

Abstract
Bioluminescence imaging with luciferase enzymes requires access to light-emitting, small molecule luciferins. Here, we describe a rapid method to synthesize d-luciferin, the substrate for firefly luciferase (Fluc), along with a novel set of electronically modified analogs. Our procedure utilizes a relatively rare, but synthetically useful dithiazolium reagent to generate heteroaromatic scaffolds in a divergent fashion. Two of the luciferin analogs produced with this approach emit light with Fluc in vitro and in live cells. Collectively, our work increases the number of substrates that can be used for bioluminescence imaging and provides a general strategy for synthesizing new collections of luciferins.
Bioluminescence imaging is among the most popular methods for visualizing biological processes in vitro, in live cells, and even in whole organisms.1,2 At the core of this technology are enzymes (luciferases) that catalyze the oxidation of small molecule substrates (luciferins) to release visible light. Since cells and tissues do not normally emit significant numbers of visible photons, bioluminescence provides extremely high signal-to-noise ratios, making it well-suited for sensitive imaging applications.3 Indeed, this technology is routinely used to monitor cell trafficking networks, gene expression patterns, and drug delivery mechanisms in vivo.4,5 Despite its remarkable versatility, bioluminescence has been largely limited to monitoring one cell type or biological feature at a time. This is because only a handful of luciferases are suitable for biological work and, of these, nearly all utilize the same substrate (d-luciferin). Retooling bioluminescence technology for multi-component imaging requires access to larger collections of light-emitting luciferins. Such molecules could potentially provide different colors of bioluminescent light or be utilized by novel luciferase variants. Unfortunately, luciferins have been notoriously difficult to produce, owing to a lack of rapid and reliable syntheses for these richly functionalized molecules. We report here an expedient method to prepare d-luciferin, along with a new class of light-emitting analogs. This chemistry is both efficient and scalable and will bolster ongoing efforts to expand the bioluminescence toolkit.
The vast majority of efforts to develop new bioluminescent tools have focused on mutating luciferase enzymes from the firefly (Fluc) and related organisms.6,7 By contrast, only a handful of studies have focused on modifying the structure of d-luciferin (1), the substrate common to all insect luciferases. This disparity is surprising, given the prominent role of the small molecule in the light-emitting reaction. During the Fluc-catalyzed oxidation of d-luciferin, an excited state version of the product (oxyluciferin) is generated; relaxation of this molecule to the ground state releases a photon of yellow-green light (Figure 1).8 Since the chemical makeup of the excited-state emitter influences light production, modifications to the aromatic core can alter the wavelength and intensity of photons released. Miller and others have shown that luciferin variants containing a nitrogen atom in place of the exocyclic oxygen are efficiently processed by Fluc and emit red light.9–11 In related work, Branchini and others have replaced the entire benzothiazole core of d-luciferin with quinoline, naphthalene, and coumarin units. These analogs emitted different colors of light with Fluc, but robust emission was only observed at elevated pH values.12,13 Although these luciferins have somewhat limited utility in biological assays, they remain the only examples of Fluc substrates that do not contain a benzothiazole moiety.
Figure 1.
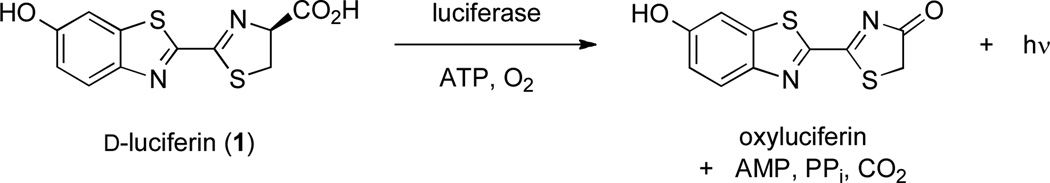
Luciferase-catalyzed oxidation of D-luciferin releases visible light.
We aimed to expand the repertoire of modified heteroaromatic luciferins suitable for biological studies. In particular, we were attracted to luciferins with benzimidazole and imidazoline rings (the nitrogenous counterparts to the benzothiazole and thiazoline units in d-luciferin) (2–4). Heterocycles of this sort are capable of absorbing and emitting light, an important criterion for bioluminescent substrates.14,15 White and McElroy have also shown that benzimidazole and other heterocycles are competitive inhibitors of Fluc, suggesting that 2–4 would be able to access the substrate binding pocket.16 Last, since benzimidazole and imidazoline motifs are present in numerous pharmaceutical agents, we felt that the electronically modified analogs would possess reasonable bioavailability and metabolic stability for use in cells and animal models.17,18
Analogs 2–4, like d-luciferin, contain a unique 2-2´ linkage of heteroaromatic rings. This connectivity is scarcely observed in known natural products, and facile methods to prepare such linkages are rare.19 In fact, the first synthesis of d-luciferin reported by White in 1963 is basically the same route used to produce nearly all luciferins today.20 This synthesis proceeds through a cyanobenzothiazole intermediate (5), which, upon protecting group removal, can be condensed with d-cysteine to provide the native luciferin. This condensation is both mild and high-yielding, making it an appealing method for late-stage introduction of the luciferin stereocenter. Unfortunately, the White synthesis of 5 requires seven steps and is not amenable to heteroatom substitutions.
Recognizing the utility of cyano heterocycles for luciferin production, we aimed to identify a more expedient route to 5, along with the analogous cyanobenzimidazole 6. Condensation of these scaffolds with either d-cysteine or d-amino propionic acid could provide the entire set of luciferin analogs (2–4). To access 5 and 6 in tandem, we were drawn to the dithiazolium chloride 9 (Scheme 1). This reagent, also known as Appel’s salt, has been previously used to synthesize both benzothiazole and benzimidazole scaffolds from anilines.21 Appel’s salt condenses readily with arylamines, and the resulting iminodithiazoles can be easily opened with a variety of nucleophiles.22 If the nucleophile is present on the aniline itself (as in the case of ortho-amino anilines), cyanobenzimidazole structures can be isolated directly. In the absence of intramolecular nucleophiles, the dithiazole adduct can be fragmented with a variety of exogenously supplied reagents. When amidine bases are used, dithiazole cleavage provides thioformamides; molecules of this sort can be readily cyclized to cyanobenzothiazoles.22
Scheme 1.
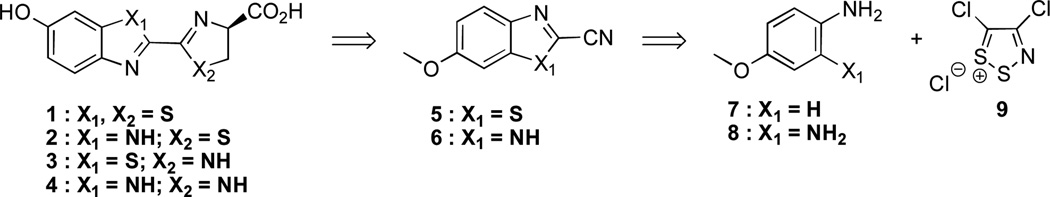
Retrosynthetic analysis of luciferin analogs.
To investigate the utility of Appel’s salt for luciferin synthesis, we first used the reagent to prepare the cyanobenzothiazole 5 (en route to d-luciferin, Scheme 2a). p-Anisidine was treated with 9 to provide the expected dithiazole adduct 10. This intermediate was isolable using standard chromatographic techniques and found to be remarkably shelf stable. Treatment of 10 with excess DBU produced cyanothioformamide 11 in excellent yield. Palladium- and copper-mediated cyclization of this compound generated the key cyanobenzothiazole 5. Notably, this route to 5 is four steps shorter than the sequence employed by White, and provides the compound in markedly better yield (84% versus 10% overall). The desired luciferin 1 was eventually isolated by removal of the methyl protecting group from 5, followed by condensation with d-cysteine under mild conditions. The functional activity of the isolated luciferin was also confirmed in light emission assays with Fluc (Figure S1).
Scheme 2.

Synthesis of luciferin scaffolds: (a) d-Luciferin (1) and the imidazoline analog 3. (b) Benzimidazole analogs 2 and 4.
Encouraged by these results, we next investigated whether Appel’s salt could provide access to the benzimidazole intermediate 6 (Scheme 2b). Gratifyingly, this molecule was isolated in a single step upon incubation of bis-aniline 8 with 9. In this reaction, the initial dithiazole adduct is likely trapped by the ortho-amino substituent of 8, providing the cyclized product. Cyanobenzimidazole 6 was ultimately de-methylated and condensed with d-cysteine as above to isolate luciferin 2. Multi-gram quantities of both luciferins 1 and 2 have been produced using the routes outlined in Scheme 2, highlighting the scalability of the approach.
The cyano heterocycles produced with Appel’s salt can be condensed with a variety of other 1,2-disubstituted nucleophiles in addition to d-cysteine. We exploited this mode of reactivity to generate the imidazoline rings present in luciferins 3 and 4. First, intermediates 12 and 14 were converted into the corresponding imidates using standard conditions. The imidates were not isolated, but treated directly with diamino propionic acid to afford the desired luciferins in reasonable yield.
Our initial efforts to characterize luciferins 2 and 4 were complicated by tautomerism. Benzimidazole scaffolds are known to undergo rapid N-H isomerization in solution (Figure S2), resulting in significantly broadened 1H- and 13C-NMR signals. We were also concerned that such rapid tautomerization would suppress bioluminescent light emission from the analogs. Such quenching behavior has been observed with other electronically excited benzimidazoles.23,24 To mitigate against potential quenching effects and aid our structural characterization efforts, we prepared a methylated version of 2 (Scheme S1). Interestingly, only one N-methyl regioisomer (15, Scheme S1) was formed in reasonable yield from intermediate 6.
With the nitrogenous analogs in hand, we assayed the compounds for light emission with Fluc. Luciferins 2–4 and 15 were incubated with the enzyme, ATP and coenzyme A (which has been shown to reduce product inhibition) at pH 7.4.25 Light emission was measured using a cooled CCD camera, and representative images are shown in Figure 2. No photons were detected for analog 3, and only minimal light emission was observed with the related imidazoline 4 at low substrate concentrations. These reduced intensities may be attributed to poor binding to Fluc, lower efficiencies of light production, or a combination of factors (Figure S3). By contrast, robust emission was observed with the benzimidazole variants 2 and 15, suggesting that these molecules can be converted to light-emitting species in the enzyme active site (Figure 2). Both analogs are weaker emitters than the native substrate (~100-fold reduced emission intensities in the µM range, Figure S1B), but on par with other luciferin scaffolds used in biological assays.10 Additional improvements in light output may also be obtained using the analogs in combination with mutant luciferases.9,26 Importantly, the bioluminescence emissions from 2 and 15 were long-lived (Figure S4).10 Prolonged light release is necessary for numerous imaging applications in vivo, and has been difficult to achieve with other luciferins.9
Figure 2.

Light production from luciferin analogs. (A) Bioluminescence images from analogs 2–4 and 15 (0–500 µM) incubated with Fluc or no enzyme. (B) Quantification of the images from (A). (C) Bioluminescence emission spectra for luciferins 1–2, 4, and 15
We next analyzed the bioluminescence emission profiles for 2, 4, and 15. The spectra for these analogs, like most luciferins, are quite broad and indicate the presence of tautomers in aqueous solution (Figure 2C). Benzimidazole analog 2 was found to emit maximally at 578 nm, slightly red-shifted from d-luciferin (λmax = 557 nm) at room temperature. The bioluminescence spectrum of 2 is also substantially different from the analog’s fluorescence profile, indicating a potential role for Fluc in modulating the color of light released (Table S1). Interestingly, the bioluminescence spectrum for benzimidazole analog 15 is substantially blue-shifted from luciferins 1 and 2. With peak emission near 460 nm, analog 15 emits the largest percentage of blue light among the known Fluc substrates. This result also implies that 15 may be useful for multi-component imaging applications, as its emission can be readily resolved from other luciferins using appropriate filter sets.
To probe whether the light-emitting luciferins would also be useful for cell studies, we incubated 2, 4, and 15 with Fluc-expressing HEK 293 cells. Photon emission was measured using a cooled CCD camera, and sample images are shown in Figure 3. Dose-dependent light emission was observed for both 2 and 15, with photon intensities peaking around 10–20 minutes post-substrate addition (Figure S5). No emission was observed from the nitrogenous analog 4 in this assay, even at high substrate concentrations. This is likely due to the poor cell permeability of the compound. It should also be noted that no light was observed in the absence of the analogs, or when the compounds were incubated with non-luciferase expressing cells (Figure 3). These results are consistent with the light-emitting behavior of d-luciferin in whole cells, and suggest that the benzimidazole scaffolds are sufficiently biocompatible for use in cellular imaging studies.2,10
Figure 3.

Cellular imaging with luciferin analogs. (A) Bioluminescence images from 2, 4, and 15 (250 µM-1 mM) incubated with luciferase-expressing HEK 293 cells or wild type cells (wt HEK). (B) Quantification of the images from (A).
In summary, we have developed a facile method to prepare luciferins from aniline starting materials and Appel’s salt. This procedure was used to synthesize d-luciferin, the native substrate for Fluc, along with a series of nitrogenous analogs. Two of the analogs were found to emit light with purified Fluc and in live cells, and these scaffolds will be generally useful for imaging studies. More broadly, the chemistry reported here provides a gateway to access additional luciferin architectures. For example, the adducts formed upon aniline condensation with Appel’s salt can be selectively fragmented to access quinazolines, benzoxazoles, and a variety of other heterocycles in addition to the benzothiazole and benzimidazole scaffolds examined here.22 This diverse manifold of reactivity will likely be exploited for synthesizing new classes of heteroaromatic luciferins in the near future. The ability to rapidly access novel luciferin substrates will expand the bioluminescence toolkit along with the scope of bioluminescence imaging.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the American Cancer Society (IRG-98-279-07 to J.A.P.) and the UC Irvine School of Physical Sciences. Some experiments reported in this paper were performed at the Laboratory for Fluorescence Dynamics (LFD) at the University of California, Irvine (UCI). The LFD is supported jointly by the National Institute of General Medical Sciences of the National Institutes of Health (8P41GM103540) and UC Irvine. We also thank members of the Weiss, Chamberlin, and Overman laboratories for providing reagents and experimental assistance.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental details, full spectroscopic data for all new compounds, and additional images are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Contag CH. Annu. Rev. Pathol. 2007;2:277. doi: 10.1146/annurev.pathol.2.010506.091930. [DOI] [PubMed] [Google Scholar]
- 2.Prescher JA, Contag CH. Curr. Opin. Chem. Biol. 2010;14:80. doi: 10.1016/j.cbpa.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Dothager RS, Flentie K, Moss B, Pan MH, Kesarwala A, Piwnica-Worms D. Curr. Opin. Biotechnol. 2009;20:45. doi: 10.1016/j.copbio.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thorne N, Inglese J, Auld DS. Chem. Biol. 2010;17:646. doi: 10.1016/j.chembiol.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, Mitsiades N, Schlossman RL, Munshi NC, Kung AL, Griffin JD, Richardson PG, Anderson KC, Mitsiades CS. Nat. Med. 2010;16:483. doi: 10.1038/nm.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Branchini BR, Ablamsky DM, Davis AL, Southworth TL, Butler B, Fan F, Jathoul AP, Pule MA. Anal. Biochem. 2010;396:290. doi: 10.1016/j.ab.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Nakatsu T, Ichiyama S, Hiratake J, Saldanha A, Kobashi N, Sakata K, Kato H. Nature. 2006;440:372. doi: 10.1038/nature04542. [DOI] [PubMed] [Google Scholar]
- 8.Fraga H. Photochem. Photobiol. Sci. 2008;7:146. doi: 10.1039/b719181b. [DOI] [PubMed] [Google Scholar]
- 9.Reddy GR, Thompson WC, Miller SC. J Am Chem Soc. 2010;132:13586. doi: 10.1021/ja104525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinde R, Perkins J, Contag CH. Biochemistry. 2006;45:11103. doi: 10.1021/bi060475o. [DOI] [PubMed] [Google Scholar]
- 11.White EH, Worther H. J. Org. Chem. 1966;31:1484. doi: 10.1021/jo01343a039. [DOI] [PubMed] [Google Scholar]
- 12.Branchini BR, Hayward MM, Bamford S, Brennan PM, Lajiness EJ. Photochem. Photobiol. 1989;49:689. doi: 10.1111/j.1751-1097.1989.tb08442.x. [DOI] [PubMed] [Google Scholar]
- 13.Takakura H, Sasakura K, Ueno T, Urano Y, Terai T, Hanaoka K, Tsuboi T, Nagano T. Chem. Asian J. 2010;5:2053. doi: 10.1002/asia.201000219. [DOI] [PubMed] [Google Scholar]
- 14.Henary MM, Wu Y, Cody J, Sumalekshmy S, Li J, Mandal S, Fahrni CJ. J. Org. Chem. 2007;72:4784. doi: 10.1021/jo070433l. [DOI] [PubMed] [Google Scholar]
- 15.Vazquez SR, Rodriguez MC, Mosquera M, Rodriguez-Prieto F. J. Phys. Chem. A. 2008;112:376. doi: 10.1021/jp076634a. [DOI] [PubMed] [Google Scholar]
- 16.Denburg JL, Lee RT, McElroy WD. Arch. Biochem. Biophys. 1969;134:381. doi: 10.1016/0003-9861(69)90297-5. [DOI] [PubMed] [Google Scholar]
- 17.Baumann M, Baxendale IR, Ley SV, Nikbin N. Beilstein J. Org. Chem. 2011;7:442. doi: 10.3762/bjoc.7.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daniel-Mwambete K, Torrado S, Cuesta-Bandera C, Ponce-Gordo F, Torrado JJ. Int. J. Pharm. 2004;272:29. doi: 10.1016/j.ijpharm.2003.11.030. [DOI] [PubMed] [Google Scholar]
- 19.Meroni G, Rajabi M, Santaniello E. Arkivoc. 2009:265. [Google Scholar]
- 20.White EH, McCapra F, Field GF. J. Am. Chem. Soc. 1963;85:337. [Google Scholar]
- 21.Akhavan-Tafti HSRD, Guoping W, Eickholt RA, Gupta RK, Kaanumalle LS. 2011/0014599A1 US. 2011 Jan.
- 22.Cuadro AM, Alvarezbuilla J. Tetrahedron. 1994;50:10037. [Google Scholar]
- 23.Ingram AJ, Dunlap AG, Dipietro R, Muller G. J. Phys. Chem. A. 2011;115:7912. doi: 10.1021/jp201209k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukherjee TK, Panda D, Datta A. J. Phys. Chem. B. 2005;109:18895. doi: 10.1021/jp052917w. [DOI] [PubMed] [Google Scholar]
- 25.da Silva LP, da Silva JCGE. Photoch. Photobio. Sci. 2011;10:1039. [Google Scholar]
- 26.Harwood KR, Mofford DM, Reddy GR, Miller SC. Chem. Biol. 2011;18:1649. doi: 10.1016/j.chembiol.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.