

Abstract
Recently, we described the first small-molecule inhibitor, (E)-ethyl 2-cyano-3-(3,4-dichlorophenyl)acryloylcarbamate (1), of the PDZ domain of protein interacting with Cα-kinase 1 (PICK1), a potential drug target against brain ischemia, pain and cocaine addiction. Herein, we explore structure–activity relationships of 1 by introducing subtle modifications of the acryloylcarbamate scaffold and variations of the substituents on this scaffold. The configuration around the double bond of 1 and analogues was settled by a combination of X-ray crystallography, NMR and density functional theory calculations. Thereby, docking studies were used to correlate biological affinities with structural considerations for ligand–protein interactions. The most potent analogue obtained in this study showed an improvement in affinity compared to 1 and is currently a lead in further studies of PICK1 inhibition.
Introduction
The inhibition of specific protein–protein interactions involved in signal transduction, cell–cell communication, apoptosis, and many other vital cellular processes with small organic molecules has great potential in probing biological questions and pursuing therapeutic modalities.1–3 PSD-95/Discs-large/ZO-1 (PDZ) domains are involved in several potentially therapeutic relevant protein–protein interactions,4–6 and they often function as scaffolding modules in proteins that are involved in trafficking and assembling of large protein complexes in the cell. PDZ domains generally recognize the C-terminal part of the interacting protein and they are highly abundant in eukaryotic organisms.7–10
The PDZ domain of protein interacting with Cα-kinase (PICK1) was first shown to interact with protein kinase Cα (PKCα) to mediate contacts between PKCα and integral membrane proteins in the central nervous system,11 whereby phosphorylation of these proteins is regulated.12–15 By comprising both the PDZ domain and the Bin/amphiphysin/Rvs (BAR) domain, a membrane-binding and curvature-sensing module,17,18 PICK1 can generate diverse protein complexes and regulate their membrane clustering and trafficking.19–22 Several membrane bound transporters and receptors have been found to interact with the PDZ domain of PICK1, including the monoaminergic (dopamine, serotonin, norepinephrine) transporters, and several subunits of the ionotropic glutamate receptors including the α-amino-3-hydroxy-5-methyl-isoxazole-4-propionic acid (AMPA), kainate, and the metabotropic glutamate receptors.4,23,24 PICK1 was found to be important for various forms of synaptic plasticity,20,25–27 including long-term depression (LTD) and long-term potentiation (LTP) presumably via its interaction with the AMPA receptors, and has been suggested as a therapeutic target for treating brain ischemia,28,29 pain,4,30,31 and cocaine addiction.32 Hence, specific small-molecule inhibitors of the PDZ domain of PICK1 could support the study of a variety of physiological processes and provide leads for potential therapeutic interventions.
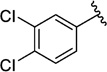
PDZ domains are considered difficult to target with small-molecules because of their shallow and elongated binding pocket,33,34 and only relatively few examples of potent (Ki <50 µM) PDZ inhibitors are found in the literature,35–47 most of which are peptide-derived.40–44,48 However, by screening ~43 000 compounds we have recently identified a small-molecule inhibitor, (E)-ethyl 2-cyano-3-(3,4-dichlorophenyl)acryloylcarbamate (1, Fig. 1), of the PICK1 PDZ domain with Ki = 9.6 µM. This affinity is in the same range as observed for the endogenous C-terminal peptide ligands.49 Biochemical studies demonstrated that 1 binds reversibly to PICK1 and exhibits selectivity with respect to cognate PDZ domains such as the three PDZ domains of postsynaptic density protein-95 (PSD-95), or PDZ4-5 of glutamate receptor interacting protein-1 (GRIP1). Additionally, 1 is membrane permeable and binds the PICK1 PDZ domain in living cells, as demonstrated by intracellular fluorescence resonance energy transfer (FRET). Furthermore, 1 affected AMPA receptor trafficking and inhibited induction of LTD and LTP in hippocampal CA1 neurons.49 Structurally, 1 is promising for further studies because it does not contain a carboxylic acid group as found in most PDZ ligands,36–47 which could partially explain its ability to cross membranes. Here, we explore structure–activity relationships (SARs) of 1, in order to elucidate the structural requirements of 1 and analogues for PICK1 affinity (Fig. 1). A fluorescence polarization assay, which quantifies the ability to displace a fluorescently labelled undecapeptide representing the extreme C-terminal of the human dopamine transporter (hDAT), was used to determine affinities between compounds and PICK1. The results from key compounds was confirmed in a glutathione S-transferase (GST) fusion protein pull-down assay.49 The compounds were also tested against the three PDZ domains of PSD-95 to examine selectivity,42 and finally, we analyzed the SAR in a structural context with computational docking studies.
Fig. 1.
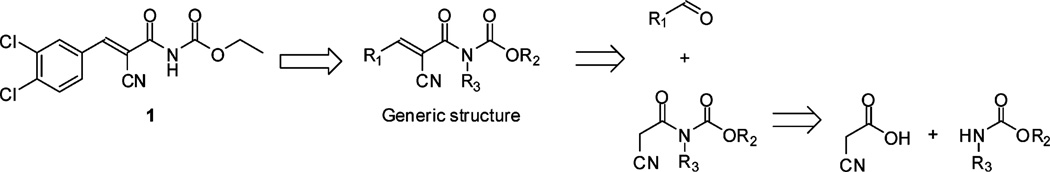
Structure of lead compound 1 and the concordant generic structure together with its retrosynthetic analysis. Substituents explored in this SAR study are indicated R1–3.
Results and discussion
Chemistry
The synthetic route for compound 1 and its analogues was identified by a retrosynthetic analysis of the generic acryloylcarbamate structure (Fig. 1). A condensation reaction with 2-cyanoacetic acid and carbamates was chosen to obtain the intermediate cyanoacetylcarbamates, which could subsequently be reacted with aromatic aldehydes in a Knoevenagel condensation reaction to furnish the target compounds. Besides being efficient, this route has the advantage of being versatile and allows convenient synthesis of analogues, as several building blocks are commercially available.
To prepare 1, the cyanoacetylcarbamic acid ethyl ester (2) intermediate was first synthesized from 2-cyanoacetic acid and ethyl carbamate (urethane) using phosphorus chloride oxide (POCl3) as a condensation agent (Scheme 1). By using POCl3 and small amounts of DMF to catalyze the reaction, in aprotic solvents, side reactions are limited and yields increased compared to using acetic acid anhydride as condensation agent and/or protic solvents.50 The Knoevenagel reaction between 2 and 3,4-dichlorobenzaldehyde to provide 1 was initially carried out at room temperature using piperidine as a catalyst.51 However, we found that adding small amounts of acetic acid improved both yield and purity, as previously shown for other Knoevenagel reactions,52,53 while refluxing did not improve the reaction.
Scheme 1.
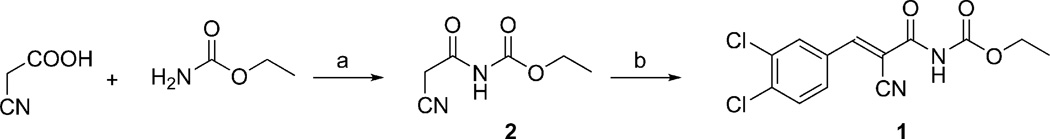
(a) POCl3, DMF, toluene, 70 °C, 1.5 h (82%); (b) 3,4-dichlorobenzaldehyde, piperidine, AcOH, DMF, 2 h (43%).
To investigate the biological importance of the acryloylcarbamate scaffold we introduced subtle modifications by individually modifying the alkene, the cyano, the ester, and the carbonyl group as in compounds 3, 5, 8 and 14 (Scheme 2). Ethyl 2-cyano-3-(3,4-dichlorophenyl)propanoylcarbamate (3) was obtained by hydrogenation of the double bond of 1, which was a highly selective reaction as neither the cyano group or halogens were affected (Scheme 2a). The des-cyano analogue of 1, (E)-ethyl 3-(3,4-dichlorophenyl)acryloylcarbamate (5), was synthesized by amidolysis of 4 with the anion of urethane (Scheme 2b). However, presumably a self-condensation of urethane anions leading to ethoxide, which reacted with 4, was responsible for the low yield of the reaction as the undesired ethyl (E)-3,4-dichlorocinnamate (6; electronic supplementary information†) was observed as the major product. The ester functionality was replaced with an alkyl moiety by aminolysis of 2 with butylamine (expelling urethane), providing 7 in high yield. A Knoevenagel condensation of 7 with 3,4-dichlorobenzaldehyde resulted in (E)-N-butyl-2-cyano-3-(3,4-dichlorophenyl)acrylamide (8) (Scheme 2c). Finally, in order to investigate the significance of the carbonyl moiety we prepared (Z)-ethyl 2-cyano-3-(3,4-dichlorophenyl)allylcarbamate (14) (Scheme 2d). The precursor 9 was produced by a Baylis–Hillman reaction, followed by treatment with phosphorus tribromide to provide 10 and 11 in a 1 : 1 mixture (GC-MS). As these compounds were inseparable for all practical purposes, we treated the mixture with methanolic ammonia to furnish 12 and 13, which were isolated by standard flash chromatography. Compound 12 was readily acylated with ethyl chloroformate affording the desired 14. TheZ geometry in 14 (as depicted in Scheme 2d) was verified by NOESY experiments and supported by density functional theory (DFT) calculations (electronic supplementary information†).
Scheme 2.

(a) H2 (1 atm.), Pd/C (3.5 mol%), EtOAc, rt, 24 h (76%); (b) SOCl2, reflux, 2 h (73%); (c) sodium (ethoxycarbonyl)amide, DMF, rt, 1 h (14%); (d) BuNH2, MeOH, rt, 1 h (91%); (e) 3,4-dichlorobenzaldehyde, piperidinium acetate (cat.), DMF, rt, 48 h (47%); (f) acrylonitrile, DABCO, dioxane–water (1 : 1), rt, 24 h (68%); (g) PBr3, Et2O, rt, 2 h (69%); (h) NH3, MeOH, rt, 2 h (12: 19%; 13: 65%); (i) EtOCOCl, Et3N, THF, rt, 1 h (64%).
In order to probe the importance of the terminal alkyl tail of 1 and to study the possibilities of introducing substituents on the central nitrogen atom, a series of cyanoacetylcarbamate intermediates were prepared (Scheme 3). Compounds 15–18 were synthesized from commercially available building blocks using the same protocol as for the synthesis of 2 (Scheme 3a). For the preparation of intermediates 20, 23 and 24, the starting carbamate had to be prepared. Compound 20 was synthesized by converting adamantan-1-ol into carbamate 19 by reaction with trichloroacetyl isocyanate and basic hydrolysis,54,55 followed by condensation with 2-cyanoacetic acid (Scheme 3b). Ethyl butylcarbamate (21) and ethyl benzylcarbamate (22) were prepared by aminolysis of ethyl chloroformate with butylamine or benzylamine, respectively, and 21 and 22 were subsequently reacted with 2-cyanoacetic acid to furnish 23 and 24 (Scheme 3c).
Scheme 3.

(a) POCl3, DMF, toluene, 70 °C, 1–2 h; (b) Cl3CCONCO, CH2Cl2, 0 °C→23 °C, 2 h; (c) K2CO3,MeOH, 50 °C, 16 h (87%); (d) 2-cyanoacetic acid, POCl3, DMF, toluene, 70 °C, 1.5 h; (e) Et3N, THF, rt, 2 h
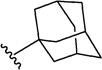
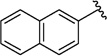
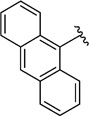
Analogues with a range of acryloylcarbamate substituents, R1–3 (Fig. 1), were prepared applying the same synthetic method as described for 1, where cyanoacetylcarbamate intermediates and commercially available aromatic aldehydes were reacted generally in good yields and purities after recrystallization. In the first class of analogues, 25–35 (Table 1) and 36 (Table 2), the aromatic and the ethyl moieties were varied. Subsequently, N-alkylated carbamates, compounds 37–40 (Table 2), were prepared and finally subtle changes of the chloro substituents of the phenyl group were made as in compounds 41–49 (Table 3).
Table 1.
 |
||||
|---|---|---|---|---|
| R2 | ||||
| R1 |  |
 |
||
 |
1 | 28 | 32 | |
| 9.6 ± 0.22 | 11 ± 1.7 | NA | ||
 |
25 | 29 | 33 | |
| 12 ± 2.5 | 15 ± 4.2 | NA | ||
 |
26 | 30 | 34 | |
| NA | NA | NA | ||
 |
27 | 31 | 35 | |
| 12 ± 2.3 | 14 ± 2.9 | NA | ||
Ki values are shown as mean ± SEM in µM based on at least three individual measurements.
NA: no affinity.
Table 2.
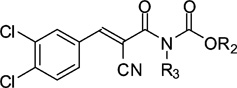
Structures and affinities of compound 1 and analogues 36–40
 | |||
|---|---|---|---|
| Compound | R2 | R3 | Ki/µMa |
| 1 | Ethyl | H | 9.6 ± 0.22 |
| 36 | Butyl | H | 12 ± 2.8 |
| 37 | Ethyl | Methyl | 55 ± 13 |
| 38 | Ethyl | Ethyl | 40 ± 3.1 |
| 39 | Ethyl | Butyl | 19 ± 4.7 |
| 40 | Ethyl | Benzyl | 14 ± 1.7 |
Ki values are shown as mean ± SEM based on at least three individual measurements.
Table 3.
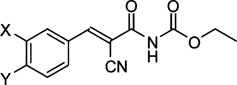
Structures and affinities of compound 1 and analogues 41–49
 | |||
|---|---|---|---|
| Compound | X | Y | Ki/µMa |
| 1 | Cl | Cl | 9.6 ± 0.22 |
| 41 | H | H | 84 ± 2.2 |
| 42 | H | Cl | 23 ± 0.7 |
| 43 | CH3 | CH3 | 23 ± 3.0 |
| 44 | H | CH3O | 89 ± 20 |
| 45 | F | CF3 | 17 ± 2.7 |
| 46 | Br | NO2 | 26 ± 2.3 |
| 47 | NO2 | Cl | 22 ± 6.1 |
| 48 | NO2 | Br | 11 ± 1.2 |
| 49 | CF3 | Cl | 7.2 ± 0.7 |
Ki values are shown as mean ± SEM based on at least three individual measurements.
Structure–activity relationship studies
All final compounds were investigated for their ability to bind the PICK1 PDZ domain in a fluorescence polarization assay.23,49 Compounds 3, 5, 8 and 14 were all devoid of affinity at up to 500 µM concentrations (data not shown) thus demonstrating the importance of an intact acryloylcarbamate scaffold. A general concern of 1 is that it binds PICK1 irreversibly as a Michael acceptor allowing nucleophilic attack from PDZ amino acid side chains on the alkene bond.56 But biochemical studies demonstrate a reversible binding mode,49 and the close analogue 8, which also possesses a Michael acceptor site, is inactive. Finally, as 1 shows no affinity for PDZ domains from PSD-95 or GRIP1,49 1 is considered a specific and reversible inhibitor of PICK1.
Since SAR studies of 1 had not been carried out thus far, we explored the steric requirements of the aromatic and the ethyl moieties (R1 and R2, respectively, Fig. 1), and introduced relatively large and hydrophobic modifications in these two regions providing a matrix of 12 analogues (1, 25–35, Table 1). The analogues yielded unambiguous information regarding the substitutions allowed in these regions, as an anthracene in the R1 position (analogues 26, 30 and 34), and adamantane in the R2 position (analogues 32–35), abolished affinity. On the other hand, introducing a naphthalene (25 and 29) or a biphenyl group (27 and 31), instead of dichlorophenyl, did not affectKi values significantly compared to 1. Similarly, an isopropyl group instead of ethyl in the R2 position, as in compounds 28–31, did not influence the affinity either (Table 1). We therefore probed the possibility of increasing the length of the alkyl group (as in 36), but the affinity remained equal to that of 1 (Table 2). Taken together, these results suggest that modifying the ethyl moiety by increasing length and/or bulk do not affect affinity.
In order to evaluate the effect of increasing the bulk in the central part of 1, substitutions were introduced on the nitrogen in the acryloylcarbamate moiety (R3). Specifically the nitrogen was alkylated with methyl, ethyl, butyl, and benzyl groups, leading to analogues 37–40. Compounds 37 and 38, with methyl and ethyl substitutions, respectively, had reduced affinity, but affinity was regained by increasing size and bulkiness of the N-substitution, as in compounds 39 and 40 (Table 2). Accordingly, affinity is not improved by adding any hydrophobic functionality in this region, but larger alkyl or aryl groups lead to compounds that are almost equipotent to 1, which indicates that these groups occupy an area in the PDZ domain where variations are tolerated.
Finally, we focused on the chloro substituents on the aromatic ring in relation to affinity towards PICK1. In the screening that had led to the discovery of 1,49 a related structure without chloro substituents on the phenyl ring (41) was identified as a weak inhibitor. To confirm this, compound 41 was synthesized together with the analogue comprising a 4-chloro substituent (42) and tested in the fluorescence polarization assay (Table 3). This revealed a stepwise reduction of affinity when removing one or both chlorine atoms, since 42 and 41 demonstrated a 2.4- and 9-fold reduction in affinity compared to 1, respectively. Also, it was seen that methyl or methoxy groups could not substitute for the chlorines, as the 3,4-dimethyl substituted analogue (43) showed ~2-fold higher Ki value compared to 1, and the 4-methoxy substituted analogue (44) demonstrated a 4-fold higher Ki compared to 42. Thus, chloro substituents are favoured, but whether this was due to the electronic properties (electronegativity) of the chlorines, their lipophilicity, or a combination of these properties was not obvious. Therefore, we explored more subtle modifications in the 3- and 4-positions inspired by the Topliss ‘decision tree’.57–60 This approach explicitly addresses the situation where a 3,4-dichloro analogue is more potent than a 4-chloro analogue, which is more potent than the non-substituted analogue. Hence, we synthesized compound 45, where the size and lipophilicity of the 3-substituent were reduced while the electronic properties were preserved with fluorine, concurrent with increasing the inductive effect and lipophilic properties of the substituent in the 4-position by introducing a trifluoromethyl substituent. Introducing a 4-nitro group similarly increased the negative polarity of the region while a simultaneous substitution with 3-bromo increased lipophilicity, as in 46. However, none of these combinations of substitutions provided increased affinity compared to 1 (Table 3). When the 3-position was substituted with a nitro group, while the 4-chloro group was preserved, as in 47, a 2.3 fold affinity decrease was seen compared to 1. Substitution of the 4-chloro in compound 47 with the larger and more lipophilic bromo, leading to 48, increased affinity relatively to 47, thus yielding an affinity similar to 1. This indicates that a large lipophilic substituent in the 4-position is favourable. Finally, a small but statistical significant increase in affinity compared to 1 was observed, when the negative polarity and lipophilicity in the 3-position were increased by introducing a trifluoromethyl group as in compound 49. This substitution was specifically suggested in the Topliss ‘decision tree’ to possess increased affinity compared to the 3,4-dichloro analogue, and accordingly 49 is observed to be the most potent compound in this series with Ki = 7.2 µM (Table 3).
A recurring problem in studies of protein–protein interactions is the possibility of false positives and specifically in fluorescence-based assays where artefacts due to autofluorescence or absorbance are common phenomena.1,61–63 The results of the fluorescence polarization assay were therefore verified for selected analogues in a semi-quantitative and non-fluorescence based, pull-down assay. As seen in the fluorescence polarization assay, compound 26 had no activity towards the PICK1 PDZ domain and 37 showed impaired affinity compared to 47 (Fig. 2). For the remaining compounds tested, the results showed no statistically significant differences compared to 1, but the pull-down assay was generally in agreement with results from the fluorescence polarization assay. In order to verify that modifying compound 1 did not alter the selectivity profile for PICK1 compared to the three individual PDZ domains of PSD-95,49 analogues 3, 5, 8, 14 and 25–49 were tested in a concentration of 500 µM towards PDZ1, PDZ2 and PDZ3 of PSD-95, respectively, in a fluorescence polarization assay as previously described.42 The results demonstrated that none of the compounds showed affinity towards any of the PDZ domains of PSD-95 (data not shown).
Fig. 2.

Semi-quantitative pull-down assay of selected compounds towards PICK1. DMSO indicates the amount of protein pulled down when no compound is added but only the equivalent amount of DMSO. The columns represent the average values and the error bars represent standard deviations based on three individual experiments.
Molecular modelling and docking studies
In the absence of NMR or X-ray crystal structures of 1 in complex with PICK1, docking studies were used to gain detailed insights into the molecular recognition responsible for PICK1 binding. However, the regiochemistry of 1 needed to be settled in order to carry out docking studies. Commercial vendors suggest a Z-configuration around the double bond, but whether this applies to our synthesis procedure of 1 was not certain. NMR clearly showed that only one regioisomer was formed in the Knoevenagel reactions independently of the character of the aromatic moiety, but because there is only one hydrogen atom on the double bond, NMR techniques cannot provide sufficient evidence for either the E- or Z-isomer. Several attempts to prepare crystals of 1 for single crystal X-ray experiments failed, but instead we obtained suitable crystals of the des-chloro analogue, 41, and the structural determination unequivocally demonstrated that compound 41 possesses the E-configuration (Fig. 3). In order to exclude discrepancies due to the difference in the aromatic moieties between 1 and 41, we performed quantum mechanical modelling using density functional theory (DFT), demonstrating that for both compounds the E-configuration is stabilized by 23–25 kJ mol−1 compared to the Z-configuration.
Fig. 3.

Perspective drawing (ORTEP-3)68 of compound 41. Displacement ellipsoids of the non-hydrogen atoms are shown at the 50% probability level. Hydrogen atoms are represented by spheres of arbitrary size.
The docking pose of (E)-1 using the crystallographic structure of the human PDZ domain of PICK164 suggested three structural areas as the primary contributors to binding (Fig. 4): (i) The dichloro- and aromatic moieties establish hydrophobic interactions with the P0 hydrophobic pocket created by four isoleucines from the PICK1 PDZ domain. This area is normally occupied by the first (P0) amino acid from peptide ligands (Fig. 4a and 4b); (ii) the cyano group facilitates ligand–protein shape-complementarity, as it is found buried in a small cavity created by the αB helix and βB beta-sheet of the PDZ domain (Fig. 4c); (iii) the alkyl tail binds near the P−2 region, which for peptide ligands is important for affinity and selectivity within PDZ domains,9,23,42,65 and mediates hydrophobic interactions with Lys83 and Val84 (Fig. 4d). For the des-cyano analogue 5, the pose of 1 correlates with the observed inactivity of 5, since the important contribution to affinity from the cyano group is missing. The modified regions of compounds 3, 8 and 14 do not mediate direct contacts with the PDZ domain, however, the lack of affinity of these compounds can be explained by introduction of increased flexibility that apparently is unfavourable for binding.
Fig. 4.

The binding of compound 1 in the PDZ domain of PICK1. The protein is rendered in green and the ligand in stick representation with the chlorine atoms shown in deep blue, oxygens in red, carbons in purple, aromatic carbons in white and nitrogen atoms in blue. Panels (a)–(d) show magnifications of the corresponding regions indicated by the labelled frames in the main figure. These (a)–(d) framed regions contain the ligand moieties that contribute most to binding, i.e., the chloro substituents, the phenyl moiety, the cyano group and the alkyl tail, respectively. In panel (a) the positions of the 3-trifluoromethyl and the 4-methoxy moieties of compound 49 and 44, are shown in orange and cyan, respectively; in (b) the naphthyl and biphenyl analogues, compounds 25 and 27 are shown in yellow and wheat; in (c) the ethyl and benzyl analogues, compounds 38 and 40 are shown in white and orange; in panel (d) compounds 28 and 29 with the isopropyl substituents are shown in blue and pink.
Next, we evaluated the effects of variations in the aromatic and the ethyl groups, compounds 25–36. The anthracene moiety in 26, 30 and 34 is too large to fit the P0 binding pocket and leads to a change in orientation in the binding pocket that is associated with the loss of interactions and hence affinity. In contrast, compounds 25 and 27 superimpose well with 1 in the binding pocket (Fig. 4b), and their binding affinities are indeed very similar (Table 1). Modification of the alkyl tail, which interacts with the P−2 region, to isopropyl and butyl (compounds 28–31 and 36) does not alter affinity significantly, probably because this part points away from the PDZ domain and further extensions do not introduce additional interactions (Fig. 4d). However, introduction of adamantyl (32–35) at this place abolished affinity, due to its large size and bulkiness, which is not favourable in this solvent-exposed part of the PDZ domain.
Upon alkylation of the carbamate nitrogen, the conformation of the compound changes thereby preventing steric clashing between the N-substituent and the cyano group, as established from conformational analysis of compounds 37–40 calculated in the absence of protein. Docking studies of 37 suggested that the aromatic region was positioned at P0 similar to 1, and that the N-methyl group pointed towards the exterior, but the changed conformation of 37 prevented favourable interactions of the cyano and ethyl groups. A similar observation was made for 38, whereas the increased length of the alkyl group in 39 was found to compensate for the lost interactions by making hydrophobic contacts with Val84 and Ala87 from the αB helix of PDZ. The same hydrophobic interactions were observed for the N-benzyl group of 40. This computational evaluation correlates well with the biological data showing that N-methylation, as in 37, impaired binding affinity, while larger and more hydrophobic N-substituents gradually lead to increased potency (38–40, Fig. 4c).
Compounds 41–49 probe the role of the 3- and 4 substitutions on the aromatic ring (Table 3). The compound with the unsubstituted phenyl, 41, appears to be buried deeper in the P0 pocket, and this affects the position of the cyano and ethyl groups that can no longer interact with PDZ in the same favourable way as for 1. The 4-chloro analogue (42) presents the same orientation as 1, but the lack of the 3-chloro substituent decreases the number of favourable interactions with the P0 pocket. In the fluorescence polarization assay, compound 43 with the 3,4-dimethyl substituents demonstrated lower affinity than 1. This correlates with the docking studies, where it is seen that 43 penetrates deeper into the P0 pocket, hence affecting the position of the cyano and ethyl groups as also seen for 41. For compound 44, the model indicates that the methoxy group induces a different orientation of the ligand that prevents the interaction between the cyano group and the PDZ domain (Fig. 4a).
Compounds 46–48 bind in similar orientations, with the nitro group interacting with backbone amides from a conserved Gly-Leu-Gly-Phe (GLGF) motif in the PDZ domain, which coordinates the C-terminal carboxylate group from peptide ligands via hydrogen bonds.66,67 However, the halogens either fit into this pocket (47, 48), or do not fit (46). Although the overall conformation of these compounds is very similar to compound 1, the cyano group presents a different orientation and cannot establish interactions with the PDZ domain, which results in a lower affinity for compound 46 and 47. For compound 48, the bromine fills out the P0 cavity more efficiently than chlorine, explaining the higher affinity compared to 47. For compound 45, the interaction with the GLGF loop involves the 4-trifluoromethyl group, and 45 is found in a conformation that prevents interaction between the cyano group and the PDZ domain. Compound 49, with a 3-trifluoromethyl and 4-chloro substituent, completely superimposes with 1 (Fig. 4a), and hydrogen bond interaction between the fluorines of the 3-trifluoromethyl group and the GLGF loop, is in agreement with the observed higher affinity of 49 towards PICK1 relative to 1.
Conclusion
PICK1 is suggested as a drug target against severe neurological disorders and possesses intriguing roles in neurobiology. Thus, pursuing inhibitors of PICK1 is important to aid biological studies and possibly develop new therapeutic modalities. In order to understand the molecular requirements for PICK1 activity and improve affinity, the SAR study presented here characterizes the first small-molecule PICK1 PDZ inhibitor, (E)-ethyl 2-cyano-3-(3,4-dichlorophenyl)acryloylcarbamate (1). To the best of our knowledge, this work constitutes the first medicinal chemistry assessment of any small-molecule PDZ domain inhibitor – a class of potentially therapeutic relevant protein domains well-known for their abundance and central roles in cell biology, but also for being difficult to target with small-molecules.
Analogues of 1 were prepared by a condensation reaction between 2-cyanoacetic acids and carbamates to generate cyanoacetylcarbamates, which were subsequently reacted with aromatic aldehydes in a Knoevenagel condensation reaction to provide the final compounds in good yields. Subtle modifications of the acryloylcarbamate backbone as in 3, 5, 8 and 14 revealed that preservation of this scaffold is an essential prerequisite for affinity. The aromatic and ethyl moieties were explored, in compounds 25–36, providing basic information on the steric requirements for biological activity. N-Alkylated carbamates, compounds 37–40, demonstrated that the size of potential alkyl substituents on the carbamate nitrogen is critical for maintaining affinity to PICK1. The analogues with subtle changes of the phenyl substituents, compounds 41–49 led to the conclusion that electronegative and lipophilic substituents are favoured in both the 3- and 4-positions of the phenyl moiety. Strengthening both of these properties in the 3-position by substituting chlorine with trifluoromethyl resulted in the most potent compound (49) with a small but significant improvement in affinity compared to 1.
X-ray crystallography, NMR and DFT calculations settled the configuration around the double bond, a premise for conducting docking studies, and revealed that the aromatic and the cyano groups are found on the same side of the double bond in 1 and analogues. Docking of all the compounds in the PICK1-PDZ binding site provided a structural context for the results of the experimental SAR study, and in general, there was a good correlation between the suggested poses and the affinity trends. Based on these investigations, it is concluded that modifications of the aromatic moiety is the most promising strategy for improving affinity, whereas other regions such as the carbamate nitrogen are more sensitive to structural changes. In addition, this work demonstrated that specificity is a robust feature for the lead compound and not easily affected by modifications. Therefore, this family of compounds should be useful in further studies probing the biological importance of PICK1.
Supplementary Material
Acknowledgements
This work was supported by the Lundbeck Foundation (K.S.) and the Drug Research Academy, Faculty of Pharmaceutical Sciences, University of Copenhagen, Denmark (Ph.D. scholarship to A.B), by University of Copenhagen Program of Excellence (BioScaRT) (U.G., A.B., K.S., K.L.M) and by the National Institutes of Health grant P01 DA012408 (to H.W., U.G.). The technical assistance of Mr. Flemming Hansen with the X-ray data collection is gratefully acknowledged. We thank Dr. Tejs Vegge and the Danish Center for Scientific Computing and the Cofrin Center for Biomedical Information in the HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine at Weill Cornell Medical College for computational time and resources.
Footnotes
Electronic supplementary information (ESI) available: General chemistry procedures. Procedures and data for purified compounds. Experimental for biochemical assays and computational docking studies. Analysis of X-ray crystallographic data, NMR data and density functional theory (DFT) calculations for determining the double bond configuration. See DOI: 10.1039/c0ob00025f
References
- 1.Arkin MR, Wells JA. Nat. Rev. Drug Discovery. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 2.Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 3.Berg T. Curr. Opin. Drug Discovery Dev. 2008;11:666–674. [PubMed] [Google Scholar]
- 4.Dev KK. Nat. Rev. Drug Discovery. 2004;3:1047–1056. doi: 10.1038/nrd1578. [DOI] [PubMed] [Google Scholar]
- 5.Blazer LL, Neubig RR. Neuropsychopharmacology. 2009;34:126–141. doi: 10.1038/npp.2008.151. [DOI] [PubMed] [Google Scholar]
- 6.Houslay MD. Br. J. Pharmacol. 2009;158:483–485. doi: 10.1111/j.1476-5381.2009.00359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 8.Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M. Nature. 1995;378:85–88. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- 9.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 10.Kim E, Sheng M. Nat. Rev. Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 11.Staudinger J, Zhou J, Burgess R, Elledge SJ, Olson EN. J. Cell Biol. 1995;128:263–271. doi: 10.1083/jcb.128.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. J. Neurosci. 2001;21:5417–5428. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang WL, Yeh SF, Chang YI, Hsiao SF, Lian WN, Lin CH, Huang CY, Lin WJ. J. Biol. Chem. 2003;278:37705–37712. doi: 10.1074/jbc.M304619200. [DOI] [PubMed] [Google Scholar]
- 14.Dev KK, Nakajima Y, Kitano J, Braithwaite SP, Henley JM, Nakanishi S. J. Neurosci. 2000;20:7252–7257. doi: 10.1523/JNEUROSCI.20-19-07252.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baron A, Deval E, Salinas M, Lingueglia E, Voilley N, Lazdunski M. J. Biol. Chem. 2002;277:50463–50468. doi: 10.1074/jbc.M208848200. [DOI] [PubMed] [Google Scholar]
- 16.Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 17.McMahon HT, Gallop JL. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 18.Khelashvili G, Harries D, Weinstein H. Biophys. J. 2009;97:1626–1635. doi: 10.1016/j.bpj.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia J, Zhang X, Staudinger J, Huganir RL. Neuron. 1999;22:179–187. doi: 10.1016/s0896-6273(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 20.Gardner SM, Takamiya K, Xia J, Suh JG, Johnson R, Yu S, Huganir RL. Neuron. 2005;45:903–915. doi: 10.1016/j.neuron.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 21.Hanley JG, Henley JM. EMBO J. 2005;24:3266–3278. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madsen KL, Eriksen J, Milan-Lobo L, Han DS, Niv MY, Ammendrup-Johnsen I, Henriksen U, Bhatia VK, Stamou D, Sitte HH, McMahon HT, Weinstein H, Gether U. Traffic. 2008;9:1327–1343. doi: 10.1111/j.1600-0854.2008.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madsen KL, Beuming T, Niv MY, Chang CW, Dev KK, Weinstein H, Gether U. J. Biol. Chem. 2005;280:20539–20548. doi: 10.1074/jbc.M500577200. [DOI] [PubMed] [Google Scholar]
- 24.Torres GE, Yao W-D, Mohn AR, Quan H, Kim K-M, Levey AI, Staudinger J, Caron MG. Neuron. 2001;30:121–134. doi: 10.1016/s0896-6273(01)00267-7. [DOI] [PubMed] [Google Scholar]
- 25.Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT. Neuron. 2000;28:873–886. doi: 10.1016/s0896-6273(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 26.Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ. Neuron. 2000;28:499–510. doi: 10.1016/s0896-6273(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 27.Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, McBain CJ, Isaac JT. Neuron. 2008;57:872–882. doi: 10.1016/j.neuron.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bell JD, Park E, Ai J, Baker AJ. Cell Death Differ. 2009;16:1665–1680. doi: 10.1038/cdd.2009.106. [DOI] [PubMed] [Google Scholar]
- 29.Dixon RM, Mellor JR, Hanley JG. J. Biol. Chem. 2009;284:14230–14235. doi: 10.1074/jbc.M901203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dev KK. Curr. Top. Med. Chem. 2007;7:3–20. doi: 10.2174/156802607779318343. [DOI] [PubMed] [Google Scholar]
- 31.Garry EM, Moss A, Rosie R, Delaney A, Mitchell R, Fleetwood-Walker SM. Mol. Cell. Neurosci. 2003;24:10–22. doi: 10.1016/s1044-7431(03)00134-9. [DOI] [PubMed] [Google Scholar]
- 32.Bellone C, Luscher C. Nat. Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- 33.Hajduk PJ, Huth JR, Fesik SW. J. Med. Chem. 2005;48:2518–2525. doi: 10.1021/jm049131r. [DOI] [PubMed] [Google Scholar]
- 34.Fry DC, Vassilev LT. J. Mol. Med. 2005;83:955–963. doi: 10.1007/s00109-005-0705-x. [DOI] [PubMed] [Google Scholar]
- 35.Joshi M, Vargas C, Boisguerin P, Diehl A, Krause G, Schmieder P, Moelling K, Hagen V, Schade M, Oschkinat H. Angew. Chem., Int. Ed. 2006;45:3790–3795. doi: 10.1002/anie.200503965. [DOI] [PubMed] [Google Scholar]
- 36.Fujii N, Haresco JJ, Novak KA, Stokoe D, Kuntz ID, Guy RK. J. Am. Chem. Soc. 2003;125:12074–12075. doi: 10.1021/ja035540l. [DOI] [PubMed] [Google Scholar]
- 37.Fujii N, Haresco JJ, Novak KA, Gage RM, Pedemonte N, Stokoe D, Kuntz ID, Guy RK. Bioorg. Med. Chem. Lett. 2007;17:549–552. doi: 10.1016/j.bmcl.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Mayasundari A, Ferreira AM, He L, Mahindroo N, Bashford D, Fujii N. Bioorg. Med. Chem. Lett. 2008;18:942–945. doi: 10.1016/j.bmcl.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 39.Shan J, Zheng JJ. J. Comput.-Aided Mol. Des. 2009;23:37–47. doi: 10.1007/s10822-008-9236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hammond MC, Harris BZ, Lim WA, Bartlett PA. Chem. Biol. 2006;13:1247–1251. doi: 10.1016/j.chembiol.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 41.Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- 42.Bach A, Chi CN, Olsen TB, Pedersen SW, Røder MU, Pang GF, Clausen RP, Jemth P, Strømgaard K. J. Med. Chem. 2008;51:6450–6459. doi: 10.1021/jm800836w. [DOI] [PubMed] [Google Scholar]
- 43.Piserchio A, Salinas GD, Li T, Marshall J, Spaller MR, Mierke DF. Chem. Biol. 2004;11:469–473. doi: 10.1016/j.chembiol.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 44.Udugamasooriya DG, Sharma SC, Spaller MR. Chem-BioChem. 2008;9:1587–1589. doi: 10.1002/cbic.200800126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belmares MP, Peter LU, Mendoza KA. 2007 PCT 079406 A1. [Google Scholar]
- 46.Lee H-J, Wange NX, Shi D-L, Zheng JJ. Angew. Chem., Int. Ed. 2009;48:6448–6452. doi: 10.1002/anie.200902981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grandy D, Shan J, Zhang X, Rao S, Akunuru S, Li H, Zhang Y, Alpatov I, Zhang XA, Lang RA, Shi DL, Zheng JJ. J. Biol. Chem. 2009;284:16256–16263. doi: 10.1074/jbc.M109.009647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bach A, Chi CN, Pang GF, Olsen L, Kristensen AS, Jemth P, Strømgaard K. Angew. Chem., Int. Ed. 2009;48:9685–9689. doi: 10.1002/anie.200904741. [DOI] [PubMed] [Google Scholar]
- 49.Thorsen TS, Madsen KL, Rebola N, Rathje M, Anggono V, Bach A, Moreira IS, Stuhr-Hansen N, Dyhring T, Peters D, Beuming T, Huganir R, Weinstein H, Mulle C, Strømgaard K, Rønn LCB, Gether U. Proc. Natl. Acad. Sci. U. S. A. 2010;107:413–418. doi: 10.1073/pnas.0902225107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Murchu C. 3810934. US Patent. 1974
- 51.Kharas GB, Fuerst AM, Feitl EL, Pepper ME, Prillaman FC, Pyo JR, Rogers GM, Tadros AZ, Umek LG, Watson K. J. Macromol. Sci., Part A: Pure Appl. Chem. 2004;41:629–635. [Google Scholar]
- 52.Cantello BC, Cawthorne MA, Cottam GP, Duff PT, Haigh D, Hindley RM, Lister CA, Smith SA, Thurlby PL. J. Med. Chem. 1994;37:3977–3985. doi: 10.1021/jm00049a017. [DOI] [PubMed] [Google Scholar]
- 53.Giles RG, Lewis NJ, Quick JK, Sasse MJ, Urquhart MWJ, Youssef L. Tetrahedron. 2000;56:4531–4537. [Google Scholar]
- 54.Rohde JJ, Pliushchev MA, Sorensen BK, Wodka D, Shuai Q, Wang J, Fung S, Monzon KM, Chiou WJ, Pan L, Deng X, Chovan LE, Ramaiya A, Mullally M, Henry RF, Stolarik DF, Imade HM, Marsh KC, Beno DW, Fey TA, Droz BA, Brune ME, Camp HS, Sham HL, Frevert EU, Jacobson PB, Link JT. J. Med. Chem. 2007;50:149–164. doi: 10.1021/jm0609364. [DOI] [PubMed] [Google Scholar]
- 55.Kocovský P. Tetrahedron Lett. 1986;27:5521–5524. [Google Scholar]
- 56.Potashman MH, Duggan ME. J. Med. Chem. 2009;52:1231–1246. doi: 10.1021/jm8008597. [DOI] [PubMed] [Google Scholar]
- 57.Topliss JG. J. Med. Chem. 1972;15:1006–1011. doi: 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- 58.Martin YC, Dunn WJ., 3rd J. Med. Chem. 1973;16:578–579. doi: 10.1021/jm00263a045. [DOI] [PubMed] [Google Scholar]
- 59.Patt WC, Edmunds JJ, Repine JT, Berryman KA, Reisdorph BR, Lee C, Plummer MS, Shahripour A, Haleen SJ, Keiser JA, Flynn MA, Welch KM, Reynolds EE, Rubin R, Tobias B, Hallak H, Doherty AM. J. Med. Chem. 1997;40:1063–1074. doi: 10.1021/jm9606507. [DOI] [PubMed] [Google Scholar]
- 60.Topliss JG. J. Med. Chem. 1977;20:463–469. doi: 10.1021/jm00214a001. [DOI] [PubMed] [Google Scholar]
- 61.Turconi S, Shea K, Ashman S, Fantom K, Earnshaw DL, Bingham RP, Haupts UM, Brown MJ, Pope AJ. J. Biomol. Screening. 2001;6:275–290. doi: 10.1177/108705710100600502. [DOI] [PubMed] [Google Scholar]
- 62.Pope AJ, Haupts UM, Moore KJ. Drug Discov. Today. 1999;4:350–362. doi: 10.1016/s1359-6446(99)01340-9. [DOI] [PubMed] [Google Scholar]
- 63.Owicki JC. J. Biomol. Screening. 2000;5:297–306. doi: 10.1177/108705710000500501. [DOI] [PubMed] [Google Scholar]
- 64.Elkins JM, Papagrigoriou E, Berridge G, Yang X, Phillips C, Gileadi C, Savitsky P, Doyle DA. Protein Sci. 2007;16:683–694. doi: 10.1110/ps.062657507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chi CN, Engström A, Gianni S, Larsson M, Jemth P. J. Biol. Chem. 2006;281:36811–36818. doi: 10.1074/jbc.M607883200. [DOI] [PubMed] [Google Scholar]
- 66.Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. Cell. 1996;85:1067–1076. doi: 10.1016/s0092-8674(00)81307-0. [DOI] [PubMed] [Google Scholar]
- 67.Hung AY, Sheng M. J. Biol. Chem. 2002;277:5699–5702. doi: 10.1074/jbc.R100065200. [DOI] [PubMed] [Google Scholar]
- 68.Farrugia LJ. J. Appl. Crystallogr. 1997;30:565–565. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.