

Abstract
Radiation therapy is a mainstay in the treatment of glioblastomas, but these tumors are often associated with radioresistance. Activation of the phosphatidylinositol-3-OH kinase (PI3K)/Akt pathway, which occurs frequently in glioblastomas due to inactivation of the tumor suppressor phosphatase and tensin homologue (PTEN), correlates with radioresistance. To directly test the link between Akt activation and radioresistance, we utilized PTEN-deficient U251 glioblastoma cells engineered to inducibly restore PTEN upon exposure to doxycycline. These cells showed high basal levels of Akt activation (i.e. high levels of phospho-Akt), but induction of PTEN led to substantially decreased phospho-Akt and was associated with radiosensitization. To investigate whether the PTEN-induced radiosensitization was attributable to impaired sensing versus repair of DNA damage, we assessed levels of γ-H2AX after ionizing radiation in U251 cells induced for PTEN. Initial post-radiation levels of γ-H2AX foci were not decreased in PTEN-induced cells; however, the resolution of these foci was significantly delayed. In contrast to these results, induction of phosphatase-dead PTEN showed no appreciable effect. Finally, exposure of cells to the PI3K inhibitor LY294002 did not decrease the occurrence of γ-H2AX foci after irradiation but did markedly delay their resolution. These results together support a direct link between Akt activation, repair of DNA damage, and radioresistance in glioblastoma. Targeting the PI3K/Akt pathway may modulate DNA repair to improve the efficacy of radiation therapy.
Glioblastoma multiforme, the most common primary adult brain tumor, has a dismal prognosis. Even with aggressive surgery, radiotherapy, and chemotherapy, the median survival for patients with glioblastomas is under one year (1, 2). The phosphatidylinositol-3-OH kinase (PI3K)3 signaling pathway is commonly activated in these tumors, often by virtue of PTEN gene mutation but possibly also by epidermal growth factor receptor expression (3, 4). PTEN encodes a phosphatase that dephosphorylates phosphatidylinositol-3,4,5 triphosphate to convert it to phosphatidylinositol-4,5 bisphosphate. Therefore, inactivation of PTEN leads to increased levels of phosphatidylinositol-3,4,5 triphosphate and increased Akt activation (5). Conversely, restoration of PTEN leads to inhibition of Akt. Chakravarti et al. (3) found significantly reduced survival times in patients whose tumors showed PI3K pathway activation. These patients were treated with a combination of surgery with postoperative radiation as the only adjuvant therapy, which suggested that this pathway might play an important role in radiation resistance.
One of the factors implicated in radioresistance is activation of the Ras/PI3K/Akt pathway (6, 7). Data from numerous investigators show that inhibition of this pathway leads to radiosensitization, not just of glioblastomas but also carcinoma of the colon, bladder, prostate, head and neck, and cervix (6–15).
The precise mechanism by which the PI3K/Akt pathway leads to radioresistance has yet to be determined. Radiation is thought to kill cells by causing DNA damage, specifically double strand breaks (DSBs) (16). This leads to a DNA damage response to allow repair of the DNA strand breaks prior to undergoing cell division. We investigated whether the PI3K/Akt pathway may influence the DNA damage response. In order to manipulate activity of this pathway we tested a cell line that is normally PTEN-deficient but has been engineered so that wild-type PTEN can be induced by adding doxycycline, U251-PTEN (17). As a control we used another cell line, U251-C124S, that is inducible for a mutant form of PTEN that is devoid of phosphatase activity due to a cysteine 124 alteration to serine. In contrast to the radiosensitization and impaired DNA damage repair noted when wild-type PTEN was induced, doxycycline-mediated induction of U251-C124S showed no effect on clonogenic survival or resolution of H2AX activation. These experiments together link Akt activation with DNA damage repair, a pathway that may be usefully modulated by PTEN.
EXPERIMENTAL PROCEDURES
Tissue Culture and Reagents
U251MG, a human malignant glioma cell line that is PTEN-deficient, was obtained from the Brain Tumor Research Center Tissue Bank at the University of California San Francisco. U251MG cells were cultured in Dulbecco’s modified Eagle’s medium (4500 mg/liter glucose; Invitrogen) containing 10% fetal bovine serum (Atlanta Biologicals). U251-PTEN and U251-C124S doxycycline-inducible cells, both derivatives of U251MG cells, were a gift from M. Georgescu (M.D. Anderson Cancer Center, Houston, TX) (17). These cells were cultured in Dulbecco’s modified Eagle’s medium (4500 mg/liter glucose; Invitrogen) containing 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) with G418 (400 μg/ml) and blasticidin (2 μg/ml) added. These cells were cultured in and grown in an incubator containing 5% carbon dioxide and 21% oxygen.
Radiation Survival Determination
Cells were seeded into 60-mm dishes at defined densities and irradiated 1 h after addition of drug at a dose rate of 1.6 Gy/min using a Mark I cesium irradiator (J. L. Shepherd, San Fernando, CA). Colonies were stained and counted 10 to 14 days after irradiation. A colony by definition had >50 cells. The surviving fraction was calculated by dividing the number of colonies formed by the number of cells plated × plating efficiency. Each point on the survival curve represents the mean surviving fraction from at least three replicates.
Protein Extraction, Western Blot Analysis
Details regarding protein isolation, gel electrophoresis, and Western blotting have been published previously (18). The following antibodies were used: monoclonal anti-phospho Akt antibody that recognizes P-S473 (Cell Signaling, Danvers, MA), anti-Akt antibody (Cell Signaling) at a dilution of 1:1000, anti-b-actin antibody (Sigma) at a 1:1,000 dilution. The secondary antibody used for these blots was a goat anti-mouse antibody (Bio-Rad). Antibody binding was detected by chemiluminescence using an ECL kit (Amersham Biosciences).
Flow Cytometry
Determination of cell cycle status was performed via flow-assisted cytometric analysis as described previously (19). Cells were flash-frozen via liquid nitrogen immediately after harvesting and resuspension in citrate buffer (40 mM sodium citrate and 5% Me2SO) and stored at −70° until analysis. At analysis, all samples were thawed and stained with propidium iodide simultaneously. Flow analysis was performed on a BD Biosciences FACSCalibur flow cytometer, and the data were analyzed using CellQuest Pro cell cycle analysis program.
Assays for γ-H2AX Activation
Following irradiation, cells were assessed via immunofluorescence for unrepaired DNA damage via phosphorylation of H2AX (γ-H2AX), a standard marker of unrepaired double strand DNA damage. For these experiments, cells were grown on coverslips. All groups of cells were fixed in 4% paraformaldehyde with 0.1% Triton-X100 and probed with anti-γ-H2AX antibody (Upstate Biological, Inc., Lake Placid, NY), followed by secondary antibody (anti-mouse Alexa Fluor 594 (Molecular Probes. subsidiary of Invitrogen). After the staining with specific antibody, the coverslips were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to mark the nuclei. All treatment groups were then assessed for γ-H2AX foci via sequential imaging through each nuclei. A minimum of 300 cells in each treatment group were counted.
RESULTS
U251MG glioblastoma cells are PTEN-deficient, resulting in a high level of Akt activation (5). Treatment of these cells with the PI3K inhibitor LY294002 increases their sensitivity to radiation as determined by clonogenic survival assays (Fig. 1A). Because pharmacologic agents may exert nonspecific effects, we wished to employ a genetic approach to verify these findings. To manipulate the level of PTEN and consequently of phospho-Akt in U251MG cells, we used a derivative cell line containing a tetracycline-inducible wild-type PTEN transgene (17). We confirmed that addition of doxycycline to this cell line increased expression of PTEN and decreased the level of phospho-Akt, as expected (Fig. 1B). An isogenic control cell line was also used in which a mutant, phosphatase-dead form of PTEN (124S) was induced with doxycycline, which did not result in a decrease in phospho-Akt expression (Fig. 1B). U251-PTEN cells were treated with doxycycline, irradiated, and then assessed for clonogenic survival. Induction of wild-type PTEN in these cells led to radiosensitization (Fig. 1C), whereas no such effect was seen in U251-C124S, the control counterpart cell line in which mutant PTEN was induced (Fig. 1D).
FIGURE 1. PI3K/Akt pathway inhibition radiosensitizes U251MG glioblastoma cells.

A, clonogenic survival assays indicate radiosensitization of U251MG after pharmacologic inhibition of PI3K/Akt. U251MG cells were seeded as single cell suspensions. Three hours after seeding, LY294002 (10 μM) or vehicle only was added and then followed 1 h later by mock-irradiation or exposure to ionizing radiation at the indicated doses. After 24 h of exposure, the LY294002 was removed and the cells left to grow undisturbed for 7–10 days; the resultant colonies were stained and counted. B, induction of wild-type or mutant PTEN. U251MG cells engineered to be inducible for either wild-type PTEN or phosphatase-dead PTEN (C124S) were exposed to doxycycline (1 μg/ml). After 24 h, cells from each group were harvested and the resultant lysates immunoblotted for the proteins indicated. Doxycycline-induced PTEN protein diminishes activated Akt as indicated by reduced levels of Akt phosphorylated at serine 473. C, radiosensitization of U251MG after induction of wild-type PTEN. Clonogenic assays performed on U251-PTEN in the presence or absence of doxycycline. Cells were exposed to doxycycline or vehicle for 24 h, followed by reseeding onto fresh plates and mock-irradiation or irradiation with the indicated doses. D, lack of radiosensitization with induction of mutant PTEN. Clonogenic assays performed on U251 cells engineered to be inducible for catalytically inactive PTENC124S in the presence or absence of doxycycline. Cells were exposed to doxycycline or vehicle for 24 h, followed by reseeding onto fresh plates and mock-irradiation or irradiation with the indicated doses. After 4 h to allow for attachment, they were irradiated. Doxycycline was removed after 24 h. Survival fraction is plotted on the y-axis versus dose of radiation on the x-axis. All surviving fractions with radiation were normalized to these base-line plating efficiencies.
These experiments established that down-regulation of the PI3K/Akt pathway in these cells led to increased sensitivity to radiation. Because radiosensitivity varies with the cell cycle, it was important to assess that induction of wild-type PTEN did not dramatically alter the cell cycle profile. Fig. 2 shows that the addition of doxycycline led to a minimal change in the percentage of cells in G1, G2, and S phase in these cells at the time of irradiation (T = 0 h). Therefore, changes in cell cycle distribution do not account for the radiosensitization seen with PTEN induction.
FIGURE 2. Cell cycle status after radiation of cells induced for wild-type PTEN.
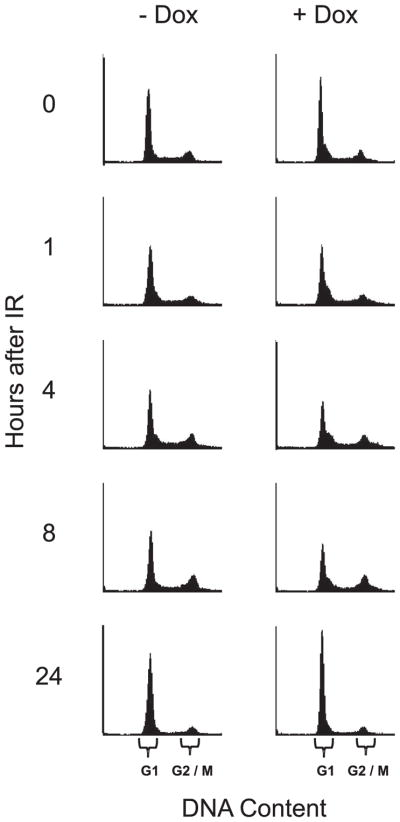
U251-PTEN cells engineered to be inducible for wild-type PTEN in response to doxycycline were exposed to vehicle only (−Dox) or doxycycline (+Dox) for 24 h, followed by irradiation (IR) with 4 Gy. Replicate plates of cells were harvested either immediately before IR (0) or at the indicated hours after IR. Cells from all time points and treatment groups were assessed for cell cycle status via fluorescent cell sorting. The resultant histograms are shown, with the portions of the x-axis (DNA Content) corresponding to either G1 or G2/M as indicated by the brackets.
We investigated next whether PTEN induction might modulate unrepaired DNA DSB damage, via assessment of γ-H2AX. At sites of DSBs, γ-H2AX is recruited to megabase domains detectable as nuclear foci, the number of which correlates with the amount of unrepaired damage (20). Fig. 3A shows representative photographs of the U251-PTEN cell line treated with combinations of doxycycline and ionizing radiation (4 Gy). These data are quantitated in Fig. 3B. Radiation-induced γ-H2AX foci were readily visible within 1 h of irradiation in both doxycycline-treated and control groups. These foci had substantially decreased in number by 8 h post-irradiation in the control group and had disappeared by 24 h. In contrast, the addition of doxycycline to U251-PTEN cells led to a substantial percentage of cells still retaining these foci at 24 h (Fig. 3B). In further support of the idea that these foci represent sites of unrepaired DSBs, we tracked the fate of the persistent foci in surviving cells. At extended times after irradiation, many γ-H2AX foci were retained within micronuclei, which are fragments of chromatin that are widely considered to represent unrepaired DNA damage (21–23) (Fig. 4).
FIGURE 3. Induction of wild-type PTEN protein leads to persistence of γ-H2AX foci.
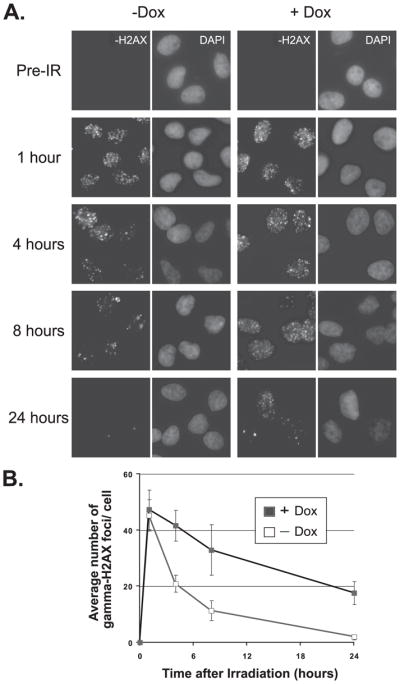
U251 cells engineered to be inducible for wild-type PTEN grown on coverslips were exposed to vehicle only (−Dox) or doxycycline for 24 h (+Dox), followed by irradiation (IR) with 4 Gy. Replicate groups of cells were fixed either immediately before IR (0) or at the indicated hours after IR. All cells were then stained for γ-H2AX or nuclei (DAPI) via immunofluorescence. Representative images of cells of each treatment group are shown in panel A. The number of γ-H2AX foci was counted for each cell and the average number of foci/cell recorded. A minimum of 300 cells in each treatment group were counted. The quantitation of the average number of γ-H2AX foci is shown in panel B. Error bars indicate S.E.
FIGURE 4. Persistence of γ-H2AX foci in micronuclei in U251 cells induced for wild-type PTEN protein.
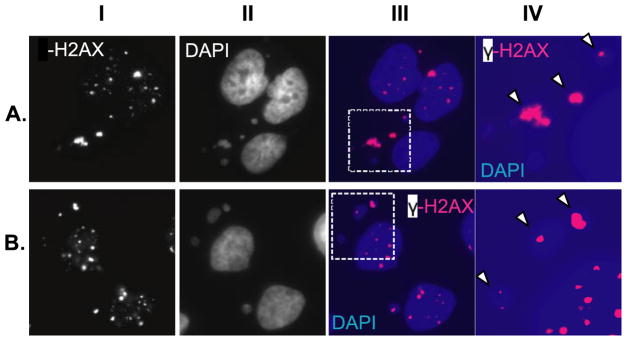
U251 cells were prepared and treated as described for Fig. 3 but fixed for immunofluorescence at 36 h after irradiation. Surviving cells were stained for γ-H2AX and then counterstained with DAPI to visualize chromatin. Images show representative groups of cells, with the panels in either A or B representing images of the same group of cells. Panels IA and B show staining for γ-H2AX, while panels IIA and B show DAPI. Panels IIIA and B show both γ-H2AX and DAPI pseudocolored, respectively, for magenta and blue and then merged. The portions of the panels in III indicated by the dashed boxes are shown at higher magnification in panels IVA and B. Arrowheads denote examples of γ-H2AX staining coinciding with micronuclei, indicating that micronuclei contain unrepaired double strand DNA damage.
To exclude the possibility that exposure to doxycycline in itself may affect the kinetics of DNA damage repair, we assessed the isogenic U251-C124S cell line in which catalytically inactive PTEN was induced by the addition of doxycycline. (Fig. 1B). In contrast to the striking difference seen with the U251-PTEN line (Fig. 3), induction of mutant PTEN showed near-identical rates of resolution of γ-H2AX foci (Fig. 5), indicating that exposure to doxycycline or the induction of an inactive PTEN protein does not influence DNA damage repair.
FIGURE 5. Induction of phosphatase-dead PTEN by doxycycline does not alter kinetics of double strand DNA damage repair.

U251-C124S cells were exposed to doxycycline (+Dox) or vehicle (−Dox) for 24 h, followed by irradiation with 4 Gy. At the indicated times after radiation, replicate groups of cells were fixed and stained for γ-H2AX and then counterstained with DAPI to visualize nuclei. The number of γ-H2AX foci was counted for each cell and the average number of foci/cell recorded. A minimum of 300 cells in each treatment group were counted. Representative cells fixed at the indicated times and stained for γ-H2AX are shown in panel A; the quantification of the average number of γ-H2AX foci/cell for each treatment group is shown in panel B. Error bars indicate S.E. γ-H2AX foci noted within micronuclei were not counted.
Our observations suggest that PTEN likely modulates its effects on radiosensitivity through down-regulation of PI3K activity and suppression of efficient DNA damage repair. As an additional test of this hypothesis, we assessed the effect of the PI3K inhibitor LY294002 on DNA damage repair. These experiments also tested whether the radiosensitization noted with LY294002 (Fig. 1A) was attributable to deficient DNA damage sensing. U251 cells were treated with LY294002 and then irradiated in the presence of the drug. At 1 h after IR, the number of γ-H2AX foci was similar in the presence or absence of the drug. The largest differences were seen at subsequent time points, in which LY294002 resulted in persistence of γ-H2AX foci (Fig. 6, A and B). These results suggest that inhibition of PI3K activity in U251 cells probably does not abolish the sensing of DNA damage but leads rather to a reduced rate of repair of this damage. Consequently, down-regulation of a pathway that results in diminished PI3K activity leads to persistence of a marker of unrepaired double strand DNA damage.
FIGURE 6. Pharmacologic inhibition of PI3K leads to impaired DNA damage repair.

U251 cells were exposed to LY294002 or vehicle for 24 h, followed by irradiation with 4 Gy. At the indicated times after radiation, replicate groups of cells were fixed and stained for γ-H2AX and then counterstained with DAPI to visualize nuclei. The number of γ-H2AX foci was counted for each cell and the average number of foci/cell recorded. A minimum of 300 cells in each treatment group were counted. Representative cells fixed at the indicated times are shown in panel A; the quantification of the average number of γ-H2AX foci/cell for each treatment group is shown in panel B. Error bars indicate S.E. γ-H2AX foci noted within micronuclei were not counted.
DISCUSSION
Numerous studies have shown that activation of the PI3K/Akt pathway is associated with resistance to radiation in many cell lines (6–15). We show in this report using U251, a PTEN-deficient cell line, that either treatment with the PI3K inhibitor LY294002 or conditional PTEN expression using a doxycycline-inducible system leads to radiosensitization. How does this occur? One possibility is that there might be redistribution of cells into more radiosensitive phases of the cell cycle. However, flow cytometry showed that induction of wild-type PTEN caused minimal changes in the cell cycle at the time of irradiation (T = 0 h); therefore, this is unlikely to be the explanation for the radiosensitization.
H2AX is a histone that is phosphorylated on Ser-129 (24) by ataxia telangiectasia mutated (ATM) and DNA-protein kinase (25–27) in response to DNA DSBs. This reaction occurs extremely rapidly with half-maximal amounts generated within a minute and maximal amounts by 10 min (24). Hence, measurement of γ-H2AX is a sensitive and specific assay for unrepaired DSBs. γ-H2AX regulates the recruitment of repair factors such as Nbs1, 53BP1, and BRCA1 to foci located at DSB sites (28, 29). It is hypothesized that the formation of these foci increases the local concentration of repair proteins and facilitates repair of DSBs. Therefore, γ-H2AX is not merely a marker for DNA damage repair but plays an essential role in the repair process. Consistent with this notion, mice deficient for both copies of the gene are viable but display genomic instability and are exquisitely sensitive to ionizing radiation (30, 31).
In the studies described here, we combined complementary genetic and pharmacologic approaches to show that a potential mechanism for the radiosensitization by PTEN is disruption of the DNA damage repair process. Both approaches indicated that PTEN modulated primarily the repair of DNA damage repair. Other studies have investigated the effects of PI3K inhibitors on γ-H2AX foci. At nanomolar concentrations, wortmannin irreversibly inhibits the lipid kinase activity of PI3K (32). Burma et al. (26) found that 1 μM of the drug did not prevent the induction of γ-H2AX after DNA damage. However, at 100 μM, γ-H2AX activation was abolished. These results are consistent with the inhibition of kinases other than PI3K, perhaps including DNA-protein kinase, mTOR, mitogen-activated protein kinase, ATM at extremely high levels of wortmannin (33–36). Stiff et al. (27) found that LY294002 did not decrease γ-H2AX foci formation in cells unless they were deficient for ATM, implicating ATM as the target of the drug. Likewise, Paull et al. (28) found that preincubation of cells with a high concentration of wortmannin (200 μM) interfered with activation of γ-H2AX but, interestingly, addition of the same concentration of wortmannin after irradiation did not prevent γ-H2AX foci formation. Unfortunately, the kinetics of foci resolution were not assessed.
A genetic approach can help avoid nonspecific effects that sometimes accompany pharmacologic interventions. An advantage to using a PTEN-inducible cell line is that this results in specific down-regulation of the PI3K/Akt pathway rather than inhibition of other phosphatidyl 3-kinase-related kinase family members such as DNA-protein kinase or ATM. Our results are more consistent with those of Toulany et al. (37), who measured γ-H2AX foci in A549 lung carcinoma cells 24 h after irradiation and found that the number of foci/cell was greater in cells treated with LY294002 and radiation than in cells treated with radiation alone. However, this result is difficult to reconcile with their other finding, that LY294002 blunted γ-H2AX induction following radiation as determined by Western blotting in these cells (37).
Of note, we found that inhibition by PTEN induction or by LY294002 did not abolish expression of γ-H2AX foci. Quite the contrary, the number of foci/cell 1 h after radiation was the same in control cells as in the PTEN-induced or LY294002-treated cells. However, the γ-H2AX foci persisted much longer in the PTEN-induced or LY294002-treated cells. By 24 h, all the foci had disappeared from control cells but they were still present in the treated cells. The fact that many of the persistent foci were seen in cells with micronuclei (Fig. 5) is consistent with the idea that unrepaired DNA damage in these cells leads to cell death. Ionizing radiation produces dicentrics that then lead to the formation of both micronuclei and anaphase bridges. Cells containing a micronucleus at the first division after irradiation or subsequently generally cannot form colonies (38).
In conclusion, we found that down-regulation of PI3K/Akt signaling by PTEN induction or by treatment with the PI3K inhibitor LY294002 led to persistence of unrepaired double strand DNA damage. These observations also imply that the PI3K/Akt pathway can modulate DNA damage repair in response to radiation and may have relevance to modulating the efficacy of radiation therapy in other types of human cancers in which this pathway has been activated.
Acknowledgments
We thank Melissa Dowling for excellent technical assistance, especially in performing flow cytometry, and Nabendu Pore for characterizing the U251-PTEN and U251-C124S cells.
Footnotes
This work was supported by Public Health Service Grants R01 CA093638 (to A. M.), R01 CA107956 (to G. D. K.), and P01CA075138 (to G. D. K.).
The abbreviations used are: PI3K, phosphatidylinositol-3-OH kinase; PTEN, phosphatase and tensin homologue; DSB, double strand break; Gy, gray; IR, irradiation; DAPI, 4′,6-diamidino-2-phenylindole; ATM, ataxia telangiectasia mutated.
References
- 1.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Chakravarti A, Zhai G, Suzuki Y, Sarkesh S, Black PM, Muzikansky A, Loeffler JS. J Clin Oncol. 2004;22:1926–1933. doi: 10.1200/JCO.2004.07.193. [DOI] [PubMed] [Google Scholar]
- 4.Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL, Mischel PS. Cancer Res. 2003;63:2742–2746. [PubMed] [Google Scholar]
- 5.Haas-Kogan D, Shalev N, Wong M, Mills G, Yount G, Stokoe D. Curr Biol. 1998;8:1195–1198. doi: 10.1016/s0960-9822(07)00493-9. [DOI] [PubMed] [Google Scholar]
- 6.Gupta AK, Bakanauskas VJ, Cerniglia GJ, Cheng Y, Bernhard EJ, Muschel RJ, McKenna WG. Cancer Res. 2001;61:4278–4282. [PubMed] [Google Scholar]
- 7.Grana TM, Rusyn EV, Zhou H, Sartor CI, Cox AD. Cancer Res. 2002;62:4142–4150. [PubMed] [Google Scholar]
- 8.Edwards E, Geng L, Tan J, Onishko H, Donnelly E, Hallahan DE. Cancer Res. 2002;62:4671–4677. [PubMed] [Google Scholar]
- 9.Gottschalk AR, Doan A, Nakamura JL, Stokoe D, Haas-Kogan DA. Int J Radiat Oncol Biol Phys. 2005;63:1221–1227. doi: 10.1016/j.ijrobp.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 10.Gupta AK, Cerniglia GJ, Mick R, Ahmed MS, Bakanauskas VJ, Muschel RJ, McKenna WG. Int J Radiat Oncol Biol Phys. 2003;56:846–853. doi: 10.1016/s0360-3016(03)00214-1. [DOI] [PubMed] [Google Scholar]
- 11.Gupta AK, McKenna WG, Weber CN, Feldman MD, Goldsmith JD, Mick R, Machtay M, Rosenthal DI, Bakanauskas VJ, Cerniglia GJ, Bernhard EJ, Weber RS, Muschel RJ. Clin Cancer Res. 2002;8:885–892. [PubMed] [Google Scholar]
- 12.Kim IA, Bae SS, Fernandes A, Wu J, Muschel RJ, McKenna WG, Birnbaum MJ, Bernhard EJ. Cancer Res. 2005;65:7902–7910. doi: 10.1158/0008-5472.CAN-05-0513. [DOI] [PubMed] [Google Scholar]
- 13.Lee CM, Fuhrman CB, Planelles V, Peltier MR, Gaffney DK, Soisson AP, Dodson MK, Tolley HD, Green CL, Zempolich KA. Clin Cancer Res. 2006;12:250–256. doi: 10.1158/1078-0432.CCR-05-1084. [DOI] [PubMed] [Google Scholar]
- 14.Li B, Yuan M, Kim IA, Chang CM, Bernhard EJ, Shu HK. Oncogene. 2004;23:4594–4602. doi: 10.1038/sj.onc.1207602. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura JL, Karlsson A, Arvold ND, Gottschalk AR, Pieper RO, Stokoe D, Haas-Kogan DA. J Neuro-oncol. 2005;71:215–222. doi: 10.1007/s11060-004-1718-y. [DOI] [PubMed] [Google Scholar]
- 16.Willers H, Dahm-Daphi J, Powell SN. Br J Cancer. 2004;90:1297–1301. doi: 10.1038/sj.bjc.6601729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM. Mol Cell Biol. 2003;23:6139–6149. doi: 10.1128/MCB.23.17.6139-6149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A. Cancer Res. 2006;66:3197–3204. doi: 10.1158/0008-5472.CAN-05-3090. [DOI] [PubMed] [Google Scholar]
- 19.Vindelov LL, Christensen IJ. Cytometry. 1990;11:753–770. doi: 10.1002/cyto.990110702. [DOI] [PubMed] [Google Scholar]
- 20.Aten JA, Stap J, Krawczyk PM, van Oven CH, Hoebe RA, Essers J, Kanaar R. Science. 2004;303:92–95. doi: 10.1126/science.1088845. [DOI] [PubMed] [Google Scholar]
- 21.Stopper H, Full M, Helbig R, Speit G. Mutat Res. 1997;383:107–112. doi: 10.1016/s0921-8777(96)00049-3. [DOI] [PubMed] [Google Scholar]
- 22.Benner SE, Wargovich MJ, Lippman SM, Hong WK. J Cell Biochem Suppl. 1993;17F:250–254. doi: 10.1002/jcb.240531037. [DOI] [PubMed] [Google Scholar]
- 23.Fenech M. Mutat Res. 2000;455:81–95. doi: 10.1016/s0027-5107(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 24.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 25.Redon C, Pilch D, Rogakou E, Sedelnikova O, Newrock K, Bonner W. Curr Opin Genet Dev. 2002;12:162–169. doi: 10.1016/s0959-437x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 26.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 27.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- 28.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 29.Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. Biochem Cell Biol. 2003;81:123–129. doi: 10.1139/o03-042. [DOI] [PubMed] [Google Scholar]
- 30.Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R, Alt FW. Proc Natl Acad Sci U S A. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, Zalkow L, Matter WF, Dodge J, Grindey G, Vlahos CJ. Cancer Res. 1994;54:2419–2423. [PubMed] [Google Scholar]
- 33.Izzard RA, Jackson SP, Smith GC. Cancer Res. 1999;59:2581–2586. [PubMed] [Google Scholar]
- 34.Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Clin Cancer Res. 1997;3:1149–1156. [PubMed] [Google Scholar]
- 35.Goodarzi AA, Lees-Miller SP. DNA Repair (Amst ) 2004;3:753–767. doi: 10.1016/j.dnarep.2004.03.041. [DOI] [PubMed] [Google Scholar]
- 36.Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC, Jr, Abraham RT. EMBO J. 1996;15:5256–5267. [PMC free article] [PubMed] [Google Scholar]
- 37.Toulany M, Kasten-Pisula U, Brammer I, Wang S, Chen J, Dittmann K, Baumann M, Dikomey E, Rodemann HP. Clin Cancer Res. 2006;12:4119–4126. doi: 10.1158/1078-0432.CCR-05-2454. [DOI] [PubMed] [Google Scholar]
- 38.Forrester HB, Albright N, Ling CC, Dewey WC. Radiat Res. 2000;154:625–639. doi: 10.1667/0033-7587(2000)154[0625:cvtlao]2.0.co;2. [DOI] [PubMed] [Google Scholar]