

Abstract
Two approaches to prepare amino acids with catalytically active organometallic side chains are presented. These methods are notable in that they provide access either free or N-protected compounds that are structurally analogous to naturally occurring amino acids. The N-protected organo-metallic amino acids are compatible with standard peptide coupling conditions and can be used to prepare catalytically active metallopeptides.
Keywords: organometallic amino acids, metallopeptides, 9-borabicyclononane
The design and synthesis of catalytically active metallopeptides1 and metalloproteins2 has received considerable attention due to the potential for peptides and proteins to impart selectivity to metal catalysts using molecular recognition.3 Most methods developed thus far involve metal binding to a peptide backbone or side chains,4 covalent or non-covalent bioconjugation of a peptide with a metal-binding ligand or metal complex,5 or incorporation of amino acids with metal binding ligands6 into a peptide. Using these methods, researchers have developed a range of catalytically active metallopeptides and artificial metalloenzymes for synthetic applications.1,2
Herein, we describe two approaches for the synthesis of organometallic amino acids that enable direct incorporation of catalytically active metal complexes into peptides (Scheme 1). Importantly, the organometallic amino acids were designed to mimic, as closely as possible, natural amino acids in order to maximize their compatibility with potential in vivo incorporation efforts.6 Preserving the native amino acid structure was also expected to minimize structural perturbations to peptides including these amino acids and to ensure close proximity of the metal center and the chiral peptide scaffold. Prior to this work, no general method existed for the preparation of unprotected amino acids with side chains containing catalytic active transition metal centers,7 and most protected compounds include extended tethers8 between the amino acid moiety and the organometallic fragment. Because both of these limitations arise primarily from the well-established metal-binding ability of the amino acid moiety,9 protecting group strategies that would enable installation of a range of metal centers while still allowing amino acid deprotection without compromising the activity of the metal center were required.
Scheme 1.
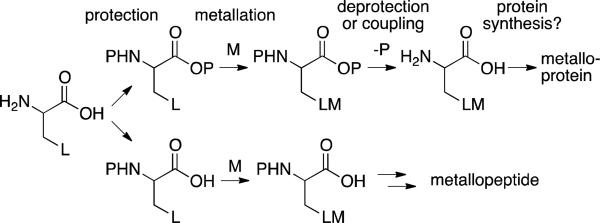
Approaches to synthesize organometallic amino acids for preparation of metalloproteins and peptides.
Particularly attractive in this regard was the 9-borabicyclononanyl (9-BBN) group, which enables simultaneous protection of both the amine and carboxylic acid functionalities and can be readily removed using ethylenediamine.10 The 9-BBN aducts of para- and meta-acetylphenylalanine (1a and 1b, respectively) were therefore prepared and purified by washing with hexanes (Scheme 2).
Scheme 2.
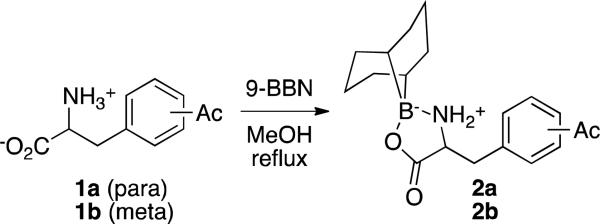
Amino acid protection using 9-BBN.
The resulting compounds, 2a and 2b, were then reacted with a range of amines and hydrazines to generate imines and hydrazines, 3. Reaction of these compounds with either NaPdCl4 or [Cp*IrCl2]2 according to standard literature procedures led to the formation of 9-BBN protected organometallic amino acids, 4.11 These were typically isolated as crude solids, and the free amino acids, 5, could then be readily prepared by simply stiring these complexes in THF solutions of ethylenediamine.10 The amino acids precipatated from solution and could be further purified by precipitation from methanol/ether solution if necessary. The ethylene diamine was typically observed to displace chloride from the metal center, binding in either a mono- or bidentate fashion depending on the number of coordination sites available.
To demonstrate the generality of the BBN protecting group for the synthesis of unprotected organometallic amino acids, two additional compounds were prepared by analogous routes. Specifically, a 9-BBN-protected hydroxyquinoline-based amino acid was reacted with [Cp*IrCl2]2 to provide the corresponding Cp*Ir-hydroxyquinoline chloride complex, which was deprotected to yield amino acid 9. In addition, the known 9-BBN-protected lysine, 10, was acylated with 2-pyridinecarboxylic acid chloride and reacted with [CpRuCO(ACN)2]PF6 to provide complex 12, which was deprotected to yield amino acid 13.12
Thus, a structurally diverse set of Pd-, Ir-, and Ru-based amino acids were prepared. The Pd and Ir compounds were designed to mimic the planar aromatic side chains of phenylalanine and tyrosine, while the last is a pyrolysine13 mimic. These compounds are water-soluble and are readily taken up by E. coli as judged by their inhibition of cell growth (see supporting information). While these compounds are therefore highly promising candidates for aqueous organometallic catalysis or in vivo incorporation in E. coli,6 their potential for use in standard peptide synthesis procedures14 required installation of either the Boc or Fmoc protecting group.
To avoid introducing additional protection steps to the syntheses outlined above and potential for decomposition of the metal centers in these compounds, we instead pursued a mono-protecting group strategy that would mask the amine functionality with either a Boc or Fmoc group based on recent work by several groups using tethered amino acid-metal complexes.8 For in vitro peptide synthesis, enantiopure amino acids were also required (E. coli only incorporates S amino acids into proteins, so racemic samples can be used for in vivo studies6), so an enzymatic resolution was used to prepare enantiopure acetylphenylalanine, 15.15 This material was then Boc-protected, converted to the corresponding methoxyimine, and reacted with either NaPdCl4 or [Cp*IrCl2]2 to yield the Boc-protected metallacyclic amino acids 17 and 18.
To demonstrate the utility of these compounds for the site-specific introduction of metal centers into peptide scaffolds, standard peptide coupling conditions14 were used to prepare tetrapeptides 20 and 21 (Scheme 4). The 2-aminoisobutyric acid (Aib)-proline motif in peptide 19 is known to enforce a beta turn structure and has been used by many groups, notably Miller and coworkers,16 to generate highly selective catalysts for a rang of chemical transformations using organic amino acids. Peptide-based coordination complexes based on beta-turn peptides have also been generated.17 We hypothesized that incorporating organometallic catalysts into this platform would greatly expand the range of catalytic transformations possible using unique selectivity afforded by peptide catalysts. By using preformed organometallic complexes as side chains, catalysts not available via simple coordination of metal ions, such as the metallacycles described herein, can be selectively introduced into peptides. NMR and mass spectroscopic analysis of 20 and 21 indicated that the metal centers remained unperturbed by the peptide coupling conditions. Several analogues of 20 and 21, involving substitution of L-proline with D-proline, variation of the terminal phenylalanine residue, and addition of an additional amino acid to form pentapeptides, were also prepared (data not shown). Attempted removal of the Boc group from 20 and 21 using TFA led to decomposition of the metallopeptides, and while these compounds were stable to the piperidine conditions typically used to remove the Fmoc group, synthesis of Fmoc-protected amino acids as shown in Scheme 4 was complicated by Fmoc removal during the methoxyamine condensation.
Scheme 4.

Synthesis of resolved, protected Pd and Ir amino acids 17 and 18.
All peptides remained catalytically active toward known reactions for analogous non-peptide metallacycle complexes, including Ir-catalyzed transfer hydrogenation11b and Pd-catalyzed imidate rearrangements18 (Scheme 5). Unfortunately, this small set of compounds investigated thus far provided no enantioinduction in either of these reactions. Control experiments using either [Cp*IrCl2]2 or PdCl2 or as catalysts for these reactions provided no conversion, indicating that release of metal complexes from the peptides was probably not responsible for the observed non-selective reactions. Presumably, the metal fragment in each case adopted a conformation that placed it outside the influence of the peptide. Further studies are underway to confirm the secondary structure (conformation) of these peptides and to identify alternate peptide architectures to provide selectivity in a range of chemical transformations.
Scheme 5.
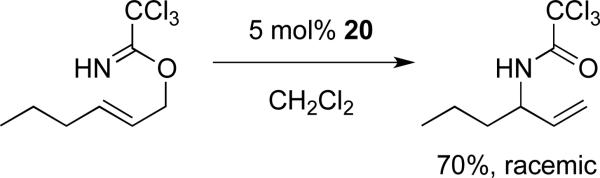
Representative imidate rearrangement catalyzed by palladacycle peptide 20.
In summary, we have described synthetic approaches for the preparation of a range of unprotected and N-protected organometallic amino acids. These compounds are structural analogues of natural amino acids making them ideal vehicles for incorporation of organometallic complexes into peptides and potentially proteins. The compatibility of these amino acids with standard peptide coupling conditions allowed facile preparation of a small set of catalytically active metallopeptides. The ability of peptide-based catalysts to provide enantio- and site selectivity in a range of organo-catalyzed reactions16 suggests that the metallopeptides analogous to those prepared hold great promise in similar selectivities in metal catalyzed reactions.
Supplementary Material
Scheme 3.

Synthesis of hydroxyquinoline- and lysine-based amino acids 9 and 13.
Scheme 2a.

Amino acid protection using 9-BBN.
Scheme 4a.

Peptide coupling using amino acids 17 and 18.
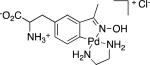
Table 1.
Representative organometallic amino acids.
| Entry | Structure (5) | Yield (%) | |
|---|---|---|---|
| 3 | 51 | ||
| 1 |
|
3a (90) | 5a (58) |
| 2 |
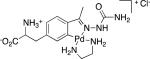
|
3b (89) | 5b (20) |
| 3 |
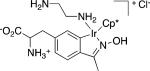
|
3c (93) | 5c (30) |
Cumulative yield over metallation and deprotection steps.
Acknowledgments
Funding Sources
This work was supported by an NIH Pathways to Independence Award (5R00GM087551-03) and a Packard Fellowship in Science and Engineering to JCL.
Footnotes
Supporting Information. Synthetic procedures and full characterization of all synthetic compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.a Francis MB, Jamison TF, Jacobsen EN. Curr. Opin. Chem. Biol. 1998;2:422–428. doi: 10.1016/s1367-5931(98)80019-7. [DOI] [PubMed] [Google Scholar]; b Roelfes G. ChemCatChem. 2011;3:647–648. [Google Scholar]
- 2.a Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ringenberg MR, Ward TR. Chem. Commun. 2011;47:8470–8476. doi: 10.1039/c1cc11592h. [DOI] [PubMed] [Google Scholar]; c Reetz MT. Top. Organomet. Chem. 2009;25:63–92. [Google Scholar]
- 3.Chen Z, Vohidov F, Coughlin JM, Stagg LJ, Arold ST, Ladbury JE, Ball ZT. J. Am. Chem. Soc. 2012;134:10138–10145. doi: 10.1021/ja302284p. [DOI] [PubMed] [Google Scholar]
- 4.a Sambasivan R, Ball ZT. J. Am. Chem. Soc. 2010;132:9289–9291. doi: 10.1021/ja103747h. [DOI] [PubMed] [Google Scholar]; b Costopoulos B, Benaki D, Pelecanou M, Mikros E, Stassinopoulou CI, Varvarigou AD, Archimandritis SC. Inorg. Chem. 2004;43:5598–5602. doi: 10.1021/ic049519c. [DOI] [PubMed] [Google Scholar]; c Ueno T, Yokoi N, Unno M, Matsui T, Tokita Y, Yamada M, Ikeda-Saito M, Nakajima H, Watanabe Y. Proc. Nat. Acad. Sci. U.S.A. 2006;103:9416–9421. doi: 10.1073/pnas.0510968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a Mayer C, Gillingham DG, Ward TG, Hilvert D. Chem. Commun. 2011;47:12068–12070. doi: 10.1039/c1cc15005g. [DOI] [PubMed] [Google Scholar]; b Reetz MT, Rentzsch M, Pletsch A, Taglieber A, Hollmann F, Mondiere RJG, Dick-mann N, Hocker B, Cerrone S, Haeger MC, Sterner R. Chem-BioChem. 2008;9:552–564. doi: 10.1002/cbic.200700413. [DOI] [PubMed] [Google Scholar]
- 6.a Xie J, Liu W, Schultz PG. Angew. Chem. Int. Ed. 2007;46:9239–9242. doi: 10.1002/anie.200703397. [DOI] [PubMed] [Google Scholar]; b Coquiere D, Bos D, Beld J, Roelfes G. Angew. Chem. Int. Ed. 2009;48:5159–5162. doi: 10.1002/anie.200901134. [DOI] [PubMed] [Google Scholar]; c Laungani AC, Slat-tery JM, Krossing I, Breit B. Chem. Eur. J. 2008;14:4488–4502. doi: 10.1002/chem.200800359. [DOI] [PubMed] [Google Scholar]; d Christensen CA, Meldal MJ. Comb. Chem. 2007;9:79–85. doi: 10.1021/cc0600627. [DOI] [PubMed] [Google Scholar]; e Agarkov A, Greenfield S, Xie D, Pawlick R, Starkey G, Gilbertson SR. Biopolymers. 2006;84:48–73. doi: 10.1002/bip.20395. [DOI] [PubMed] [Google Scholar]
- 7.For protected amino acids, see: a 5e Xu G, Gilbertson SR. Org. Lett. 2005;7:4605–4608. doi: 10.1021/ol0516521.Dialer H, Steglich W, Beck W. Tetrahedron. 2001;57:4855–4861.Ranganathan S, Tamilarasu N. Inorg. Chem. 1999;38:1019–1023. doi: 10.1021/ic980263i.Sokolov VI, Bulygina LA, Borbulevych OY, Shishkin OV. J. Organomet. Chem. 1999;582:246–251.
- 8.a Vairaprakash P, Ueki H, Tashiro K, Yaghi OM. J. Am. Chem. Soc. 2011;133:759–761. doi: 10.1021/ja1097644. [DOI] [PubMed] [Google Scholar]; b Bartholoma M, Valliant J, Maresca KP, Babich J, Zubieta J. Chem. Commun. 2009:493–512. doi: 10.1039/b814903h. [DOI] [PubMed] [Google Scholar]
- 9.Severin K, Bergs R, Beck W. Angew. Chem. Int. Ed. 1998;37:1634–1654. doi: 10.1002/(SICI)1521-3773(19980703)37:12<1634::AID-ANIE1634>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 10.Dent WH, Randal Erickson W, Fields SC, Parker MH, Tromiczak EG. Org. Lett. 2002;4:1249–1251. doi: 10.1021/ol017241b. [DOI] [PubMed] [Google Scholar]
- 11.a Alonso DA, Najera C, Pacheco MC. Adv. Synth. Catal. 2002;344:172–183. [Google Scholar]; b Wang C, Pettman A, Basca J, Xiao J. Angew. Chem. Int. Ed. 2010;49:7548–7552. doi: 10.1002/anie.201002944. [DOI] [PubMed] [Google Scholar]
- 12.In analogy to Bregman H, Williams DS, Atilla GE, Carroll PJ, Meggers E. J. Am. Chem. Soc. 2004;126:13594–13595. doi: 10.1021/ja046049c.
- 13.Hao B, Zhao G, Kang PT, Soares JA, Ferguson TK, Gallucci J, Krzycki JA, Chan MK. Chem. Biol. 2004;11:1317–1324. doi: 10.1016/j.chembiol.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 14.Kolundzic F, Noshi MN, Tjandra M, Movassaghi M, Miller SJ. J. Am. Chem. Soc. 2011;133:9104–9111. doi: 10.1021/ja202706g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chenault HK, Dahmer J, Whitesides GM. J. Am. Chem. Soc. 1989;111:6354–6364. [Google Scholar]
- 16.Miller SJ. Acc. Chem. Res. 2004;37:601–610. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- 17.Francis MB, Jamison TF, Jacobsen EN. Curr. Opin. Chem. Biol. 1998;2:422–428. doi: 10.1016/s1367-5931(98)80019-7. [DOI] [PubMed] [Google Scholar]
- 18.Overman Larry E., Owen Carolyn E., Pavan Mary M. Org. Lett. 2003;5:1809–1812. doi: 10.1021/ol0271786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.