

Abstract
Tumor-derived mutant forms of p53 compromise its DNA binding, transcriptional, and growth regulatory activity in a manner that is dependent upon the cell-type and the type of mutation. Given the high frequency of p53 mutations in human tumors, reactivation of the p53 pathway has been widely proposed as beneficial for cancer therapy. In support of this possibility p53 mutants possess a certain degree of conformational flexibility that allows for re-induction of function by a number of structurally different artificial compounds or by short peptides. This raises the question of whether physiological pathways for p53 mutant reactivation also exist and can be exploited therapeutically. The activity of wild-type p53 is modulated by various acetyl-transferases and deacetylases, but whether acetylation influences signaling by p53 mutant is still unknown. Here, we show that the PCAF acetyl-transferase is down-regulated in tumors harboring p53 mutants, where its re-expression leads to p53 acetylation and to cell death. Furthermore, acetylation restores the DNA-binding ability of p53 mutants in vitro and expression of PCAF, or treatment with deacetylase inhibitors, promotes their binding to p53-regulated promoters and transcriptional activity in vivo. These data suggest that PCAF-mediated acetylation rescues activity of at least a set of p53 mutations. Therefore, we propose that dis-regulation of PCAF activity is a pre-requisite for p53 mutant loss of function and for the oncogenic potential acquired by neoplastic cells expressing these proteins. Our findings offer a new rationale for therapeutic targeting of PCAF activity in tumors harboring oncogenic versions of p53.
The presence of p53 missense mutations is an overwhelming characteristic of solid tumors and likely represents a critical step in the oncogenic process (reviewed in Brown et al., 2009; Vousden and Prives, 2009). In its wild-type configuration, p53 is predominantly a nuclear protein, which exerts anti-proliferative effects by regulating a variety of genes that in turn, induce G1 arrest, senescence, or apoptosis, depending upon the cellular context and the type of stress. Since p53 is a sequence-specific DNA-binding transcription factor, and the majority of mutations occur within the region encoding the DNA-binding domain, it has been argued that a prominent consequence of p53 mutations consists in disabling the sequence-specific DNA binding and transcriptional activity. However, in addition to loss of function, it is clear that the majority of p53 mutants also gain novel pro-oncogenic activity relatively to the wild-type protein, a feature that explains why one mutated copy of the p53 allele in the absence of a wild-type allele is often maintained even in genomic unstable, advanced forms of neoplasias (Brosh and Rotter, 2009; Brown et al., 2009).
Based on crystallographic studies the mutations more frequently found in human tumors have been classified into two main categories: type I mutations, which affect amino acid residues directly involved in the DNA interaction (R248 and H273), and class II mutations involving residues responsible for the stabilization of the three-dimensional structure of p53 (Cho et al., 1994). This latter category, defined as structural mutants, includes the majority of p53 proteins found in human tumors, such as the V143, R175, G245, R249, D281, and R282 mutants, all of which destabilize p53 conformation and the p53–DNA-binding inter-phase. A number of studies in the past revealed that DNA-binding capacity can be artificially restored for several of these mutants via incubation with anti-p53-specific antibodies, via phosphorylation of the p53 C-terminus, or by introducing amino acid substitutions (Hupp et al., 1993; Niewolik et al., 1995; Nikolova et al., 2000; Joerger and Fersht, 2007). These latter, called “second site” mutations, rescue activity by creating novel DNA contacts, by correcting local distortion, or by increasing the thermodynamic stability of the DNA-binding domain (Joerger et al., 2005). Similarly, a number of artificial compounds have been identified that can reactivate mutant p53 by directly stabilizing the interaction with DNA and/or by preventing misfolding or aggregation. In fact, the structure of wild-type p53 itself naturally comprises unfolded regions and displays high tendency to aggregation (Sakaguchi et al., 1998; Bell et al., 2002; Veprintsev et al., 2006). The prototype of these reactivating agents are CP-31398 (Foster et al., 1999) ellipticine (Shi et al., 1998), (North et al., 2002), MIRA-1 (Bykov et al., 2005), RITA (Grinkevich et al., 2009), and PRIMA-1 (Lambert et al., 2009). A third category of reactivating molecules is represented by short peptides encompassing the C-terminal region of p53 that, when introduced into tumor cells harboring p53 mutants, lead to induction of p53-regulated genes and apoptosis (Selivanova et al., 1997). In this case, however, the mechanism(s) of activation is less clear. It is noteworthy that transitions from p53 mutant to wild-type activities and conformations are likely physiologically relevant. In fact, very early studies showed that certain p53 mutants retain the ability to activate transcription of genes known to be regulated by wild-type p53 and to induce cell cycle arrest or apoptosis in some cell types but not in others (Chumakov et al., 1993; Miller et al., 1993; Niewolik, 1995; Ludwig et al., 1996; Aurelio et al., 2000; Campomenosi et al., 2001). These multiple lines of evidence suggest that p53 mutants possess a certain degree of conformational flexibility that allows for restoration of function. Because the presence of p53 mutations is often a negative prognostic factor and a predictor of poor radio- and chemotherapy outcome, the identification of the pathways that influence the phenotypic outcome of p53 mutations would have therapeutic implications (Campling and El-Deiry, 2003; Geisler et al., 2003).
We and others have shown that in the case of wild-type p53, the interaction with acetylases and acetylation of p53 itself regulates the DNA binding and transcription activation, as well as the ability of p53 to trigger cell cycle arrest or apoptosis (Sakaguchi et al., 1998; Liu et al., 1999; Feng et al., 2005; Chao et al., 2006; Knights et al., 2006; Tang et al., 2006). In past years various types of acetyl-transferases, such as p300/CBP, PCAF, and TiP60, have been shown to acetylate p53 at different residues (Kruse and Gu, 2009). The current model proposes that acetylation has multiple effects on p53 activities, acting by enabling it to interact with specific sets of promoters, by regulating the interaction with co-activators and co-repressors, as well as by modulating p53 stability and the interaction with MDM2 family members. Acetylation also competes with other lysine-dependent modifications that have inhibitory effects on p53, such as methylation, ubiquitination, and neddylation, thereby regulating a variety of p53 functions dependent upon these modifications (Berger, 2008; Kruse and Gu, 2009). Furthermore, at least two different deacetylases, specifically HDAC1 and Sir2/SIRT1, inhibit p53-dependent transcription and apoptosis (Luo et al., 2000; Vaziri et al., 2001). These studies have identified acetylation as an important point of control for the function of wild-type p53. However, it is currently unknown whether the activity of p53 mutant(s) can be modulated by this important post-translational modification. In this study we provide the first line of evidence that the acetylation pathway, and particularly PCAF, is likely a key determinant of the growth properties of tumor cells expressing certain types of conformational mutant forms of p53.
Materials and Methods
Cells, plasmids, and antibodies
The cell lines employed in this study were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS). The methods for generating H1299 cell lines expressing p53 mutants have been previously described (Knights, 2006). The murine cell lines were described previously (Tzeng et al., 1998). The PCAF and PCAF-ΔHAT vectors were a kind gift from Dr. Keiko Ozato. The anti-PCAF polyclonal goat antiserum was purchased from Santa Cruz (Santa Cruz, CA). The anti-p53 antibodies were purchased from Oncogene Science (Cambridge, MA) (DO1) or from Santa Cruz (FL393). Anti-acetyl–p53 antibodies directed against acetylated K320 and acetylated K373 were from Upstate Biotechnology (Lake Placid, NY) and the p300/CBP polyclonal antibody was described previously (Avantaggiati et al., 1997). Other antibodies were as follows: p21 (WAF1/Ab-5; Calbiochem, San Diego, CA), Bax (anti-Bax/NT; Upstate Biotechnology), and actin (I19; Santa Cruz Biotechnology, Inc.). Proteins were detected by using a chemiluminescence-based system (Pierce Chemical Co., Rockford, IL) according to the manufacturer’s instructions.
Transfections and reporter assays
Transfections were performed by using either lipofectamine (GIBCO, BRL, Carlsbad, CA) or a calcium phosphate-based method (Pharmacia, Pittsburgh, PA). Early-passage cell cultures were employed for both reporter assays and immunoprecipitation experiments. For transactivation assays, cells were plated at 20–40% confluence 12–18 h prior transfection. Unless otherwise indicated, transfections were carried out by using a ratio between reporter and activators of 1:2. Usually, 1μg of reporter plasmid and 0.5–2μg of activator were transfected in the combinations indicated in the legends of the figures. When necessary the total content of transfected DNA was equalized with either the control backbone plasmid, CMVO or with pUC19. Cells were exposed to lipofectamine or to calcium phosphate for about 8–12 h, washed twice with PBS, and re-fed with complete medium. Equal amounts of cell lysates were employed for detection of luciferase activity (Promega, Madison, WI).
Immunoprecipitations and immunoblots
Total cell extracts were prepared from either transfected or untransfected cells by incubating the cells in buffer A (20mM NaPO4 pH 7.8; 240mM NaCl; 0.1% NP40; 5mM EDTA; 1mM DTT), supplemented with freshly prepared protease and phosphatase inhibitors (10mM sodium fluoride, 1mM sodium orthovanadate, 1mM phenylmethylsulfonylfluoride, and leupeptin, aprotinin, and pepstatin at 10μg/ml each). After incubation (10′, 4°C) extracts were centrifuged at 12,000 rpm, and the supernatants were collected. Pellets were re-suspended in 2 vol. of buffer A, vortexed, and incubated on ice for additional 10 min. Extracts from these two extractions were combined and precleared twice with an excess of Protein A. For immunoprecipitation experiments typically, 1–3mg of cell extracts were used. Immunoprecipitations were carried out at 4°C for 45–90 min, precipitates were washed three to six times in lysis buffer, eluted in 2× SDS sample buffer, loaded on 7.5% SDS–PAGE gels (30:0.7 Acryl/Bis), and run overnight. After transfer at 65 V for 4.5 h at room temperature, membranes were incubated in blocking solution (10% horse serum, 0.1% Tween 20, in 1× PBS) for 1 h. Immunocomplexes were detected by using a chemiluminescence-based system (Amersham, Pittsburgh, PA) according to the manufacturer’s instructions.
Gel retardation and acetylation assays
Methods for infection and purification of baculovirus-infected SF21 cells were described before (Knights, 2006). Baculoviruses expressing unflagged p53 wild-type, p53V143A, and p53-R175H (Fig. 5A) were a kind gift from Dr. Carol Prives. The p21 oligonucleotide was as follows: P21-5′ (37 mer) TCT GGC CAT CAG GAA CAT GTC CCA ACA TGT TGA GCT CTG G, and its reverse complementary sequence. Cell extracts from baculovirus infected cells were prepared from a minimum of 107 cells. Cells were resuspended in five volumes of buffer A (10mM Hepes, pH 7.5; 500mM KCl; 1.5mM MgCl2; 4mM BME; 0.5mM PMSF; 10 mg/ml of aprotinin, leupeptin, and pepstatin), incubated on ice for 10 min, and centrifuged for 10 min at 4°C. Proteins were purified as previously described (Knights et al., 2006). Acetylation reactions from these purified proteins were assembled in 20 μl total volume containing acetyl-transferase buffer (50mM Hepes, pH 7.9; 50mM KCl; 10% glycerol) and acetyl-coenzyme A (Amersham) with 50–400 ng of p53 proteins and 1μg of PCAF or of p300/CBP. Samples were first incubated at 32°C for 1 h, and then reaction volumes were brought to 30 μl with 5× EMSA buffer A (100mM Hepes pH 7.9; 125mM KCl; 0.5mM EDTA; 50% glycerol; 10mM MgCl2); and 2 μl of EMSA buffer B (10μM spermidine; 40μM DTT; 1.2% NP40, 2 mg/ml BSA); 1 μl of 50μg/ml double-stranded poly[d(I-C); and 4 ng of labeled oligonucleotide. Reactions were then incubated with the probe either in ice or at room temperature for 20–40 min. Samples were applied on native 5% polyacrilamide gels which were slowly run at room temperature until the xylen-cyanol blue reached 6 cm from the bottom of the gel.
Fig. 5.

PCAF interacts with different mutant forms of p53. A,B: Insect SF21 cells were co-infected with baculoviruses expressing p53 wild-type, p53-R175H, or p53V143A, together with the PCAF baculovirus, expressing an epitope flagged PCAF protein. Cell lysates were subjected to immunoprecipitation with the anti-Flag-column at 4°C for 12 h. PCAF–p53 complexes were eluted with the flag peptide in 100μl final total elution volume, which was then divided as follows. In (A, upper part): anti-PCAF immunoprecipitation reactions (lanes 1–3) or 1/50 of total cell lysates (lanes 5–7), were analyzed in immunoblot with the anti-p53 antibody. Lane 4 contains cell extracts derived from control uninfected cells immunoprecipitated with the anti-Flag antibody. The bottom part shows the anti-Flag PCAF immunoblot on the anti-Flag immunoprecipitation. In (B) 15μl of elution material derived from the PCAF–p53 wild-type immunoprecipitation (lanes 2–4); or from the PCAF–p53–R175H(lanes 5–7); or from the PCAF–p53–V143A (lanes 8–10) immunoprecipitation, were subjected to EMSA in the absence (lanes 2, 5, and 8) or presence of pAb421 (lanes 3, 6, and 9) or of a 50-fold excess of cold-specific competitor (lanes 4, 7, and 10).
Chromatin immunoprecipitation assays
ChIP assays were performed as described previously (Di Giovanni et al., 2006; Knights, 2006). Briefly, 2×107 were grown in the absence or presence of the appropriate treatment or transfection, and subsequently exposed to 1% formaldehyde–PBS solution for 13 min at room temperature. The extracts were sonicated after lysis to shear DNA between the lengths of 300–800 bp. Chromatin solutions were precipitated overnight with rotation using 2 g of rabbit polyclonal anti-p53 antibody (FL393, Santa Cruz). On the following day, protein A agarose beads that had been previously blocked with salmon sperm DNA and BSA were added to each reaction to precipitate antibody complexes. The precipitated complexes were washed and then incubated at 65°C overnight in parallel with “input” samples to reverse the crosslinking. DNA was isolated by Qiagen-PCR purification kit. The precipitated DNA was subjected to PCR reactions for 30–35 cycles. The primers used for amplification of p53-responsive elements were: p21 forward, 5′-CAGGCTGTGGCTCTGATTGG-3′ and reverse, 5′-TTCAGAGTAAGAGGCTAAGG-3′; and PUMA: forward, 5′-CAGCGATGCGTACACAGA-3′, reverse, 5′-CCAGGTGTGATCATCAGT-3′.
Cell fractionation experiments
For each cell type analyzed three 150mm dishes of 75% confluent cells were harvested, centrifuged at 750g and the resulting pellet was resuspended in 10 ml of cold PBS. Equalization for these experiments was performed by cell count, and by keeping the ratio of cells to reagents equal between all samples. The cells were centrifuged at 750g and resuspended in 10 packed cell volumes of buffer A (10mM Tris pH 7.9, 1.5mM MgCl2, 10mM KCl, freshly spiked with protease cocktail and BME). Cells were then incubated on ice for 20 min, and Triton X-100 was then added to the cell mixture to a final concentration of 0.2% following incubation for 30–45 min on ice with occasional mixing. Cells were centrifuged at 750g for 10 min at 4°C. The resulting supernatant was collected as cytoplasmic extract and the salt concentration was adjusted with NaCl at a final concentration of 200 mM. The cell pellet was washed one time in buffer A and resuspended in buffer B (10mM Tris pH 7.9, 1.5mM MgCl2, 10mM KCl, 400mM NaCl, 0.4% Triton X-100, freshly prepared protease inhibitor cocktail and beta-mercaptoethanol) using half of the original packed cell volume. Cell pellets were kept on ice for additional 30 min and occasionally vortexed, then centrifuged at 20,000g for 15 min at 4°C. The resulting supernatant was collected as nuclear extract and the NaCl concentration was diluted to 200mM by adding an equal volume of buffer A.
Patient specimens and analysis
Tumor and adjacent normal colon tissues were obtained from 10 sporadic colorectal cancer patients who underwent surgical resections at Roswell Park Cancer Institute from 1991 to 1998. Samples were extracted by homogenization in RIPA buffer (50mM Tris–HCl pH 7.4, 150mM NaCl, 2mM EDTA, 1% NP-40, 0.1% SDS), and analyzed as described in the legend of the figures. All patients gave written consent to obtain tissue specimens, as approved by the Roswell Park Institute Review Board.
Results
The presence of p53 mutants sensitizes to TSA-mediated cell killing
It was noted by others that in tumor cell lines of different origin treatment with deacetylase inhibitors (HDACi) induces activation of transcription of p53-regulated genes (Blagosklonny et al., 2005). However, the molecular mechanisms by which such activation occurred could not be fully elucidated, because in the cells studied TSA also induced rapid p53 mutant degradation. To eliminate the influence of variations in the cellular genetic background and to gain insights about the mechanisms by which the acetylation pathway affects the activity of p53 mutants, we employed three isogenic derivatives of the p53 null, H1299 lung carcinoma cell line, each harboring a tumor-derived p53 mutant, specifically p53-R175H, p53-G245A, and p53-D281G. These are conformational “hot spot” mutants found with high frequency in human tumors (Joerger and Fersht, 2007). The cell cycle distribution of cells expressing these p53 proteins was studied in the absence of any treatment, as well as in the presence of the deacetylase inhibitor TSA, and was then compared to that displayed by a cell line expressing wild-type p53 (Knights, 2006). As shown in Figure 1A, expression of wild-type p53 produced a strong arrest at the G1-phase of the cell cycle accompanied by a reduction of cells traversing S-phase, as expected. In contrast, and not surprisingly, H1299 cells tolerated constitutive expression of various p53 mutants without undergoing cell cycle arrest or apoptosis. However, the cell cycle profile of various p53-expressing cell lines changed remarkably in the presence of TSA. While in naïve H1299 cells TSA treatment at a 500 nM concentration had almost insignificant consequences on cell growth, in cells expressing p53 wild-type a net increase in the apoptotic fraction was observed. Furthermore, cells harboring p53-R175H, p53-G245A, or p53-D281G also displayed a stark increase in the number of apoptotic cells in these conditions. These results were confirmed with cell viability assays, where naïve H1299 cells or p53-expressing cells were treated with TSA and growth rates were assessed 1 week after treatment. We clearly observed that TSA reduced the number of surviving cells expressing mutant forms of p53, while growth inhibition was less prominent in p53-null H1299 cells (Fig. 1B).
Fig. 1.

The presence of p53 mutations sensitizes to TSA-mediated cell killing. A: Cell cycle profile of various H1299 cell lines untreated (upper parts), or treated with 500 nM TSA (lower parts). The type of cell line is indicated at the top of the figure. The percentage of cell at each phase of the cell cycle is indicated in each part. B: Cells were treated with TSA for 1 week, stained with trypan blue, and cell viability was assessed. Experiments were performed in triplicate and histograms represent average cell number of viable cells.
Thus, TSA treatment seems to unveil a cryptic growth-inhibitory activity of p53 mutants, suggesting restoration of function.
TSA induces p53 mutant acetylation and binding to chromatin
Deacetylase inhibitors act by inhibiting the activity of enzymes that reverse acetylation, thereby leading to hyper-acetylation of chromatin and of various cellular factors. We and others have previously shown that in the case of the wild-type protein, acetylation regulates p53 stability, DNA binding, and transcription (Kruse and Gu, 2009). It is currently unknown, however, whether tumor-derived p53 mutants can also be acetylated. To test this, untreated and TSA-treated cells were subjected to immunoblot with antibodies recognizing acetylated p53. As shown in Figure 2A,B, TSA treatment rapidly enhanced the extent of acetylation of both p53-R175H and p53-G245A at two target residues, specifically K320 and K373. Cell fractionation experiments further revealed that although basal levels of p53 mutant acetylation could be detected in untreated cells, this pre-existing acetylated fraction was more prominent in the cytoplasm compared to the nucleus (Fig. 2C compare lane 1 with lane 4). One noticeable effect of TSA treatment consisted in a time-dependent enrichment of the acetylated p53 population not only in the cytoplasmic, but also in the nuclear compartment (compare lanes 4–6 with lanes 1–3). Whether this enrichment is due to accumulation of pre-existing acetylated populations, or to de novo synthesis of acetylated p53 is currently unclear. Further, such increase in acetylation coincided with an enhancement in the expression levels of the acetyl-transferase PCAF (Fig. 2D, compare lanes 2 and 5 with lanes 1 and 4).
Fig. 2.

Molecular characteristics of TSA-treated cells. A,B: Cells expressing p53-R175H (part A) or p53-G245A (part B), were left untreated (indicated as U), or were treated with 500 nM TSA for 6 or 16 h. Cell extracts were prepared and probed with antibodies recognizing specifically acetylated p53 at position K320 or K373, as indicated at the side of each part. Approximately 1/50 of total cell extracts was probed in direct immunoblot with the p53 monoclonal antibody DOI. C: Cells treated as described in A and B were fractionated in cytoplasmic (lanes 1–3) and nuclear extracts (lanes 4–6; see Materials and Methods Section), and processed with anti-K320 or anti-K373 antibodies as described before and as indicated at the side of each part. The fractionation procedure was verified with anti-tubulin or anti-lamin A/C antibodies. D: Cells expressing p53-R175H (lanes 1–3), or p53-G245A (lanes 4–6), were left untreated (lanes 1 and 4) or were treated with 500 nM of TSA (lanes 2–3 and 5–6). Cell extracts were immuno-precipitated with the anti-PCAF antibody (lanes 1–2 and 4–5), or with a control isotype-matched antibody (lanes 3 and 6). After SDS–PAGE and transferring, membranes were probed with the indicated antibodies in immunoblot.
To then determine whether TSA influences the ability of p53 to bind to chromatin embedded p53-regulated promoters, chromatin immunoprecipitation assays (ChIP) were performed. We found that TSA induced the interaction of p53-R175H and p-53 G245A mutants with two different target genes, specifically the p21 and PUMA promoters, at levels similar to those observed with native p53 (Fig. 3A, compare lanes 3 and 4 and lanes 6 and 7 with lane 5, respectively). This stimulation was not seen with control IgG-matched antibodies (not shown) or in p53 negative H1299 cells (lanes 1 and 2), thus providing evidence of specificity of the amplification reaction. Mirroring such enhancement of DNA-binding activity, TSA also increased the expression levels of p21 and PUMA proteins in p53-R175H and p53-G245A expressing cells, but not in naïve H1299 cells (Fig. 3B, compare lanes 3–4 with lane 1).
Fig. 3.
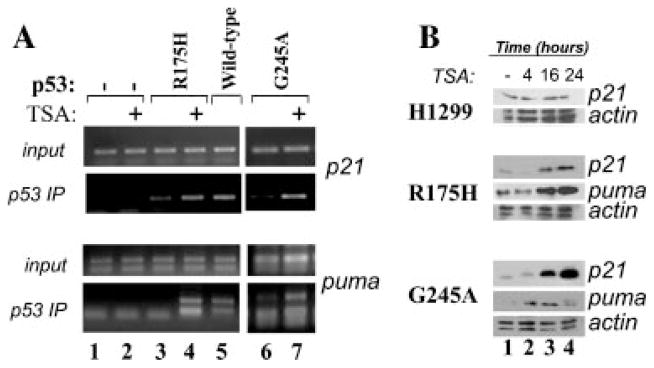
TSA restores p53 mutant DNA binding activity. A: Naïve H1299 cells (lanes 1 and 2), or cells expressing p53-R175H(lanes 3 and 4) or p53 wild-type (lane 5) or p53-G245A (lanes 6 and 7) were left untreated (lanes 1,3,5, and 6), or were treated with TSA(lanes 2, 4, and 7) for 16 h. Chromatin immunoprecipitation assays were then performed with the anti-p53 polyclonal antibody, followed by amplification with primers amplifying the p53 binding regions of the p21 or PUMA promoter (indicated at the side of each part). Note that the same anti-p53 antibody-agarose immunoprecipitation mixture was used in native H1299 cells (lanes 1 and 2), and served as a control of specificity for the anti-p53 antibody. Input levels shown at the top of each part corresponded to 1% of the total chromatin extract used for the immunoprecipitation. B: Cells as described in A were left untreated(lane 1), or were treated with TSA for 4,16, and 24 h(lanes 2–4). Cell extracts derived from these cells were probed with the antibody recognizing p21, PUMA, or actin, as indicated at the side of each part.
PCAF rescues DNA-binding activity of p53-G245A
These results led us to explore whether acetylation can rescue DNA-binding activity from mutant forms of p53. To directly test this hypothesis, we turned to electrophoretic mobility shift assays (EMSA), performed in vitro and by using p53 and PCAF proteins purified from baculovirus-infected SF21 insect cells. In the experiments shown in Figure 4A, the effects of acetylation were assessed on p53-G245A, but similar results were obtained with p53-R175H (not shown and see Fig. 5B). p53-G245A was incubated with PCAF in the absence or presence of acetyl coenzyme A (Ac-CoA), followed by subsequent incubation with a labeled oligonucleotide containing the p53-consensus site of the p21 promoter. As also shown by others (Friedler et al., 2003), replacement of glycine at position 245 has a very disruptive effect on DNA-binding capacity such that no activity was seen in the absence of PCAF (Fig. 4A, lane 2). Similarly, addition of PCAF without Ac-CoA had no affect on DNA-binding capacity (Fig. 4A, lane 3). By contrast, incubation of PCAF together with Ac-CoA increased specific binding to the probe (Fig. 4A, lane 5). This DNA-binding activity was specific, as indicated by competition experiments with oligonucleotides containing or lacking the p53 consensus site (Fig. 4A, lane 6 vs. lane 7, respectively).
Fig. 4.

PCAF restores DNA-binding ability from mutant p53 in vitro. PCAF and p53 proteins were purified from baculovirus-infected cells as described in the Materials and Methods Section. A:50 ng of PCAF were pre-incubated with 200 ng of p53-G245A in the absence (lane 3) or presence (lane 5) of acetyl-coenzyme A (Ac-CoA), or with Ac-CoA alone (lane 3). Lane 2 contains purified p53-G245A without PCAF or Ac-CoA, and lane 1 the probe alone. Acetylation reactions were assembled in 20μl final volume and in acetyl-transferase buffer for 1 h at 32°C. Reactions volumes were brought to 30μl with the addition of DNA-binding buffer and of a 32P-radiolabeled oligonucleotide containing the p53-binding sequences of the p21 promoter (lanes 1–5). Reactions in lane 6 and 7 were carried out in the presence of 50-fold excess of cold p21 oligonucleotide or of a control random oligonucleotide, respectively. p53-DNA complexes were allowed to form for 20 min at room temperature or in ice and then resolved on native 5% polyacrylamide gels, followed by autoradiography. The position of DNA–p53 complexes is indicated by the arrow. B: p53-G245A (lanes b–i) or p53 wild-type (lanes j–o) were incubated with either PCAF (lanes c, f, h, k, l, m, o) or with p300 (lanes d, g, i, n) in the absence (lanes a–e; lanes j–l) or presence (lanes f–i; lanes m–o) of Ac-CoA, as indicated at the bottom of the part. Following acetylation, reactions volumes were adjusted with DNA-binding buffer and probe as described in A. Parallel reactions were incubated with 0.5μg of the pAb421 antibody (lanes e, h, i, l, o) during the entire time of incubation with the probe. The position of the antibody-DNA–p53 complexes is indicated by the upper arrow. C: Non-acetylated p53-G245A(lane 1)or p53-G245A subjected to an acetylation reaction with p300 (lane 2) or PCAF (lane 3), was probed in direct immunoblot with the anti-p53 monoclonal antibody (upper part), or with the anti-acetyl-K320 antibody, or with the anti-acetyl-K373 antibody (bottom part).
To test whether PCAF is specific in this respect, similar experiments were performed with the catalytic domain of p300/CBP. These showed that p300/CBP was capable of inducing p53-G245A DNA-binding activity (Fig. 4B, lane g). However, no additive or synergistic activity between these two enzymes were seen (not shown), indicating that the effects of acetylation overlap, at least in vitro. Consistent with this view both p300 and PCAF were able to acetylate p53 at multiple residues, particularly at K320 and at K373, with surprisingly similar pattern of affinity for each of these residues (Fig. 4C).
Another noticeable result was that PCAF-mediated acetylation had less significant effects on wild-type p53, as we also noticed in our previous work (Knights, 2006). In fact, the DNA-binding ability of native p53 was only modestly stimulated by acetylation via either PCAF or p300 (Fig. 4B, compare lanes m and n with j and k), indicating that when in a normal conformation, p53 does not rely upon acetylation for its interaction with the p21 promoter (Knights, 2006).
Previous studies showed that the DNA-binding ability of several mutant forms of p53 can be rescued in vitro by anti-p53-specific antibodies. Therefore, in the next set of experiments we sought to determine whether the effects of PCAF overlap with the well known antibody-mediated stimulation of DNA-binding capacity. For these assays, we used the pAb421 antibody that recognizes a p53 C-terminal epitope comprised between amino acid 370 and 382. In our experimental conditions incubation of p53-G245A with the pAb421 in the absence of an acetylation reaction was not able to rescue DNA binding (Fig. 4B, lane e). However, a very strong cooperative stimulation was seen when p53-G245A was first acetylated and then incubated with pAb421. In this case, distinguishable higher molecular weight antibody–p53 complexes that strongly interacted to DNA were detected (lanes h and i). We believe that this result is important, as it suggests that acetylation is not only a pre-requisite for the ability of mutant p53 to bind to DNA, but it may also promote further stabilization of binding by factors that interact with- or modify the p53 C-terminus once acetylation has occurred. Again in contrast with p53 mutant, in the case of native p53 the pAb421 was able to enhance DNA binding activity regardless of acetylation (compare lane l, with lane o).
PCAF interacts with different types of p53 mutants
Various mutant forms of p53 have a misfolded and altered conformation relatively to the wild-type protein that impairs interactions with intracellular partners (Dey et al., 2010). Therefore, it was relevant to determine whether structurally and functionally different p53 mutants interact with PCAF as efficiently as native p53. To test this, insect cells were co-infected with recombinant baculoviruses expressing either p53, or p53-R175H, or p53V143A together with a Flag-PCAF-expressing baculovirus (Yang et al., 1996). Cell extracts from infected cells were prepared and immunoprecipitated with the anti-Flag antibody. With these experiments we determined that p53-R175H and p53V143A were retained in the anti-PCAF immunoprecipitation as efficiently as wild-type p53 (Fig. 5A, compare lanes 1 and 2 with lane 3). Furthermore, these PCAF-bound fractions of p53 were all competent for DNA interaction (Fig. 5B, lanes 5 and 8). Such DNA-binding activity was specific, as inferred based on competition experiments with the cold competitor (lanes 4, 7, and 10) and, as shown before, was super-inducible by the pAb421 antibody (lanes 3, 6, and 9).
Based on the combination of these data we conclude that in vitro, PCAF restores the affinity for DNA of at least a set of p53 mutants via acetylation.
PCAF expression restores p53 mutant transcription ability
In the subsequent set of experiments we then asked whether PCAF rescues p53 mutant activity in vivo. To this end, luciferase and ChIP assays were performed in H1299 cells lacking or expressing p53 mutant proteins. First, as shown in Figure 6A,B, transfection of the PCAF expressing vector in cells expressing either p53-R175H or p53-G245A stimulated the activity of a luciferase reporter driven by the p21 promoter. Such stimulation was again specific for the presence of p53, as it was not seen in naïve H1299 cells (Fig. 6C). Second, a PCAF construct lacking enzymatic activity (PCAF-ΔHAT, Fig. 6A–C) was unable to stimulate the reporter, in spite of the fact that such mutant was expressed at higher levels compared to the native protein (Fig. 6D, compare lanes 4–6 with lanes 1–3). Furthermore, expression of PCAF induced p53-G245A acetylation (Fig. 6E) and promoted its binding to the endogenous p21 promoter (Fig. 6F, compare lane 6 with lane 4). Such induction of DNA binding was not seen with PCAF-ΔHAT (Fig. 6F, lane 5). Finally, by employing a polyclonal antibody directed against acetylated p53, we determined that it is the acetylated p53-G245A fraction that binds to DNA in PCAF-expressing cells (Fig. 6F, lane 7). Viewed together with previous results, these data provide strong evidence that PCAF-mediated acetylation can restore the activity of p53-R175H or p53-G245A.
Fig. 6.

PCAF restores DNA binding and transcription activity from p53 mutants in cells. A–D: Cells expressing p53-G245A (part A), p53-R175H (part B), or naïve H1299 cells (part C)were transfected using a calcium phosphate-based method with 1μg of the p21 luciferase reporter and with the vectors expressing either wild-type PCAF(plain triangular boxes in all parts), or a PCAF mutant lacking the acetyl-transferase domain both of which are tagged with an N-terminal Flag epitope (indicated as PCAF-ΔHAT; striped triangular boxes in all parts), at increasing concentrations (0.25, 0.5, and 1μg). Luciferase levels were measured 24 h after transfection. At the side of each part luciferase activity is expressed in relative lights units and bars represent standard deviations. Note that in some histograms bars are too small to be seen. In (D), cell extracts of p53-R175H-expressing cells transfected as described above, were subjected to direct immunoblot with anti-Flag antibody, to determine the relative expression levels of PCAF or of PCAF-ΔHAT. E: Cells transfected with the control pCMV0 plasmid (indicated as −), or with the epitope-Flag PCAF vector (indicated as +), were probed with the anti-Flag antibody (indicated as PCAF, upper part), or with the anti-acetyl-p53 antibody (Ac-p53, middle part), or with the anti-p53 polyclonal antibody (lower part). F: p53-G245A-expressing cells were transfected with pCDNA empty vector (lanes 1 and 4), or with the vector expressing PCAF (lanes 3,6–8) or PCAF-ΔHAT (lanes 2 and 5), and then subjected to chromatin immunoprecipitation assays. Immunoprecipitation reactions were carried out with the polyclonal anti-p53 antibody (lanes 4–6) or with the polyclonal antibody directed against acetylated p53 (lane 7), or with an isotype matched polyclonal control antibody (lane 8). Lanes 1–3 contain 1% of input DNA from prior to the immunoprecipitation. After reversal of cross-linking, the immunoprecipitated material was subjected to PCR by using the p21 primers. Note that the same anti-p53 antibody-agarose immunoprecipitation mixture was used in native H1299 cells (lanes 1 and 4), and served as a further control of specificity for the amplification reaction.
PCAF is down-regulated in several human tumors harboring p53 mutations
To investigate in further depth the significance of this newly established relationship between PCAF and mutant p53, different approaches were employed to ask the question of whether PCAF levels change in tumors. First, we conducted a meta-analysis of existing micro-array data of normal and human tumor tissues available through the Oncomine database (http://www.oncomine.org). Multi-array studies derived from colon, lung, head/neck, and bladder cancers show that PCAF mRNA levels are down-regulated in these tumors relatively to normal samples in a statistically significant fashion (Fig. 7A). Second, PCAF expression was studied in a set of 10 matched tumor-normal colorectal samples, where p53 is mutated with a frequency of approximately 50% (Walther et al., 2009). Tumor and adjacent normal samples from patients were obtained at the time of surgical resection of the tumor, and analyzed for PCAF, p300/CBP, and p53 levels that when elevated are viewed as diagnostic of the presence of mutations (Fig. 7B). Consistent with the high frequency of p53 mutations in colorectal tumors, overtly high p53 expression was seen in six of the 10 tumors examined (tumors 2,4,5,6,8, and 10), and a aberrantly migrating form of p53 consistent with a deletion was detected in one of these tumors (tumor 8). In four of these tumors, PCAF was either undetectable or down-regulated relatively to the normal tissue (samples 2, 4, 6, 10). Down-regulation of PCAF was also seen in two tumors where p53 was undetectable (tumors 3 and 9). By contrast, there were no meaningful differences in the total expression levels of p300/CBP relatively to p53 expression.
Fig. 7.

PCAF is down-regulated in tumors. A: Expression of PCAF mRNA in colon, lung, head and neck, and bladder cancer was analyzed from published data using the Oncomine database (Rhodesetal., 2004). The study and number of samples from normal and tumor tissues are indicated in the table, along with the P-value for the respective comparison. B: Analysis of PCAF levels in tumors. Cell extracts derived from normal (N) or tumor(T) samples were subjected to immunoblot with the anti-p300/CBP (bottom part), anti-PCAF (middle part), or anti-p53 antibody (top part). Numbers on the top of the part refer to a pair normal/tumor samples from the same patient. C: Low PCAF levels in syngenic mammary tumor cell lines. Total cell extracts were prepared from cell lines derived from the mammary gland of mice containing a wild-type gene (p53) or from tumor cells expressing a p53 mutant harboring the p53-G242A mutant. After SDS–PAGE electrophoresis and transfer, the membranes were probed in immunoblot with the anti-p300/CBP-, anti PCAF-, or anti-p53 polyclonal antibodies, as indicated at the left side of each part.
To corroborate these data further, we extended these studies to isogenic cell lines (Fig. 7C). Two types of mammary tumor cells were analyzed for this purpose expressing either a wild-type p53 gene (lane 1, indicated as wt.) or a p53 mutant at codon 242 which substitutes an alanine for a glycine (p53-G242A, lane 2) (Tzeng et al., 1998). Codon 242 in mice is equivalent to codon 245 in humans and, therefore, this p53 protein is the murine ortholog of the p53-G245A that we have characterized before. Cell extracts from these cells were prepared and analyzed in immunoblot with different antibodies. A reduction of PCAF protein was observed in cells harboring p53-G242A as compared to isogenic cells derived from mammary tissues expressing wild-type p53. By contrast, the expression levels of p300/CBP displayed an opposite behavior.
By viewing our data together, an argument can be made that down-regulation of PCAF and mutations of the p53 gene occur simultaneously in tumors, likely as a result of a selective pressure for dismantling PCAF signaling that would otherwise preserve some degree of wild-type activity incompatible with proliferation.
PCAF suppresses proliferation and anchorage-dependent growth of tumor cells expressing p53 mutations
The above findings prompted us to investigate the effects of restoring PCAF levels in cells harboring p53 mutants. In a first set of experiments, the vectors encoding the PCAF or the PCAF-ΔHAT were transfected into the p53-G242A the murine cell line (Fig. 8A,D). These plasmids also contain the neomycin-resistence gene that allows selection of transfected cells with G418. An independent transfection with the backbone plasmid carrying the CMV promoter alone (CMV0) provided the control for these experiments. Cells were kept under selection for about 2 weeks and arising clones were either stained with Coomassie brilliant blue (Fig. 8A), or isolated and expanded for further analysis. Colonies derived from experiments described above were pulled together and tested for PCAF protein levels in immunoblot (Fig. 8D). Figure 8B shows the typical morphological characteristics of p53-G242A cells and of the isolated PCAF-expressing clones. The difference in the morphology was rather compelling. The p53-G242A have a spindle-like morphotype and no defined orientation, resulting in a randomly organized architecture. Moreover, due to loss of contact inhibition, they grow in overlapping multiple layers, and after a few days of culturing form dense aggregates and spontaneously detach from tissue culture dishes. By contrast, PCAF expressing cells displayed polygonal and flat shape, grew in a monolayer and were more resistant to detachment by trypsin compared to the parental cells, indicating a greater capacity to adhere to a solid support (not shown). Eventually, and not surprisingly, after 2 weeks of culturing PCAF expression was completely lost from these surviving clones. The characteristics of PCAF-ΔHAT expressing cells were instead identical to the parental p53-G242A cell line. To assess whether the observed morphological changes seen in the presence of PCAF reflect a reversion of the transformed phenotype, isolated clones were grown in soft-agar (Fig. 8C and quantified in E). This assay measures anchorage-dependent growth, and together with morphotype, saturation density and contact inhibition is a reliable criterion of the oncogenic potential of a given cell population. We found that PCAF but not PCAF-ΔHAT produced a significant reduction of the size and number of soft agar clones. Furthermore, consistent with our previous data, in the PCAF expressing cells acetylation of p53-G242A was increased, coinciding with induction of p21 protein levels (Fig. 9A). To then establish whether this growth suppressive activity exerted by PCAF relies upon the p53 acetylated residues, similar experiments were performed in the human p53 null H1299 cells transfected with p53-G245A or with a mutant where four of the known acetylation sites, K320/ K370/K372/K373 were replaced (p53-G245A-4Km). As shown in Figure 9B, PCAF was able to suppress proliferation in cells expressing p53-G245A, but only partially in cells harboring the p53-G245A-4Km or in p53 null H1299 cells.
Fig. 8.

PCAF inhibits proliferation and anchorage-dependent growth in p53G242A cells. A: Murine cell lines expressing p53-G242A were transfected with the CMV0 backbone vector, with a vector encoding full length PCAF, or with the PCAF-ΔHAT, as indicated at the top of each part. After 24 h, transfection cells were diluted and cultured in media containing G418 (400μg/ml) for approximately 2 weeks, after which time dishes were stained with Coomassie brilliant blue to visualize the surviving clones. B: Morphology of cells transfected with pCDNA, PCAF, or PCAF-ΔHAT. C: Cells transfected as described in A, were plated in soft agar, and colonies were scored after 2 weeks of selection. D: Expression levels of PCAF and PCAF-ΔHAT in isolated clones. E: The experiments shown in C were repeated three times and the results were quantified from a triplicate experiment.
Fig. 9.

The growth inhibitory effects of PCAF are p53 and acetylation-dependent. A: Murine cell lines expressing p53-G242A were transiently transfected with the CMV0 vector (lane 1) or with the epitope-flag PCAF expressing vector (lanes 2–4), and cells were harvested at the indicated time points. Cell extracts derived from these transfections were probed with the following antibodies, as indicated at the right of each part: the anti-Flag monoclonal antibody (PCAF); the anti-acetyl p53 antibody (second part); the polyclonal antibody recognizing p53; the anti-p21 polyclonal antibody. The bottom part contains a portion of the membrane stained with Coomassie brilliant blue. B: Naïve H1299 cells or cells transfected with the vector expressing p53-G245A, or with a vector harboring mutations at the acetylated residues K320/K370/K372/K373 (p53-G245A-K4m) were co-transfected with the PCDNA empty vector (solid bars) or with a vector expressing PCAF, both of which carry the G418 resistance. Cells were placed under antibiotic selection, and viable cells were counted a week after. Vertical bars represent standard deviations of the number of viable cells calculated from a triplicate experiment.
Discussion
A number of studies in recent years have focused on the design of novel anticancer drugs that target p53 mutants, demonstrating that small artificial molecules can restore activity acting with various mechanisms (Selivanova, 2010). These data raise the question of whether intracellular signals or pathways acting in a fashion similar to artificial compounds exist, that can prevent p53 mutant loss of function. In this study, we have provided strong evidence that PCAF-dependent acetylation restores activity from at least a set of p53 mutants, such as p53-R175H and p53-G245A. In the case of p53-G245A, we further established that acetylation was necessary for the ability of this otherwise DNA-binding incompetent mutant to interact with DNA, and it also acted cooperatively with anti-p53 antibodies to further enhance DNA-binding capacity. Therefore, it is possible that in vivo p53 mutant affinity for DNA might also be influenced by post-acetylation events, such as by the interaction with cellular factors or by additional posttranslational modifications that occur in an acetylation dependent manner. How does acetylation rescue DNA-binding activity? The high-resolution crystal structure of several p53 oncogenic mutants has shown that C-terminal residues beyond K291 are highly disordered (Cho et al., 1994; Wong et al., 1999; Joerger et al., 2006; Joerger and Fersht, 2008). In addition, p53 mutants destabilize the folded conformation leading to an increased tendency to form amyloid or fibrillar aggregates (Ishimaru et al., 2003). Therefore, it is possible that acetylation allows appropriate and ordered positioning of the C-terminus relatively to other structural p53 domains and by doing so, prevents misfolding and aggregation. Interestingly, a different mechanism is in action in the case of wild-type p53. In fact, we showed that in this case acetylation, particularly at K320, is not absolutely necessary for the interaction with DNA, but rather modifies the spectrum of DNA-binding activity by redistributing p53 on different promoters (Di Giovanni et al., 2006; Knights, 2006).
Evidence presented in this study suggests that PCAF provides a safe-guard mechanism against unrestrained proliferation of cells that have acquired p53 mutations. In fact, we found that PCAF levels are low in cell lines and human tumors harboring p53 mutations and that re-introduction of PCAF in some of these cells is incompatible with survival. In this respect, not only our data corroborate previous speculations implicating PCAF as an anti-oncogene and inhibitor of proliferation, but also provide new insights into the mechanisms by which this protein specifically cross-talks with mutant p53. In fact, it can be posited that neoplastic clones where p53 mutations have arisen may not acquire a gain of function phenotype and a full proliferation advantage unless PCAF signaling is dismantled such that the end result would be the emergence of clones that maintain p53 mutants but have lost PCAF. Furthermore, as the majority of human tumors harboring missense p53 mutations are almost inevitably characterized by loss of heterozygosity, it is attractive to speculate that PCAF-mediated cell killing provides a selective pressure for the emergence of tumor clones that have also lost the remaining p53 wild-type allele. On the other hand, although it appears that PCAF ability to inhibit cell growth is lessened in isogenic cells completely lacking p53 versus p53-mutant expressing cell lines, this protein performs in various biological processes that do not necessarily require p53. Therefore, it is likely that PCAF acts within and outward of the p53 pathway, perhaps making it even more advantageous for tumors to eliminate its function. It is further relevant to note that in addition to the acetyl-transferase activity, PCAF possesses intrinsic ubiquitination activity through which it indirectly controls the stability of wild-type p53 (Linares et al., 2007). It has been recently shown that p53 mutant accumulation is important for the acquisition of its gain-of-function potential and for tumor progression (Terzian et al., 2008). In knock-in mouse models or in patients with Li–Fraumeni syndrome, p53 mutants do not accumulate in normal tissues but only in tumors (Lang et al., 2004; Olive et al., 2004), suggesting that additional events occurring during tumorigenesis are necessary for p53 mutant accumulation. Therefore, it is possible that loss of PCAF also contributes to p53 mutant stabilization in tumors.
While the mechanisms by which PCAF is down-regulated in tumors remain to be fully elucidated, the region of chromosome 3p24 where the PCAF gene is located is frequently epigenetically silenced via methylation (Zhu et al., 2009). In addition, earlier studies have failed to detect mutations or deletions of the PCAF gene that impair activity. Similarly, we found that only one out of the 10 tumors had demonstrable LOH of the PCAF locus (M.L. Avantaggiati and D. Stoler, unpublished work). Thus, the available data are consistent with the possibility that PCAF is down-regulated in cancers via epigenetic or prost-translational mechanisms, likely also explaining the increase in PCAF levels seen in TSA-treated cells. An additional equally important, yet unanswered question is whether or not other acetyltransferases can influence p53 mutant activity in a manner similar to that seen for PCAF. The answer to this question will obviously require more studies. At least in vitro, we found that the catalytic domain of p300 was able to produce effects similar to those seen with PCAF on p53 mutant DNA-binding activity. Furthermore, studies conducted by other groups have reported mutations or loss of heterozygosity at the p300 or CBP locus, although it is still unclear whether they occur independently or concurrently with p53 mutations (Iyer et al., 2004). Nevertheless, we found that unlike PCAF, p300 is not able to stimulate transcription of p53 mutants when assessed in luciferase assays and, on the contrary, its over-expression induces p53 mutant misfolding and aggregation (Kiliryuk and Avantaggiati, unpublished work). It will be nevertheless important to determine whether other acetylases display effects similar to those seen with PCAF, especially considering that lysine residues located within the DNA-binding domain of p53 can also be targeted by different enzymes (Kruse and Gu, 2009).
It is finally intriguing that basal levels of p53 mutant acetylation were seen in cells independently of TSA treatment. However, such sub-population displayed a predominant cytoplasmic localization, whereas TSA enriched the nuclear compartment with acetylated p53. This finding raises the issue of whether cytoplasmic, transcription-independent activities of acetylated p53 that support proliferation exist, such that while dismantling nuclear pathways of acetylation (i.e., PCAF), tumor cells might select for such cytoplasmic functions. Given that cytoplasmic p53 mutants can affect autophagy, cytoskeleton architecture, and mitochondrial activity (Vousden, 2009), it will be relevant to determine whether acetylation in the cytoplasm has any consequence on these important biological processes.
In conclusion, we have provided the first demonstration that a physiological pathway, specifically acetylation mediated by PCAF, restores function of p53 mutants. Based on our data we infer that the development of reagents able to affect or mimic the activity of PCAF will provide an additional and distinct advantage for manipulating the environment of tumor cells wherein p53 pathway is impaired.
Acknowledgments
This study was supported by NIH R01 CA102746 to M.L.A. We thank Dr. Pat Nakatani and Dr. Keiko Ozato for the kind gift of the PCAF expressing vectors and of the PCAF baculovirus, and Dr. Carol Prives for the gift of the baculoviruses expressing p53-RH175H and p53V143A.
Literature Cited
- Aurelio ON, Kong XT, Gupta S, Stanbridge EJ. p53 mutants have selective dominant-negative effects on apoptosis but not growth arrest in human cancer cell lines. Mol Cell Biol. 2000;20:770–778. doi: 10.1128/mcb.20.3.770-778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- Bell S, Klein C, Muller L, Hansen S, Buchner J. p53 contains large unstructured regions in its native state. J Mol Biol. 2002;322:917–927. doi: 10.1016/s0022-2836(02)00848-3. [DOI] [PubMed] [Google Scholar]
- Berger SL. Out of the jaws of death: PRMT5 steers p53. Nat Cell Biol. 2008;10:1389–1390. doi: 10.1038/ncb1208-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV, Trostel S, Kayastha G, Demidenko ZN, Vassilev LT, Romanova LY, Bates S, Fojo T. Depletion of mutant p53 and cytotoxicity of histone deacetylase inhibitors. Cancer Res. 2005;65:7386–7392. doi: 10.1158/0008-5472.CAN-04-3433. [DOI] [PubMed] [Google Scholar]
- Brosh R, Rotter V. When mutants gain new powers: News from the mutant p53 field. Nat Rev Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: Drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- Bykov VJ, Issaeva N, Zache N, Shilov A, Hultcrantz M, Bergman J, Selivanova G, Wiman KG. Reactivation of mutant p53 and induction of apoptosis in human tumor cells by maleimide analogs. J Biol Chem. 2005;280:30384–30391. doi: 10.1074/jbc.M501664200. [DOI] [PubMed] [Google Scholar]
- Campling BG, El-Deiry WS. Clinical implications of p53 mutations in lung cancer. Methods Mol Med. 2003;75:53–77. doi: 10.1385/1-59259-324-0:53. [DOI] [PubMed] [Google Scholar]
- Campomenosi P, Monti P, Aprile A, Abbondandolo A, Frebourg T, Gold B, Crook T, Inga A, Resnick MA, Iggo R, Fronza G. p53 mutants can often transactivate promoters containing a p21 but not Bax or PIG3 responsive elements. Oncogene. 2001;20:3573–3579. doi: 10.1038/sj.onc.1204468. [DOI] [PubMed] [Google Scholar]
- Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T, Wang JY, Anderson CW, Appella E, Xu Y. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol Cell Biol. 2006;26:6859–6869. doi: 10.1128/MCB.00062-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor–DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Chumakov AM, Miller CW, Chen DL, Koeffler HP. Analysis of p53 transactivation through high-affinity binding sites. Oncogene. 1993;8:3005–3011. [PubMed] [Google Scholar]
- Dey A, Lane DP, Verma CS. Modulating the p53 pathway. Semin Cancer Biol. 2010;20:3–9. doi: 10.1016/j.semcancer.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Di Giovanni S, Knights CD, Rao M, Yakovlev A, Beers J, Catania J, Avantaggiati ML, Faden AI. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006;2517:4084–4096. doi: 10.1038/sj.emboj.7601292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Lin T, Uranishi H, Gu W, Xu Y. Functional analysis of the roles of posttranslational modifications at the p53C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005;25:5389–5395. doi: 10.1128/MCB.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286:2507–2510. doi: 10.1126/science.286.5449.2507. [DOI] [PubMed] [Google Scholar]
- Friedler A, Veprintsev DB, Hansson LO, Fersht AR. Kinetic instability of p53 core domain mutants: Implications for rescue by small molecules. J Biol Chem. 2003;278:24108–24112. doi: 10.1074/jbc.M302458200. [DOI] [PubMed] [Google Scholar]
- Geisler S, Borresen-Dale AL, Johnsen H, Aas T, Geisler J, Akslen LA, Anker G, Lønning PE. TP53 gene mutations predict the response to neoadjuvant treatment with 5-fluorouracil and mitomycin in locally advanced breast cancer. Clin Cancer Res. 2003;9:5582–5588. [PubMed] [Google Scholar]
- Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004;64:55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- Grinkevich VV, Nikulenkov F, Shi Y, Enge M, Bao W, Maljukova A, Gluch A, Kel A, Sangfelt O, Selivanova G. Ablation of key oncogenic pathways by RITA-reactivated p53 is required for efficient apoptosis. Cancer Cell. 2009;15:441–453. doi: 10.1016/j.ccr.2009.03.021. [DOI] [PubMed] [Google Scholar]
- Hupp TR, Meek DW, Midgley CA, Lane DP. Activation of the cryptic DNA binding function of mutant forms of p53. Nucleic Acids Res. 1993;21:3167–3174. doi: 10.1093/nar/21.14.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru D, Andrade LR, Teixeira LS, Quesado PA, Maiolino LM, Lopez PM, Cordeiro Y, Costa LT, Heckl WM, Weissmüller G, Foguel D, Silva JL. Fibrillar aggregates of the tumor suppressor p53 core domain. Biochemistry. 2003;42:9022–9027. doi: 10.1021/bi034218k. [DOI] [PubMed] [Google Scholar]
- Iyer NG, Ozdag H, Caldas C. p300/CBP and cancer. Oncogene. 2004;23:4225–4231. doi: 10.1038/sj.onc.1207118. [DOI] [PubMed] [Google Scholar]
- Joerger AC, Fersht AR. Structure–function–rescue: The diverse nature of common p53 cancer mutants. Oncogene. 2007;26:2226–2242. doi: 10.1038/sj.onc.1210291. [DOI] [PubMed] [Google Scholar]
- Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53. Annu Rev Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- Joerger AC, Ang HC, Veprintsev DB, Blair CM, Fersht AR. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J Biol Chem. 2005;280:16030–16037. doi: 10.1074/jbc.M500179200. [DOI] [PubMed] [Google Scholar]
- Joerger AC, Ang HC, Fersht AR. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci USA. 2006;103:15056–15061. doi: 10.1073/pnas.0607286103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ki DH, Jeung HC, Park CH, Kang SH, Lee GY, Lee WS, Kim NK, Chung HC, Rha SY. Whole genome analysis for liver metastasis gene signatures in colorectal cancer. Int J Cancer. 2007;121:2005–2012. doi: 10.1002/ijc.22975. [DOI] [PubMed] [Google Scholar]
- Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, Quong AA, Zhang X, Beerman T, Pestell RG, Avantaggiati ML. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173:533–544. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JM, Gorzov P, Veprintsev DB, Söderqvist M, Segerbäck D, Bergman J, Fersht AR, Hainaut P, Wiman KG, Bykov VJ. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376–388. doi: 10.1016/j.ccr.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El-Naggar AK, Lozano G. Gain of function of a p53 hot spot mutation in a mouse model of Li–Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Linares LK, Kiernan R, Triboulet R, Chable-Bessia C, Latreille D, Cuvier O, Lacroix M, Le Cam L, Coux O, Benkirane M. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Hdm2. Nat Cell Biol. 2007;9:331–338. doi: 10.1038/ncb1545. [DOI] [PubMed] [Google Scholar]
- Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, Berger SL. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19:1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig RL, Bates S, Vousden KH. Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol Cell Biol. 1996;16:4952–4960. doi: 10.1128/mcb.16.9.4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–381. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- Miller CW, Chumakov A, Said J, Chen DL, Aslo A, Koeffler HP. Mutant p53 proteins have diverse intracellular abilities to oligomerize and activate transcription. Oncogene. 1993;8:1815–1824. [PubMed] [Google Scholar]
- Niewolik D, Vojtesek B, Kovarik J. p53 derived from human tumour cell lines and containing distinct point mutations can be activated to bind its consensus target sequence. Oncogene. 1995;10:881–890. [PubMed] [Google Scholar]
- Nikolova PV, Wong KB, DeDecker B, Henckel J, Fersht AR. Mechanism of rescue of common p53 cancer mutations by second-site suppressor mutations. EMBO J. 2000;19:370–378. doi: 10.1093/emboj/19.3.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North S, Pluquet D, Maurici D, El-Ghissassi F, Hainaut P. Restoration of wild-type conformation and activity of a temperature-sensitive mutant of p53 (p53(V272M)) by the cytoprotective aminothiol WR1065 in the esophageal cancer cell line TE-1. Mol Carcinog. 2002;33:181–188. doi: 10.1002/mc.10038. [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li–Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: A cancer microarray database and integrated datamining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Carbayo M, Socci ND, Lozano J, Saint F, Cordon-Cardo C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006;24:778–789. doi: 10.1200/JCO.2005.03.2375. Epub 2006 Jan 23. [DOI] [PubMed] [Google Scholar]
- Selivanova G. Therapeutic targeting of p53 by small molecules. Semin Cancer Biol. 2010;20:46–56. doi: 10.1016/j.semcancer.2010.02.006. Epub 2010 Mar 3. [DOI] [PubMed] [Google Scholar]
- Selivanova G, Iotsova V, Okan I, Fritsche M, Ström M, Groner B, Grafström RC, Wiman KG. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat Med. 1997;3:632–638. doi: 10.1038/nm0697-632. [DOI] [PubMed] [Google Scholar]
- Shi LM, Fan Y, Myers TG, O’Connor PM, Paull KD, Friend SH, Weinstein JN. Mining the NCI anticancer drug discovery databases: Genetic function approximation for the QSAR study of anticancer ellipticine analogues. J Chem Inf Comput Sci. 1998;38:189–199. doi: 10.1021/ci970085w. [DOI] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng YJ, Zimmermann C, Guhl E, Berg B, Avantaggiati ML, Graessmann A. SV40 T/t-antigen induces premature mammary gland involution by apoptosis and selects for p53 missense mutation in mammary tumors. Oncogene. 1998;16:2103–2114. doi: 10.1038/sj.onc.1201733. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Veprintsev DB, Freund SM, Andreeva A, Rutledge SE, Tidow H, Canadillas JM, Blair CM, Fersh TAR. Core domain interactions in full-length p53 in solution. Proc Natl Acad Sci USA. 2006;103:2115–2119. doi: 10.1073/pnas.0511130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH. Functions of p53 in metabolism and invasion. Biochem Soc Trans. 2009;37:511–517. doi: 10.1042/BST0370511. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9:489–499. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- Wong KB, DeDecker BS, Freund SM, Proctor MR, Bycroft M, Fersht AR. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc Natl Acad Sci USA. 1999;96:8438–8442. doi: 10.1073/pnas.96.15.8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- Zhu C, Qin YR, Xie D, Chua DT, Fung JM, Chen L, Fu L, Hu L, Guan XY. Characterization of tumor suppressive function of P300/CBP-associated factor at frequently deleted region 3p24 in esophageal squamous cell carcinoma. Oncogene. 2009;28:2821–2828. doi: 10.1038/onc.2009.137. [DOI] [PubMed] [Google Scholar]