaV1.2 morpholinos coinjected with rabbit CaV1.2TS cRNA. (B) Mandibular area (as outlined for CON in A normalized to the area in CMO (n = 20 for each). *P < 0.001 versus CMO. (C) Mandibular area normalized to the area in MO (n = 20 for each). *P < 0.001 versus MO. (D) Alcian blue–stained embryos at 72 hpf after treatment with DMSO or nisoldipine. (E) Mandibular area normalized to the area in DMSO (n = 20 for each). *P < 0.001 versus DMSO.
We were therefore able to query the effects of rescue with informative mutants. Like CaV1.2WT cRNA, coinjection of a CaV1.2TS cRNA also doubled mandible size. Although CaV1.2TS cRNA did not increase the mandible size more than CaV1.2WT cRNA, it did affect chondrocyte size and pattern within the developing mandible (see Figure 4F). We next tested whether jaw development depended upon Ca2+ influx through CaV1.2 with a Ca2+-impermeant rabbit CaV1.2, in which 4 key aspartate residues in the channel’s pore-forming α1C subunit were mutated to glutamine. This 4EQ mutant (CaV1.24EQ) retains its ability to scaffold other signaling proteins (e.g., auxiliary subunits) and to conduct ions, but completely blocks Ca2+ permeation (11). Unlike CaV1.2WT or CaV1.2TS, CaV1.24EQ did not restore mandible size when coinjected with the CaV1.2 knockdown morpholino cocktail (Figure 3, A and C).
Figure 4. Ca2+ influx through CaV1.2 affects cellular hypertrophy and hyperplasia in the mandible.

(A) Low-power DIC image of flat-mounted, Alcian blue–stained CON embryo. The red box indicates the region from which images in B, D, and F were obtained. (B) Higher magnification of the region shown in A for an embryo treated with DMSO or NIS. (C) Cell size normalized to DMSO (n = 20 for each). *P < 0.001 versus DMSO. (D) Higher magnification of the region shown in A for a CON, CMO, or MO embryo. (E) Cell size normalized to CMO (n = 20 for each). *P < 0.001 versus CMO. (F) Higher magnification of the region shown in A for an MO + WT, MO + TS, or MO + 4EQ embryo. (G) Cell size normalized to MO (n = 20 for each). *P < 0.001 versus MO; **P < 0.001 versus MO + WT or versus MO. (H) BrdU staining and quantification of BrdU+ cells (in GFP+ cells) of sox10+ zebrafish embryos treated with CMO or MO. *P < 0.001. Note that we were unable to use 1-phenyl 2-thiourea, the tyrosinase inhibitor commonly used to block pigmentation and aid visualization (for BrdU staining) in zebrafish because it interferes with neural crest development (32): the larger red nuclei likely denote newly produced melanocytes, which migrate to the same area, and were not counted.
Finally, we validated the morpholino knockdown effects and confirmed the requirement for Ca2+ influx by two additional approaches. First, we measured mandible size in the offspring of heterozygotes of the CaV1.2 null mutant cacna1cm231 zebrafish (12). Embryos without ventricular contraction (isl phenotype) (9) at 24 hpf were separated from the rest of the clutch (beating phenotype) and mandible size was calculated at 72 hpf. The size of mandibles from embryos with the isl phenotype was reduced by more than 50% (Supplemental Figure 6). Second, we applied the CaV1.2 antagonist nisoldipine, which reduced mandible size compared with the DMSO control treatment (Figure 3, D and E). Together, these data suggest that Ca2+ influx through CaV1.2 is necessary for regulating mandible size during development.
Ca2+ influx through CaV1.2 regulates mandibular chondrocyte hypertrophy and hyperplasia.
CaV1.2 could affect mandible size by regulating cellular hypertrophy and/or hyperplasia. To determine if CaV1.2 affected cell hypertrophy, we measured cell size in the developing mandible. After staining embryos at 72 hpf with Alcian blue, flat-mounted images of the lateral aspect of Meckel’s cartilage (Figure 4A) provided an unobstructed view of individual chondrocytes, which assumed a regular and linear pattern that we exploited to quantify the individual cell area (Figure 4B). Pharmacological blockade of CaV1.2 with nisoldipine reduced cell size (Figure 4, B and C), consistent with the effects on the mandible (see Figure 3D) and also affected the linear organized pattern. Cell area was also reduced by CaV1.2 knockdown morpholinos compared with embryos injected with a control morpholino or noninjected controls. As with overall mandible size, cell size was restored by coinjection of a morpholino-insensitive CaV1.2WT but not by the Ca2+-impermeant mutant CaV1.24EQ (Figure 4, D–G). Interestingly, after coinjection of CaV1.2TS, cell size was not only restored (and even larger than rescue with CaV1.2WT), but also this region of the Meckel’s cartilage appeared to be crowded with additional cells (Figure 4F). This suggests that Ca2+ influx through CaV1.2 might also regulate cellular hyperplasia in the developing mandible. We measured cell proliferation in developing mandibles of transgenic zebrafish embryos in which GFP is driven by the sox10 promoter, a marker of the cranial neural crest (13). In these embryos, the developing mandible is GFP+ by 72 hpf, at which point we labeled embryos (treated with control morpholino or CaV1.2 morpholinos) with BrdU and calculated the percentage of cells that were both GFP+ and BrdU+ compared with the total number of GFP+ cells in the section. There was a significantly lower percentage of both BrdU+ and double-labeled cells in the CaV1.2 morpholino–treated embryos within the mandible (Figure 4H). Together, these data suggest that CaV1.2 controls chondrocyte hypertrophy and hyperplasia during mandibular development.
Calcineurin lies downstream of CaV1.2 in regulating size and hypertrophy in mandibular chondrocytes.
The regulation of cellular hypertrophy (Figure 4) suggested to us that the Ca2+-dependent phosphatase calcineurin, which regulates the nuclear factor of activated T cells (NFAT) signaling pathway, might be one candidate for a regulator downstream of CaV1.2, as this pathway regulates hypertrophy in several cell types, such as cardiac myocytes (14). We tested this hypothesis with two independent approaches. First, we inhibited calcineurin activity with FK506 and cyclosporine with doses previously used in zebrafish (15). Mandibular area and cell size were reduced compared with a DMSO control treatment (Figure 5, A and B), reminiscent of the effects after loss of Ca2+ influx through CaV1.2 (see Figures 3 and 4). To query whether calcineurin might be downstream of CaV1.2, we then bypassed the requirement for Ca2+ influx through CaV1.2 by coinjecting cRNA for a constitutively active, Ca2+-insensitive calcineurin (caCN) (16) with CaV1.2 morpholinos. This treatment partially restored mandible size and cell size (Figure 5, C and D). This second approach also helped confirm that the CaV1.2-dependent effects on mandibular development were independent of cardiac function, since the rescue of mandible and cell size did not correlate with the restoration of cardiac function in CaV1.2 morpholino–treated embryos (Supplemental Video 3). We also examined whether calcineurin affected cell size. Using analyses similar to those shown in Figure 4, we demonstrated that calcineurin inhibition with FK506 and cyclosporine reduced cell size compared with DMSO (Figure 5, E and F). On the other hand, coinjection of cRNA for a constitutively active, Ca2+-insensitive calcineurin markedly increased cell size (Figure 5, G and H).
Figure 5. Calcineurin, downstream of CaV1.2, regulates cellular hypertrophy in the developing mandible.

(A) Alcian blue–stained embryo treated with DMSO or a combination of cyclosporine and FK506 (FK/CsA). (B) Mandibular area normalized to the area in DMSO (n = 20 for each). *P < 0.001 versus DMSO. (C) Alcian blue–stained embryo treated with MO or MO plus cRNA for a constitutively active calcineurin (MO + caCN). (D) Mandibular area normalized to the area in MO (n = 20 for each). *P < 0.001 versus MO. (E) Higher magnification of the region shown in Figure 4A for an embryo treated with DMSO or FK/CsA. (F) Cell size normalized to DMSO (n = 20 for each). *P < 0.001 versus DMSO. (G) Higher magnification of the region shown in Figure 4A for an embryo treated with MO or MO + caCN. (H) Cell size normalized to MO (n = 20 for each). *P < 0.001 versus MO. Scale bar: 200 μm.
Mandibular progenitor cells express functional CaV1.2 channels.
The identification of CaV1.2 expression in cranial neural crest–derived cells (Figure 1, E and F) and the developmental effects observed after perturbing CaV1.2 (Figures 2–4), prompted us to test whether cranial neural crest cells express functional CaV1.2 channels. Previously, voltage-gated L-type Ca2+ channels have been hypothesized to regulate neural crest migration and differentiation (17), yet the contributions of specific L-type Ca2+ channels have not been identified, and analysis by electrophysiologic methods has not yet been performed. We did not find that CaV1.2 regulated cranial neural crest cell migration. Using the sox10-GFP transgenic zebrafish embryos to visualize the cranial neural crest cells that migrate dorsolaterally in distinct streams from the neural tube to form craniofacial structures, we found that cranial neural crest migration was unaffected in embryos treated with CaV1.2 morpholinos (Figure 6A). The development of a grossly intact, albeit smaller, jaw in embryos treated with CaV1.2 morpholinos (Figure 3) also suggested that cranial neural crest migration does not depend upon CaV1.2. We queried the presence of CaV1.2 Ca2+ channels in cranial neural crest cells by comparing voltage-gated Ca2+ currents in cells isolated from morpholino-treated and wild-type embryos. We isolated single GFP+ cells from decapitated heads obtained from sox10-GFP zebrafish embryos at 64 to 66 hpf (Figure 6, A and B). Cells without processes were chosen for electrophysiologic analysis to reduce the chance of recording from neuron precursors. The cells were small (average cell capacitance was 1.55 ± 0.3 pF). Resting membrane potential was –35.9 ± 2.7 mV, consistent with what has previously been recorded in cranial neural crest cells (18). Using the whole-cell patch-clamp technique, a ramp protocol in the presence of 15 mM Ba2+ elicited small inward currents in cells isolated from wild-type embryos, but never in cells isolated from embryos treated with the CaV1.2 morpholinos, which always displayed outward currents under these conditions (Figure 6, C and D).
Figure 6. sox10+ cranial neural crest–derived cells express functional CaV1.2 channels.

(A) GFP images of decapitated sox10-GFP transgenic embryos at approximately 65 hpf of a control or CaV1.2 MO embryo. Arrowhead denotes migrating cranial neural crest. (B) Sample GFP+ cell after single-cell isolation. Image on the right shows a DAPI overlay. Note the GFP– cell. (C) Whole-cell Ca2+ current from a GFP+ cell isolated from an MO or CON embryo. Voltage ramp protocol shown below. (D) Box plot showing current density for MO or CON embryos. *P < 0.01. Scale bar: 5 μm.
Discussion
Although identification of CACNA1C as the TS locus revealed unexpected roles for CaV1.2 in nonexcitable cells (1), CaV1.2 participation outside of neurons, muscle, and hormone-secreting cells has received little attention. Here, we provide to the best of our knowledge the first evidence for how CaV1.2 participates in mandibular development and thereby contributes to the dysmorphic facial features that have been reported in more than half of TS patients. Using a combination of mouse and zebrafish models, we demonstrated that Ca2+ influx through CaV1.2 is necessary for proper mandibular development.
Regulation of Ca2+ influx is the best-characterized role for CaV1.2, but voltage-gated Ca2+ channels anchor large macromolecular complexes (19) and could therefore control cellular processes independent of Ca2+ influx. Indeed, the Ca2+ channel auxiliary subunit β4 controls epiboly in zebrafish through a Ca2+-independent manner (20). The inability of the CaV1.24EQ channel to rescue the CaV1.2 morpholino effects (Figure 3, A and C) and the smaller jaws observed after pharmacologic blockade of CaV1.2 with nisoldipine (Figure 3, D and E) provide additional evidence that mandibular development depends directly on Ca2+ influx through CaV1.2. Dihydropyridines have not been shown to disrupt the CaV1.2 macromolecular complex, and the CaV1.24EQ channel remains a voltage-sensitive cation channel — though one that is not able to permeate Ca2+ due to loss of the high-affinity binding site in the pore (11). Thus, we conclude that functions other than Ca2+ permeation are unlikely to contribute to the CaV1.2-dependent regulation of mandibular development.
Our hypothesis is further reinforced by the complementary effects on mandible size with gain-of-function and loss-of-function approaches in mice: CaV1.2 haploinsufficiency reduced mandible size, while expression of a CaV1.2TS mutant channel in the developing jaw and the accompanying augmented Ca2+ influx increased mandible size (Figure 2). While we observed an increase in mandible size in mice expressing CaV1.2TS, the original description in TS patients reported a small upper jaw (maxilla) (1). We suspect that the maxilla in TS patients appears relatively small in comparison with the abnormally large mandible, as shown in the x-rays from Prx1-CaV1.2TS mice (Figure 2B). In zebrafish, the inability of CaV1.2TS cRNA to increase mandible size more than CaV1.2WT cRNA did likely reflects that the zebrafish model is best suited to inform about earlier developmental events. The cRNA was injected into a single-cell embryo along with cacna1c morpholinos. As the embryo increases in size and cell number, both the amount of morpholino and CaV1.2TS cRNA per cell become diluted. Endogenous CaV1.2 expression increases (as shown in Supplemental Figure 3D) and likely dominates the phenotype. Nevertheless, that both CaV1.2WT and CaV1.2TS cRNA can rescue cacna1c morphants provides powerful evidence that our morpholinos are on target.
The cells expressing CaV1.2 that are responsible for these developmental effects likely include the early chondrocytes of the mandible. Using a CaV1.2 reporter mouse, we first identified CaV1.2 at E11.5 within the periphery of the first branchial arch, which contains cranial neural crest–derived cells (see Figure 1). Consistent with these data, we recorded inward Ca2+ currents from zebrafish cranial neural crest–derived (Sox10+) cells, but not after CaV1.2 morpholino knockdown (Figure 6). The Prx1-CaV1.2TS mouse model also suggests a role for developing chondrocytes, since the Prx1-Cre transgene is specifically active in the first pharyngeal arch (7) and in chondrocytes (8).
The resting membrane potential (approximately –35 mV) recorded from the isolated cranial neural crest cells expressing a CaV1.2-dependent, inward Ca2+ current suggests a mechanism by which the gain-of-function TS mutant channel could contribute to disease phenotypes in nonexcitable cells. At this voltage, steady-state inactivation curves for CaV1.2WT and CaV1.2TS channels begin to diverge, so the fraction of CaV1.2TS channels inactivated would be significantly smaller than that of CaV1.2WT channels (1). Thus, after CaV1.2 channels open to participate in the Ca2+ waves that accompany neural crest migration (17, 21), additional Ca2+ likely continues to flow through CaV1.2TS channels for some time, even after a return to more negative resting membrane potentials.
Knockdown of CaV1.2 by morpholinos resulted in a smaller jaw, but one that appeared to have all of its elements, suggesting that CaV1.2 exerts its effects after cranial neural crest migration. Consistent with that, we did not observe any defect in cranial neural crest migration after CaV1.2 knockdown (Figure 6A). Rather, CaV1.2 appears to participate in the subsequent hypertrophy and hyperplasia of the developing chondrocytes.
Calcineurin appears to be one Ca2+-dependent effector downstream of CaV1.2 based on two lines of evidence. First, inhibition of calcineurin produced a phenotype similar to pharmacological blockade of Ca2+ influx through CaV1.2 or CaV1.2 knockdown. Second, expression of a constitutively active calcineurin was able to bypass the requirement for Ca2+ influx through CaV1.2 (Figure 6). It is intriguing that calcineurin is a part of the CaV1.2 macromolecular complex, having been shown to interact either directly with CaV1.2 (22) or through an A-kinase anchoring protein (23). The most likely target of activated calcineurin is NFAT. The ability of constitutively active calcineurin to induce hypertrophy in developing chondrocytes is consistent with its well-documented roles in cardiac myocyte hypertrophy (24). Indeed, transgenic overexpression of NFAT, either in chondrocytes/osteoblasts or osteoclasts, leads to abnormal craniofacial development (25, 26). Since the rescue with constitutively active calcineurin was incomplete, additional Ca2+-sensitive pathways downstream of CaV1.2 are likely necessary.
The mechanisms by which CaV1.2 affects cellular processes in other nonexcitable tissues may be similar in some cases and different in others. For example, the baldness observed in all TS patients at birth could result from excessive activation of the calcineurin/NFAT pathway (via the mutant CaV1.2), as suggested by the hirsutism side effect commonly seen in patients treated with calcineurin inhibitors for immunosuppression. On the other hand, it is not readily obvious how the mutant CaV1.2 leads to the invariant syndactyly phenotype through a calcineurin/NFAT pathway. Interdigital apoptosis has been shown to be induced by BMPs, counterbalanced by noggin, a BMP antagonist, and survival signals from FGFs, all under the master control of the secreted morphogen Sonic hedgehog (Shh) (27), but not from an NFAT-dependent process.
Our findings provide a platform to specifically define the unexpected contributions of CaV1.2 to nonexcitable tissues and to explore more generally the roles of ion channels in these tissues. Loss-of-function mutations in GJA1, the gene coding for the gap-junction protein connexin43, underlie oculodentodigital dysplasia, which shares several features with TS, including craniofacial and tooth abnormalities, as well as syndactyly (28). This study provides additional support for the concept that ion channels are critical for multiple, unexpected processes in nonexcitable tissues.
Methods
Mouse models.
CaV1.2+/LacZ mice (B6.129P2-Cacna1ctm1Dgen/J) were obtained from The Jackson Laboratory. CaV1.2WT and CaV1.2TS mice, previously described (6), were crossed with Prx1-Cre (B6.Cg-Tg[Prx1-cre]1Cjt/J; The Jackson Laboratory). Mandible and tibia measurements were performed at the times indicated either by dissection and isolation or by x-ray imaging on a Faxitron LX-60 Digital Radiography System acquired at a voltage of 26 kV and a 13-second acquisition time. All mouse experiments were approved by the Institutional Animal Care and Use Committee at Duke University.
Zebrafish embryo collection and preparation.
Zebrafish (Danio rerio) were maintained following published protocols (29) and various developmental stages were identified as described previously. wea and haf mutants (provided by Deborah Yelon, New York University School of Medicine, New York, New York, USA) were bred to generate wea;haf double heterozygotes. Wild-type Tübingen fish; wea;haf double heterozygotes; isl (cacna1cm231; provided by Calum MacRae, Harvard University, Cambridge, Massachusetts, USA); and transgenic fish expressing eGFP under control of the sox10 promoter (sox10:egfp, provided by Thomas Schilling, UC Irvine, Irvine California, USA) were used to obtain embryos where indicated in the text. All zebrafish experiments were approved by the Institutional Animal Care and Use Committee of Duke University.
Whole-mount Alcian blue staining and whole-mount cell-size imaging.
At 72 hpf, fish were anesthetized with 1X tricaine and fixed in 2% paraformaldehyde and PBS (wt/vol) for 2 to 4 hours. After a graded rinse in ethanol, they were transferred into a solution of 0.1% Alcian blue (Sigma-Aldrich), 80% ethanol, and 20% acetic acid (wt/vol/vol) and incubated overnight at 4°C. After a series of graded ethanol-PBS rinses, embryos were bleached with 1% KOH and 3% H2O2 (vol/vol) for 2 hours, followed by a mild digestion overnight at 4°C with 0.05% trypsin, EDTA, saturated sodium tetraborate, and PBS (vol/wt/vol). Stained embryos were preserved and imaged in 80% glycerol (vol/vol). Quantification on the whole mandible was performed with ImageJ software (NIH) with the observer blinded to the treatments. Following whole mandible imaging and quantification, embryos were decapitated and had excess tissue stripped before being flat-mounted between a slide and coverslip. Images were then taken on a Leica phase-contrast compound light microscope. Images were contrast and brightness enhanced. Data analysis was performed with OriginLab 8.5.
Immunohistochemistry.
Embryos (sox10:egfp) at 76 hpf were treated with bromodeoxyuracil (BrdU) for 1 hour over ice and were allowed to recover for 1 hour at 27.8°C. Embryos were then anesthetized with 1X tricaine and fixed in 4% paraformaldehyde and PBS (wt/vol) for 2 hours. Fish were then washed gradually into 100% methanol for 3 days at 4°C. Subsequently, they were rehydrated into PBS, treated with 2N HCl at 42°C for 30 minutes. Following PBST (PBS and 0.1% Tween [vol/vol]) washes, embryos were incubated with 1:200 mouse IgG anti-BrdU overnight at 4°C and then incubated with 1:200 goat anti-mouse IgG Alexa Fluor 488 overnight at 4°C. In order to visualize the jaw, embryos were incubated with 1:200 mouse IgG1 anti-GFP overnight and then with a secondary rabbit anti-mouse IgG1 Alexa Fluor 543 overnight. Embryo heads were removed, stabilized in methycelullose in the middle of a depression slide, and gently compressed by a coverslip before confocal microscopy. Quantification was performed using ImageJ software (NIH). A threshold was set and only cells containing GFP were examined for BrdU staining.
Antisense morpholino knockdown and RNA rescue analysis.
Two morpholino oligonucleotides (Gene Tools) against cacna1c were designed against the splice-site acceptor of exon 4 (CCCGTTCCTAGACAGACGAAACAGA) and the splice-site donor of exon 4 (GGATCTTGCACTCACCTACGACCA). Two scrambled 25-nt control morpholinos were designed by Gene Tools. Morpholinos were diluted and injected at 2 ng per embryo in a 2-nl bolus into single-cell zygotes. Efficacy of knockdown was assessed by quantitative PCR (qPCR) (Supplemental Figure 3) using qRT-PCR following protocols previously described. Primers used were as follows: Exon 3 forward, TTGCAAACTGTGTGGCCTTAGCTG; Intron 3 forward, CCGGTGCAAAGACTTTCAGTGTGT; Exon 4 forward, TCACCCTAATGCATACCTGCGGAA; and Exon 5 reverse, TTGCCTCCCATTGAAGTACCACCA. Rabbit Cacna1c cRNA (CaV1.2WT) was prepared as described previously from pCARDHE using T7 mMESSAGE mMACHINE (Ambion, Life Technologies) (30). The CaV1.2TS cRNA was prepared after mutating pCARDHE (G436R) with QuikChange (Agilent). The CaV1.24EQ cRNA was prepared after simultaneously mutating E393, E736, E1145, and E1446 to A using the QuikChange Multi Site–Directed mutagenesis kit (Agilent Technologies). The constitutively active calcineurin cDNA was provided by Paul Rosenberg (Duke University, Durham, North Carolina, USA) and cloned into pGHE for cRNA synthesis. Each construct was coinjected with the cacna1c morpholinos at 800 pg per embryo where indicated in the text.
Pharmacology and drugs.
Nisoldipine, FK506, and cyclosporine were dissolved in DMSO (all from Sigma-Aldrich). Embryos (24 hpf) were chemically and manually dechorionated using pronase. Drugs were applied directly into embryo medium, which was supplemented with fresh medium every 24 hours. The following concentrations were used: 1% DMSO (vol/vol), 5 μM nisoldipine, 5 μm cyclosporine, and 1 μm FK506.
Neural crest isolation and electrophysiology.
Embryos (sox10:egfp) at 64 to 66 hpf were anesthetized with 2X tricaine and then decapitated in HBSS without calcium or magnesium. Isolated heads were washed in HBSS, manually dissected, and then cells were enzymatically dissociated with a combination of collagenase and trypsin-EDTA in HBSS. Cells were spun down, washed, and resuspended in 10% FBS, 1% penicillin/streptomycin, and DMEM-F12 and incubated on glass coverslips coated with fibronectin, poly-D-lysine, and laminin. Ba2+ currents were recorded using the whole-cell patch-clamp technique. Patch pipettes were fabricated from borosilicate glass (Warner Instruments) by a P-97 Flaming-Brown micropipette puller (Sutter Instruments). Pipette resistance was between 6.0 and 8.0 MΩ. Voltage-clamp experiments were performed with an Axopatch 200B amplifier (Molecular Devices) at room temperature (20°C–22°C) 2 hours after plating. A Tyrode bath solution contained (in mM; Sigma-Aldrich) NaCl 140, KCl 5.4, CaCl2 1, MgCl2 1, HEPES 5, and glucose 10 (pH 7.3). Once the cell was ruptured, solution was quickly exchanged to a Ba2+ recording solution containing (in mM; Sigma-Aldrich) TEA-Cl 110, CsCl 5, BaCl2 15 mM, MgCl2 1, HEPES 5, glucose 10, and 4-aminopyridine 2, pH 7.2, with CsOH. IBa was recorded using a ramp protocol from –80 to +60 over 500 milliseconds. Internal solution contained (in mM; Sigma-Aldrich) CsOH-H2O 70, aspartic acid 80, CsCl 40, NaCl 10, HEPES 10, EGTA 10, MgATP 5, and Na2GTP 4 (pH 7.2). Recordings were filtered at 2 kHz and digitally sampled at 25 kHz. Whole-cell membrane capacitance was calculated by integrating the capacitive transient elicited by a 5-mV pulse from –80 mV to 75 mV. Pipette capacitance was corrected and whole-cell capacitance and series resistance were at least 80% compensated. Peak negative currents were normalized to cell capacitance and reported as current density (pA/pF). To record resting membrane potential, a perforated patch with 400 nM amphotericin (Sigma-Aldrich) was performed using the following internal solution (in mM; Sigma-Aldrich): KCl 110, NaCl 5, MgATP 5, phosphocreatine 5, Na2GTP 1, HEPES 10, pH 7.3, and Tyrode extracellular solution. Junction potential was calculated to be 5.1 mV and not corrected. Data analysis was performed using Clampfit 10.2 software (Axon Instruments) and OriginPro 8.5 (OriginLab).
Chondrocyte isolation and immunocytochemistry.
Chondrocytes were isolated from femurs and tibia as described (31). Cells were plated onto poly-D-lysine–coated coverslips and allowed to adhere for at least 2 hours. Cells were washed and fixed with 2% paraformaldehyde in PBS for 15 minutes at room temperature. After quenching with 10 mM glycine in PBS, cells were permeabilized with 0.2% Triton X-100 for 5 minutes and blocked with 10% goat serum. Cells were incubated in primary antibody (rabbit anti-α1C from Alomone Labs) overnight at 4°C in a PBS solution containing 3% goat serum, 0.1% Triton X-100, and 1% BSA. Cells were washed and incubated with goat anti–rabbit-Cy3 secondary antibody (Jackson Immunoresearch) in the same solution as the primary antibody for 45 minutes at room temperature. Coverslips were washed and mounted in Vectashield (Vector Laboratories) containing DAPI and imaged on a Zeiss Axio Imager. Exposure times were kept constant for all images and the experimenter was blinded to the genotype.
Statistics.
Differences in measured variables between genetically altered mice and the control group or between or among treatment groups were assessed using a 2-tailed Student’s t test, ANOVA, or χ-squared test, where appropriate. Data are expressed as mean ± SEM where applicable. A P value less than 0.05 was considered a statistically significant difference.
Study approval.
All animal studies were approved by the Institutional Animal Care and Use Committee of Duke University.
Supplementary Material
Acknowledgments
This work was supported by NHLBI R01 HL71165, the March of Dimes, an American Heart Association Established Investigator Award, and the Duke Chancellor’s Science Council Pilot Funds program (to G.S. Pitt).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Note regarding evaluation of this manuscript: Manuscripts authored by scientists associated with Duke University, The University of North Carolina at Chapel Hill, Duke-NUS, and the Sanford-Burnham Medical Research Institute are handled not by members of the editorial board but rather by the science editors, who consult with selected external editors and reviewers.
Citation for this article: J Clin Invest. 2013;123(4):1638–1646. doi:10.1172/JCI66903.
References
- 1.Splawski I, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119(1):19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Marks ML, Whisler SL, Clericuzio C, Keating M. A new form of long QT syndrome associated with syndactyly. J Am Coll Cardiol. 1995;25(1):59–64. doi: 10.1016/0735-1097(94)00318-K. [DOI] [PubMed] [Google Scholar]
- 3.Seisenberger C, et al. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem. 2000;275(50):39193–39199. doi: 10.1074/jbc.M006467200. [DOI] [PubMed] [Google Scholar]
- 4.Chai Y, et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127(8):1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- 5.Grenier J, Teillet MA, Grifone R, Kelly RG, Duprez D. Relationship between neural crest cells and cranial mesoderm during head muscle development. PLoS One. 2009;4(2):e4381. doi: 10.1371/journal.pone.0004381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasca SP, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17(12):1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ. Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33(2):77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 8.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–1241. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rottbauer W, Baker K, Wo ZG, Mohideen MA, Cantiello HF, Fishman MC. Growth and function of the embryonic heart depend upon the cardiac-specific L-type calcium channel alpha1 subunit. Dev Cell. 2001;1(2):265–275. doi: 10.1016/S1534-5807(01)00023-5. [DOI] [PubMed] [Google Scholar]
- 10.Auman HJ, Coleman H, Riley HE, Olale F, Tsai HJ, Yelon D. Functional modulation of cardiac form through regionally confined cell shape changes. PLoS Biol. 2007;5(3):e53. doi: 10.1371/journal.pbio.0050053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Ellinor PT, Sather WA, Zhang JF, Tsien RW. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature. 1993;366(6451):158–161. doi: 10.1038/366158a0. [DOI] [PubMed] [Google Scholar]
- 12.Panakova D, Werdich AA, Macrae CA. Wnt11 patterns a myocardial electrical gradient through regulation of the L-type Ca(2+) channel. Nature. 2010;466(7308):874–878. doi: 10.1038/nature09249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carney TJ, et al. A direct role for Sox10 in specification of neural crest-derived sensory neurons. Development. 2006;133(23):4619–4630. doi: 10.1242/dev.02668. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, et al. Calcium influx through Cav1.2 is a proximal signal for pathological cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2011;50(3):460–470. doi: 10.1016/j.yjmcc.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang CP, et al. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118(5):649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 16.O’Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O’Neill EA. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature. 1992;357(6380):692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- 17.Moran D. Voltage-dependent -L-type Ca2+ channels participate in regulating neural crest migration and differentiation. . Am J Anat. 1991;192(1):14–22. doi: 10.1002/aja.1001920103. [DOI] [PubMed] [Google Scholar]
- 18.Collazo A, Bronner-Fraser M, Fraser SE. Vital dye labelling of Xenopus laevis trunk neural crest reveals multipotency and novel pathways of migration. Development. 1993;118(2):363–376. doi: 10.1242/dev.118.2.363. [DOI] [PubMed] [Google Scholar]
- 19.Muller CS, et al. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A. 2010;107(34):14950–14957. doi: 10.1073/pnas.1005940107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebert AM, McAnelly CA, Srinivasan A, Linker JL, Horne WA, Garrity DM. Ca2+ channel-independent requirement for MAGUK family CACNB4 genes in initiation of zebrafish epiboly. Proc Natl Acad Sci U S A. 2008;105(1):198–203. doi: 10.1073/pnas.0707948105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKinney MC, Kulesa PM. In vivo calcium dynamics during neural crest cell migration and patterning using GCaMP3. Dev Biol. 2011;358(2):309–317. doi: 10.1016/j.ydbio.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tandan S, et al. Physical and functional interaction between calcineurin and the cardiac L-type Ca2+ channel. . Circ Res. 2009;105(1):51–60. doi: 10.1161/CIRCRESAHA.109.199828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oliveria SF, Dell’Acqua ML, Sather WA. AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. . Neuron. 2007;55(2):261–275. doi: 10.1016/j.neuron.2007.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322(4):1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 25.Aliprantis AO, et al. NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118(11):3775–3789. doi: 10.1172/JCI35711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Winslow MM, et al. Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev Cell. 2006;10(6):771–782. doi: 10.1016/j.devcel.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 27.Pajni-Underwood S, Wilson CP, Elder C, Mishina Y, Lewandoski M. BMP signals control limb bud interdigital programmed cell death by regulating FGF signaling. Development. 2007;134(12):2359–2368. doi: 10.1242/dev.001677. [DOI] [PubMed] [Google Scholar]
- 28.Paznekas WA, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72(2):408–418. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nüsslein-Volhard C, Dahm R. Zebrafish: A Practical Approach. New York, New York, USA: Oxford University Press; 2002. [Google Scholar]
- 30.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. . Neuron. 2004;41(5):745–754. doi: 10.1016/S0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 31.Gosset M, Berenbaum F, Thirion S, Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat Protoc. 2008;3(8):1253–1260. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]
- 32.Bohnsack BL, Gallina D, Kahana A. Phenothiourea sensitizes zebrafish cranial neural crest and extraocular muscle development to changes in retinoic acid and IGF signaling. PLoS One. 2011;6(8):e22991. doi: 10.1371/journal.pone.0022991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.