

Abstract
Obesity and type 2 diabetes are recognised risk factors for the development of some cancers and, increasingly, predict more aggressive disease, treatment failure, and cancer-specific mortality. Many factors may contribute to this clinical observation. Hyperinsulinaemia, dyslipidaemia, hypoxia, ER stress, and inflammation associated with expanded adipose tissue are thought to be among the main culprits driving malignant growth and cancer advancement. This observation has led to the proposal of the potential utility of “old players” for the treatment of type 2 diabetes and metabolic syndrome as new cancer adjuvant therapeutics. Androgen-regulated pathways drive proliferation, differentiation, and survival of benign and malignant prostate tissue. Androgen deprivation therapy (ADT) exploits this dependence to systemically treat advanced prostate cancer resulting in anticancer response and improvement of cancer symptoms. However, the initial therapeutic response from ADT eventually progresses to castrate resistant prostate cancer (CRPC) which is currently incurable. ADT rapidly induces hyperinsulinaemia which is associated with more rapid treatment failure. We discuss current observations of cancer in the context of obesity, diabetes, and insulin-lowering medication. We provide an update on current treatments for advanced prostate cancer and discuss whether metabolic dysfunction, developed during ADT, provides a unique therapeutic window for rapid translation of insulin-sensitising medication as combination therapy with antiandrogen targeting agents for the management of advanced prostate cancer.
1. Obesity, Type 2 Diabetes and Prostate Cancer
1.1. Obesity and Cancer Risk
Worldwide rates of obesity have doubled in a generation with a global estimate of ~500 million obese adults (with an additional 1.5 billion overweight) being followed by a generation of 40 million overweight children [1]. In both industrialised and developing countries these staggering numbers pose a soaring economic and health care burden as a result of chronic comorbidities including increased rates of cardiovascular disease, hypertension, stroke, and type 2 diabetes (T2DM) [2].
Obesity is also a risk factor for a growing number of cancers. Retrospective observational studies and meta-analyses, using body mass index (BMI; mass (kg)/height (m2)) as a measure of adiposity, have demonstrated an increased risk of breast, ovarian, colorectal, bladder, kidney, and endometrial cancers with increasing BMI [3–6]. Similarly, obesity attributes a 12% increased risk of prostate cancer diagnosis [3] although studies have varied, with some showing a weak association (relative risk (RR): 1.05 [7]), or a significant risk of high-grade and metastatic cancers, (RR: 1.22–1.55) but not low-grade prostate cancer [8]. These differences may be accounted for by differences in detection bias (using cohorts of closely monitored patients, for example, during diabetes screening), differences in time of “disease-free” follow-up and patient age [9]. Also, these studies may be limited by the use of BMI as a measurement of adiposity, underscored by the recent finding that specific measurement of visceral fat volume, the more metabolically compromised fat depot in obesity, may provide a much stronger statistical correlate with disease-free survival in cancer patients than BMI [10]. Adding to these statistical differences, there are more practical difficulties of prostate cancer detection in obese subjects. Measurements of prostate specific antigen (PSA), a serum biomarker used for screening, which can also be elevated in nonmalignant disease such as benign prostatic hyperplasia (BPH) or with aging, can be difficult to interpret in obese patients; both lower levels of PSA, due to increased blood volume and increased PSA levels concomitant with increased incidence of BPH [11], have been reported in obese men [12]. Notwithstanding, a recent study by Fowke et al. [9] has found that the association between obesity and prostate cancer persists when these factors are taken into account.
1.2. Obesity and Cancer Progression
What is indisputable from the epidemiology is the impact of obesity on cancer behaviour. Obesity is consistently identified as a significant risk factor for more aggressive disease and an independent predictor of recurrence and cancer-specific mortality for breast [13], endometrial, ovarian [14], and bladder cancer [5] as well as prostate cancer [9, 15]. Men with higher BMI are more likely to be diagnosed with higher-grade cancers and higher Gleason scores and suffer an increased incidence of recurrence [3, 7, 15, 16] and increased cancer-specific mortality than men with a healthy BMI [16–18].
The molecular link(s) between obesity and malignancy is the subject of current research and has been recently reviewed [19–21]. Obese adipose tissue creates a hypoxic environment, as well as an overproduction of ROS resulting in oxidative and ER stress [22]. In addition, many bioactive molecules are altered in obesity which could contribute to neoplasia and cancer progression [21] including lipid mediators, inflammatory cytokines, and hormones/growth factors. As adipose tissue expands, a proinflammatory environment develops with increased secretion of cytokines such as IL-6, TNFα, and MCP1, from both adipocytes and resident immune cells. These, in turn, act as chemoattractants for further immune cells and thus create a feed-forward loop, perpetuating immune cell infiltration and cytokine production. Cell signalling is impacted, particularly insulin signalling in local and peripheral tissue (e.g., muscle and liver) leading to systemic insulin resistance [23] and sustained elevated circulating insulin levels.
Dysregulated lipid flux in obesity results in decreased HDL cholesterol and elevated LDL, circulating levels of triglycerides and free fatty acids which have been shown to promote prostate cancer cell survival [24, 25]. Inflammatory lipid mediators such as arachidonate and downstream signalling lipids, such as eicosanoids, prostanoids, and leukotrienes, are also increased [21, 26] and could potentially impact tumour cell biology; arachidonate can also promote steroid hormone production in prostate cancer cells [27].
Altered hormonal profiles are also a hallmark of metabolic dysfunction with modulated adipokine production and secretion, including increased leptin, proportional to adipose mass, while adiponectin shows an inverse relationship [28]. Elevated leptin levels have been associated with breast, prostate and colon cancer progression, and leptin promotes in vitro cell proliferation and invasion [9, 29], as well as independently modulating inflammation. Reduced adiponectin, which purportedly puts a brake on malignant cell proliferation, has also been observed in a range of cancers [30] including prostate cancer.
However, it is hyperinsulinaemia, as a result of insulin resistance in classical metabolic tissues, which has been identified as a highly significant risk factor to progression of prostate and other cancers [14]. In prostate cancer, elevated insulin or C-peptide levels (used as a normalised surrogate) have recently been shown to significantly correlate with high-grade prostate cancer and worse patient prognosis [16, 31–36], more significantly than BMI alone, suggesting that at least part of the effect of increased BMI on prostate cancer mortality is related to coincident hyperinsulinaemia [33]. Similarly, a strong association with high C peptide and the development of high-grade prostate cancer of Gleason grade 7 or greater has been identified, independent of BMI [36]. Baseline insulin levels at time of prostate cancer diagnosis have also been shown to be the most significant predictor of lethal prostate cancer, strongly suggesting that insulin is a major factor in prostate cancer progression associated with metabolic dysfunction [35]. Chronically elevated insulin facilitates in tumour tissues increased activation of mitogenic, anabolic, and prosurvival pathways with the increased levels of insulin and concomitant elevated insulin-like growth factor 1 (IGF-1) [37, 38]. We have also recently reported that insulin regulates the expression of novel gene transcripts/splice variants in tumour tissue [39]. In addition, many tumour types, including prostate tumours, upregulate the expression of related insulin receptor (INSR), IGF-1 receptor (IGF-1R), and hybrid INSR/IGF-1Rs [31, 40] further promoting insulin-driven cancer survival pathways [41]. And notably, an increased ratio of INSR/IGF1R expression has recently been described in prostate tumours and adjacent tissue [42], which suggests specifically that insulin signalling plays a key role in these tumours.
Together, the altered hormonal and inflammatory milieu of obesity may contribute significantly to cancer growth and progression via promoting mitogenesis (e.g., leptin, IGF-1, insulin), angiogenesis (e.g., VEGF, IL6, IL8), and invasion (e.g., leptin, IL6, PAI-1, CCL5, CCL2) [19].
1.3. Diabetes and Cancer Risk
Obesity is a major risk factor for the development of T2DM, a condition defined by hyperglycaemia in a background of insulin resistance in metabolic organs (e.g., muscle and liver). Diabetes generally develops as pancreatic β-cells are unable to respond adequately to increased insulin required to maintain normoglycaemia. Diabetes has been independently associated with increased risk of several cancers including colon, pancreatic, and breast cancer [43, 44]. In contrast, some studies suggest that diabetes may be protective of prostate cancer risk [45–47], perhaps due to the typically decreased levels of androgens in these men; prostate tumours are fuelled by androgens for growth and survival, and testosterone levels are typically lower in men with diabetes [46–48]. However, this is also confounded by a lack of separation of men taking diabetic control agents, such as metformin [46, 49]. Although diabetes is associated with a lower incidence of prostate cancer, postdiagnosis, diabetes increases the risk of cancer-specific mortality [50], suggesting that similar risk factors contributing to cancer progression are involved in prostate cancer as other tumour types. Large cohort studies of diabetic patients are partly compromised by the fact that many studies reporting the relative risk of cancer with diabetic status have not separately defined the proportion of type 1 and type 2 diabetes within these studies, nor analysed the cohorts individually or based on medications [46, 49, 51]. While highlighting the association between hyperglycaemia per se and prostate cancer, metabolic risk factors associated with T2DM with potential links to cancer, such as hyperinsulinaemia and inflammation, are likely to lose statistical impact by combining diabetic subtypes; the rate of obesity in T2DM is 52% (86% overweight), compared to 16% in type I diabetes (55.3% overweight), and, as such, the metabolic contribution to malignancy, described above, would be expected to have significantly different impact [52]. Furthermore, type 1 diabetes is characterised by a loss of insulin and use of exogenous insulin analogues whereas T2DM is treated with a range of medications including metformin, sulphonylureas, and insulin which vary in cancer risk profiles [53–55], some conferring protection, (metformin) and others increasing risk (sulphonylureas and insulin analogues) [45, 53]. Indeed, this observation has led many to speculate on the potential utility of diabetes treatments as adjuvant cancer therapeutics.
2. Emergence of Insulin-Sensitising Drugs in Cancer
The observation of increased cancer risk with obesity and diabetes has led to the somewhat more hopeful speculation of potential therapeutic benefit of insulin-sensitizing drugs with the major clinical outcome of lowering systemic insulin levels. Studies have shown some cancer survival benefit in type 2 diabetes patients treated with the biguanide and metformin compared to those treated with insulin or sulphonylureas (stimulate insulin secretion from pancreatic β-cells) [53, 56–58]. In addition, the thiazolidinedione class of PPAR agonists [59], the lipase inhibitor, orlistat, and cholesterol-lowering statin class of drugs [60, 61] have also been shown to associate with lower cancer risk. A growing body of in vitro evidence is beginning to provide mechanistic detail to these observations.
2.1. Metformin
Metformin, which works in part by activating AMP-activated protein kinase (AMPK), is used clinically in obese and diabetic patients to normalise circulating insulin levels primarily via reduced hepatic glucose output and improve insulin/receptor interactions. Recent clinical studies have suggested that metformin may improve patient outcomes in prostate and other cancers [33, 53, 62–64]. Amongst diabetic populations, metformin has the lowest cancer mortality profile when compared to sulphonylurea or insulin treatment [53, 56–58, 63], in a dose and duration of treatment-dependent manner.
Apart from normalising systemic insulin, the intracellular effects of metformin in tumour cells have been the subject of recent study and review [41]. Metformin can potentiate several pathways to prevent tumour cell growth and invasion, primarily via blocking anabolic pathways such as lipognesis and protein translation, preventing mitosis and increasing sensitivity to chemotherapy.
The activation of AMPK in cancer cells blocks cell proliferation by negatively regulating mammalian target of rapamycin (mTOR) control of protein synthesis [43, 65–67]. Selective suppression of translation of mRNAs-encoding cell cycle regulators within the mTOR/EIF4E pathway by metformin has been recently described [68], and the expression of many genes involved in mitosis, such as tubulins, histones, and kinesins is decreased by metformin [69]. AMPK acts as an intracellular energy sensor, responding to fluctuations in the ratio of AMP and ATP. In times of energy deficit (high AMP:ATP ratio), the activation of AMPK blocks anabolic pathways and promotes the generation of ATP (e.g., oxidative phosphorylation, β-oxidation). Thus, metformin effectively blocks lipogenesis by inhibiting activation or expression of lipid biosynthesis enzymes such as acetyl Co-A carboxylase (ACC) and fatty acid synthase (FASN), key regulators of the metabolic reprogramming identified in prostate cancer [70]. Likewise, the enzyme responsible for steroid (estrogen) production, aromatase, is decreased in breast adipose tissue with metformin use [71].
Another proposed mechanism of metformin is preventing pathways which lead to invasion and metastasis evidenced by the demonstration of metformin to prevent invasiveness in cancer models [72, 73]. Similarly, it has been reported in pancreatic cancer cells that metformin reversed cell invasion by facilitating the reexpression of critical RNA species required for phenotypic maintenance [74]. Based on these results, targeting AMP-activated protein kinase (AMPK) has been proposed as a therapeutic strategy in cancer [75, 76].
2.2. Lipid Modulators: Orlistat and Statins
The pancreatic lipase inhibitor, orlistat, has its primary effect on reducing hydrolysis of triglycerides and preventing the absorption of dietary free fatty acids. Its major clinical benefit in obesity is preventing caloric intake, thus, reducing circulating lipid levels and improving insulin sensitivity [77]. FASN expression is upregulated in a number of cancers including ovarian, breast, and prostate [78–80] so the discovery that orlistat was a novel inhibitor of FASN [81] triggered numerous studies into the efficacy of orlistat as a therapeutic agent for cancer. orlistat has been shown to prevent in vitro and in vivo growth of the prostate cancer cell line PC3 and to induce apoptosis. [81]. The inhibition of FASN with orlistat also causes cell cycle arrest and induces caspase-mediated apoptosis [82] and has also been shown to increase sensitivity to chemotherapeutic compounds [83, 84].
Statins lower cholesterol levels by inhibiting a critical enzyme for cholesterol synthesis, HMGCoA reductase (HMGCR), which is upregulated in CaP and breast cancer tissue [85]. The role of HMGCR in cholesterol synthesis, and a precursor to steroid hormone synthesis, is likely to be an important link in the association between statin use and a lower rate of prostate cancer recurrence [26, 86, 87]. Indeed, it has been shown that statin use inversely correlated with the risk and progression of prostate cancer [88, 89]. In a large-scale study by Loeb et al. men on statins were older and had high BMI at time of radical prostatectomy, but, despite these increased risk factors, the patients on statins had lower PSA, as well as lower Gleason score, lower tumour volumes, smaller surgical margins, and less risk of biological recurrence [88]. Other studies demonstrate a significant decrease in advanced and metastatic disease [88, 90].
2.3. Thiazolidinediones
Thiazolidinediones (TZDs) are a class of insulin-lowering (insulin-sensitising) drugs which activate the PPAR family of nuclear receptors, with predominant affinity for PPARγ, highly expressed in prostate cells [91]. Several TZDs are used clinically to treat type 2 diabetes, including pioglitazone, troglitazone, rosiglitazone, and ciglitazone. Their use in other diseases with hallmark hyperinsulinaemia (e.g., polycystic ovarian syndrome and some lipodystrophies) is the subject of an ongoing research [92]. A large meta-analysis comparing TZD use and cancer incidence [93] found a decreased risk of colorectal, lung, and breast cancers with TZDs, but specifically, the use of pioglitazone—which extended protection to prostate and renal cancer as well. This finding has also been demonstrated in other studies where TZD use is associated with improved survival of diabetic prostate cancer patients [59]. In prostate cancer cell models, troglitazone reduced in vitro and in vivo cell growth via ERK-dependent regulation of p21 and cMyc [94] and inducing apoptosis [80]. Prostate-specific knockdown of PPARγ resulted in the dysregulation of cell cycle control and lipid signalling networks [95]. In ovarian models, differential efficacy of ciglitazone and troglitazone (but not rosi- or pioglitazone) was observed with respect to cell cycle arrest, increased caspase activation, and evidence is now mounting that this may occur via PPARγ-independent mechanisms [96, 97]. In prostate cancer cells TZDs induced cell senescence, via PPARγ-dependent (rosiglitazone) and -independent (ciglitazone) mechanisms [98]. Studies in lung cancer cell models have reported TZD treatment works directly on cancer cells to decrease pathways that lead to invasion such as epithelial-to-mesenchymal transition (EMT) via Smad-dependent and -independent mechanisms [99]. Taken together, there is compelling evidence that current antidiabetes treatments offer therapeutic benefit in cancer.
2.4. Exogenous Insulin Analogues
The efficacy of insulin-lowering therapies in reducing risk and progression of cancer is highlighted by the converse, that is, increased risk of cancer in diabetic patients treated with insulin analogues. These include fast-acting analogues such as Aspart and Lispro with similar INSR but reduced IGF1R-binding affinities compared to native insulin [100], and Glargine and X10 with increased IGF1R affinity compared to insulin, and sustained binding kinetics [101, 102]. Nevertheless, the clear benefit of good glucose control in diabetes is worth bearing in mind in regard to cancer risk.
3. Prostate Cancer as a Candidate for Insulin-Lowering Therapy
3.1. Advanced Prostate Cancer
At current rates, prostate cancer will affect one man in seven and is the leading cause of cancer deaths in men in western countries [103]. For rapidly advancing, localised disease, patients are generally treated with radical prostatectomy or radiation therapy with a high rate of success (>90%) [104]. However, up to 25% of patients will recur, heralded by a biochemical recurrence measured as a rising PSA, and require secondary treatment. Prostate tumours depend on androgen signalling for growth and survival, and this dependence has rationalised the standard treatment for metastatic prostate cancer, androgen-deprivation therapy (ADT), for decades [105, 106]. The most common method of ADT uses luteinising hormone-releasing hormone (LHRH) agonists or antagonists to medically disrupt hypogonadal feedback loop to inhibit testicular testosterone production, or to a lesser degree direct orchiectomy. An initial surge of testosterone in response to LHRH agonists is often blunted with coadministration of androgen receptor (AR) antagonists (e.g., bicalutamide and flutamide). ADT results in anticancer response and prolongs cancer control; however, after a median time of 12 to 33 months, the cancer resurges, with a second PSA rise, despite castrate levels of serum testosterone (<20 ng/dL) [107]. This disease stage is referred to castrate-resistant prostate cancer (CRPC) and traditionally has had limited effective treatment options, finding that effective therapy for CRPC has been the focus of intense and productive research in recent years with a surge of new agents approved and, in development, many of which re-targeting in new ways the androgen axis.
4. Current Therapy for CRPC
While chemotherapies such as docetaxel [108, 109] and cabazitaxel [110] provide some survival benefit in metastatic CRPC, therapeutic strategies are increasingly focussing on the inhibition of androgen signalling as evidence mounts that CRPC continues to utilize androgens for tumour growth and driving cell survival pathways [111–113]. There are several mechanisms by which prostate tumours may reactivate androgen signalling, including gain-of-function mutations or alternative splicing of the androgen receptor (AR) that broaden its range of ligands to other steroid hormones (e.g., progesterone, estrogen, and cortisol), antiandrogens (e.g., flutamide and bicalutamide) [114–116], or confer ligand-independent activity [117]. The development of a hypersensitive receptor, via overexpression of AR and/or receptor stabilization, provides a second mechanism that results in AR activation even at low levels of androgens [118–121]. A further mechanism by which CRPC maintains AR signalling is by producing its own androgens from both adrenal conversion and intratumoural intracrine steroidogenic pathways. The observation that, despite low serum levels, intraprostatic levels of DHT and testosterone were high in men treated with ADT [122], up to 50% of intraprostatic levels measured in eugonadal men [122–125], led to the finding by us and others that, under androgen deprived conditions, enzymes required for conversion of adrenal androgens and de novo intratumoural androgen synthesis are upregulated in prostate cancer cells [113, 126, 127]. Thus, no longer reliant on testicular androgens, AR signalling can resume within the tumour microenvironment, leading to the development of CRPC. These factors combined have rationalised the recent surge in new antiandrogens for the treatment of CRPC, which target the AR and inhibit activity, or block rate-limiting enzymes in de novo steroid synthesis.
4.1. New Inhibitors Targeting the AR
A tranche of new-generation antiandrogens have recently emerged from preliminary clinical trials with promising results; MDV3100 (Medivation Inc.), a small molecule AR antagonist, has recently been FDA approved for secondary hormone treatment for CRPC under the trade name of Enzalutamide. MDV3100 binding the AR blocks ligand binding, impairs nuclear translocation of the receptor, and induces a conformational change that prevents AR transcription and cofactor recruitment [128]. MDV3100 has from 5- to 10-fold greater binding affinity for the AR than bicalutamide or Casodex, but, unlike bicalutamide, has no partial agonist activity [128] and, excitingly, may also block constitutively active AR splice variants [129]. A second AR-antagonist, ARN509 (Aragon Pharmaceuticals), similarly binds AR, prevents nuclear translocation and transcription, and, in early clinical trials, has shown promise for efficacy at lower doses than MDV3100 [130].
4.2. New Inhibitors Targeting Steroid Synthesis
Antiandrogen therapies are also aimed at inhibiting androgen biosynthesis. Androgens are synthesized from cholesterol via steroidogenesis. Multifunctional enzyme CYP17A1 catalyzes two important steps in the steroidogenesis pathway: the conversion of progestogens to androgen precursors and subsequent conversion to androgens (DHEA and androstenedione). DHEA and androstenedione are then converted to testosterone via 17-β-hydroxysteroid dehydrogenase (17BHSD) and then to DHT via RDH5. Currently, both ketoconazole (a pan-CYP inhibitor, Figure 1) and aminoglutethimide (inhibitor of CYP11A1) are used to block key enzymes in the androgen synthesis pathway in combination with ADT.
Figure 1.
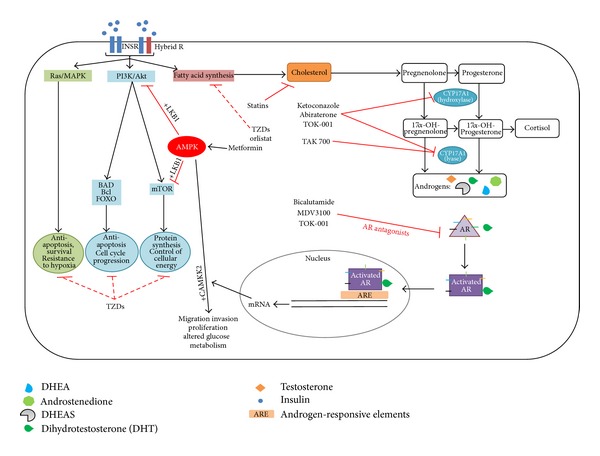
Therapeutic regimens combining insulin-sensitizing drugs and androgen-signalling inhibitors may be beneficial for treating prostate cancer progression following ADT. Androgen signalling is targeted using direct AR inhibitors. Bicalutamide (Casodex) and MDV3100 directly bind AR to prevent activity. Ketoconazole, abiraterone (Zitega), and TOK-001 have dual inhibition of CYP17A1 lyase/hydroxylase activity. In addition TOK-001 directly blocks AR activity. TAK-700 inhibits CYP17A1 lyase activity only and may not require concomitant control of rise in mineralocorticoids as is needed for abiraterone. Hyperinsulinaemia would be expected to increase insulin signalling in prostate cancer cells. Insulin can accelerate de novo androgen synthesis [153], which provides the AR with several suitable ligands (Key) and allows AR-mediated transcription of genes needed for CRPC progression. Additionally, insulin signalling may activate several AR-independent pathways (e.g., antiapoptosis and cell proliferation). Insulin-sensitizing drugs, such as metformin, orlistat, thiazolidinediones, and statins effectively block insulin-induced effects such as proliferation, lipid, and cholesterol synthesis and, hence, may be effective for the treatment of prostate cancer. The role of AR on AMPK-mediated cell response is still unclear and may be dependent on availability of cofactors (LKB1 versus CAMKK2).
Improved, specific CYP17A1 inhibitors such as abiraterone (Janssen) FDA, TAK-700 (Takeda/Millenium Pharmaceuticals), and TOK-001 (Tokai Pharmaceuticals) are now in various stages of clinical assessment for adjuvant use with LHRH agonists. abiraterone, unlike previous CYP17 inhibitors, has a 3-pyridyl substitute and 16,17 double bond, which makes it a highly specific, potent, and irreversible inhibitor of both the hydroxylase and lyase activity of CYP17A1 [131–133]. abiraterone acetate, the oral drug precursor of abiraterone, has been FDA approved with trade named Zytiga for CRPC treatment, in settings of postdocetaxel [134] and now prechemotherapy [135]. TAK-700 is a nonsteroidal imidazole, a potent inhibitor of CYP17A1 lyase activity, with weak inhibition of hydroxylase activity and therefore may not require concomitant control of rise in mineralocorticoids as is needed for abiraterone. TAK-700 is currently undergoing Phase I/II clinical trials. A third molecule, 17-benzoimidazole TOK-001, has combined CYP17/AR inhibitor activity and has shown promising results in preclinical studies including a remarkable decrease in AR protein expression and regression of in vivo xenograft tumours [136].
While much progress has been made in the emergence of new drugs suppressing AR activity in prostate cancer, the unfortunate reality is that, even in these early trials, patients have become resistant to these new therapies [137]. Both abiraterone and MDV3100, currently the only new-generation drugs with FDA approval, show survival benefits of 4.6 months [134] and 4.8 months [138], respectively, in the postdocetaxel setting. In the prechemotherapy setting, abiraterone treatment offers an increased time to biochemical recurrence of 11.1 months [134, 135]. Early trial results show that MDV3100 has improved efficacy, with an extra 5.3 months to biochemical failure compared to postdocetaxel [138]. The persistent capability of CRPC to become resistant to various means of suppression of AR signalling suggests that a multipronged approach to cancer treatment is required and leads to the research question: what drives prostate cancer aggressiveness and CRPC progression?
5. Metabolic Dysfunction Caused by ADT Accelerates CRPC
Metabolic dysfunction is a well-established side effect of ADT [34, 139]. The response to suppression of testosterone in men, irrespective of patient BMI at treatment commencement, includes gain of fat and loss of muscle mass, increased LDL and triglycerides, hypertension, and increased fasting glucose [140]—metabolic symptoms which significantly overlap the comorbidities of obesity. Abrupt withdrawal of androgens by ADT results in hyperinsulinaemia and loss of insulin sensitivity reflected by increased homeostatic model assessment (HOMA) score, within 2 weeks [141], suggesting a direct effect of ADT and is independent of fat mass and age [139, 142]. Major findings from recent studies [36, 143] and our own recent pilot study of men receiving ADT demonstrated a strong trend between elevated C-peptide and more rapid progression to CRPC. This phenomenon appears to be due to a direct inverse relationship that exists between the testosterone and insulin hormonal axes in men [144–149]; testosterone is inversely linked to insulin sensitivity and insulin-sensitising medication increases testosterone [59, 150]; however, the exact mechanisms of crosstalk between these pathways are not well understood [151, 152].
Traditionally, insulin has been considered a hormone-controlling metabolic regulation; however, the pathways activated by insulin, including phosphatidylinositol 3-kinase (PI3K)/Akt and Ras/MAPK pathways, have many well-characterised downstream effects relevant to CRPC progression, including the inhibition of apoptosis (e.g., via FOXO and BAD-mediated pathways) [118, 153] and stimulation of cell proliferation (e.g., via mTOR) (Figure 1 , insulin-signalling pathways) [40, 154]. Hyperinsulinaemia, secondary to ADT treatment, would be expected to increase the activation of these pathways in the relatively insulin-sensitive prostate tumour. Furthermore, increased insulin signalling would be facilitated by increased INSR expression in advanced prostate cancer [31] (Lubik, Gunter, Nelson, unpublished data). We have recently demonstrated that insulin accelerates intratumoural androgen synthesis [155], a major pathway contributing to progression to CRPC. But, insulin is expected to upregulate a number of pathways leading to ADT failure [156, 157]. Regulation of molecules by signalling downstream of insulin/IGF-1R has also been implicated in CRPC progression, including COX2 [158–160], and nuclear AR chaperone, Hsp27 [161, 162]. Notably, obesity and diabetes may independently regulate these molecules [163–165]. Moreover, serum from obese mice has been shown to induce an invasive phenotype in prostate cancer cell lines [166] suggesting that metabolic changes associated with obesity (including elevated insulin) may drive metastatic transformation. However, chronic insulin signalling in the context of ADT might be complex and highly differentiated from those observed in other cancers [167]. Potential crosstalk between AR and insulin-signalling cascades [168–171] emphasises the potential insulin-driven CRPC progression. And recent studies have implicated the PI3K/Akt pathway activation to inhibit AR signalling [172] resulting in androgen-independent growth. Several other growth factors and cytokines upregulated in obesity, such as IL-6, IL-8, IGF-1, and TGFβ, may also influence prostate cancer cell proliferation [173–175].
6. A Future for Combined Therapy
Epidemiological evidence, coupled with the upregulation of insulin-signalling components and insulin-mediated upregulation of CRPC pathways in prostate cancer cells, has collectively rationalised a multidisciplinary approach to treating advanced prostate cancer that incorporates antiandrogen therapy with simultaneous treatment of metabolic side effects induced by ADT.
Metformin has been the first drug to be explored in this area, with ongoing clinical trials using metformin during active surveillance (NCT01733836) and in combination with ADT (NCT01620593) testing the efficacy of metformin in stabilising/normalising circulating insulin levels. Metformin has a well-characterised safety profile and, while it effectively decreases hyperglycaemia, does not affect blood glucose levels in nondiabetic patients. Metformin may also act directly on tumour cells to reduce cancer cell proliferation via the inhibition of anabolic pathways such as lipogenesis starving the major bioenergetic pathway in prostate cancer cells. However, the ability of the major intracellular target of metformin, AMPK, to potentiate insulin action on cell growth and survival may have more complex regulation in prostate cancer cells via interaction with AR-regulated genes; activated AMPK may potentiate increased prostate cancer cell proliferation and migration when activated downstream of the androgen receptor (AR) [176, 177] under the control of a master regulator calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2) [177]. AR directly regulates CAMKK2 and is highly expressed in normal prostate with elevated expression in both AR-sensitive and CRPC models of prostate cancer [176, 177]. In studies where metformin activation of AMPK results in cessation of cancer cell growth, signalling is through LKB-1 tumour suppressor [178] suggesting that AMPK is a potentially bifunctional modulator, particularly relevant in the prostate cell, dominated by AR-regulated transcriptional landscape, and conversely may be dysregulated with AR as occurs in prostate cancer. The ability of insulin to accelerate de novo steroid synthesis in prostate cancer cells suggests that drugs targeting steroidogenesis in combination with insulin-lowering therapies would be beneficial; trials combining abiraterone and metformin are yet to commence (NCT01677897).
Atorvastatin (statin) is also currently under examination for treatment in prostate cancer in combination with ADT (NCT01555632). Statins target cholesterol synthesis, the substrate for steroidogenesis and in adrenocortical cells and the ovaries, have been shown to decrease steroidogenic enzymes CYP11A1, HSD3B, and CYP17A1 [179]. Statins were able to slow or halt progression to castrate resistance in LNCaP xenograft models [180] and inhibited prostate cancer growth with greater potency in androgen-deprived condition. The clinical benefit of metformin combined with statin for treatment of biochemical recurrence following primary treatment failure (PSA increase following prostatectomy or radiation therapy) is also under clinical investigation (NCT01561482), targeting lipid/cholesterol metabolism and hyperinsulinaemia in an effort to slow progression.
The dual activity of orlistat, reducing dietary lipid absorption and as an inhibitor of FASN, could also have an adjunct role to play in mitigating the side effects of ADT which may promote cancer progression. orlistat could potentially be used to reduce systemic dyslipidaemia, normalising free fatty acid and cholesterol levels and thereby reducing substrate for tumour metabolism and biosynthesis. However, dietary fats make up only a fraction of the lipid available to prostate cancer cells, which almost universally upregulate FASN [181].
7. Conclusion
Prostate cancer is the most common cancer in men [103] and, given its long natural history and with onset beginning in 5-6 decades of life, will continue to rise and plague our ageing population. At the same time we face the growing epidemic of obesity while the risk of aggressive prostate cancer is increased 3-fold with obesity. First-line therapies for localised cancer will fail in nearly 25% of prostate cancer patients, and these men will subsequently be treated with ADT [182]. While effectively treating prostate cancer, ADT induces hyperinsulinaemia [182–185] which independently acts on prostate cancer to upregulate lipid and steroid synthesis and contribute to CRPC progression and which may promote metastases, tumour growth, and treatment resistance. Standard chemotherapeutic agents have had limited benefit in CRPC, and there has been quite promising development of next-generation therapies targeting androgen synthesis and directly AR in combination with ADT. These new targeted agents are generally well-tolerated and significantly improve survival; however, most patients ultimately fail, highlighting the urgent need to understand mechanisms underlying treatment resistance and find rationally informed combined and/or sequential treatment options. An existing toolbox of well-tolerated insulin-lowering therapies has accumulated over several decades to treat type 2 diabetes and metabolic complications associated with obesity. Currently, ADT-induced hyperinsulinaemia is not addressed in prostate cancer patients, despite a significantly increased risk of cardiovascular and cancer-related mortality in these patients [186]; the combination of ongoing research and clinical trials will determine the benefit of adjunct antiinsulin therapy to current standard and emerging prostate cancer treatments.
References
- 1.WHO. Obesity and Overweight. Fact Sheet #311, 2012, http://www.who.int/mediacentre/factsheets/fs311/en/
- 2.Malnick SDH, Knobler H. The medical complications of obesity. Oxford Journals: Medicine. 2006;99(9):565–579. doi: 10.1093/qjmed/hcl085. [DOI] [PubMed] [Google Scholar]
- 3.Bergstrm A, Pisani P, Tenet V, Wolk A, Adami HO. Overweight as an avoidable cause of cancer in Europe. International Journal of Cancer. 2001;91(3):421–430. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1053>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 4.Beral V, Hermon C, Peto R, et al. Ovarian cancer and body size: Individual participant meta-analysis including 25,157 women with ovarian cancer from 47 epidemiological studies. PLoS Medicine. 2012;9(4) doi: 10.1371/journal.pmed.1001200.e1001200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chromecki TF, Cha EK, Fajkovic H, et al. Obesity is associated with worse oncological outcomes in patients treated with radical cystectomy. BJU International. 2012;111(2):249–255. doi: 10.1111/j.1464-410X.2012.11322.x. [DOI] [PubMed] [Google Scholar]
- 6.Navai N, Wood CG. Environmental and modifiable risk factors in renal cell carcinoma. Urologic Oncology: Seminars and Original Investigations. 2012;30(2):220–224. doi: 10.1016/j.urolonc.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 7.MacInnis RJ, English DR. Body size and composition and prostate cancer risk: systematic review and meta-regression analysis. Cancer Causes and Control. 2006;17(8):989–1003. doi: 10.1007/s10552-006-0049-z. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez C, Freedland SJ, Deka A, et al. Body mass index, weight change, and risk of prostate cancer in the cancer prevention study II nutrition cohort. Cancer Epidemiology Biomarkers and Prevention. 2007;16(1):63–69. doi: 10.1158/1055-9965.EPI-06-0754. [DOI] [PubMed] [Google Scholar]
- 9.Fowke JH, Motley SS, Barocas DA, et al. The associations between statin use and prostate cancer screening, prostate size, high-grade prostatic intraepithelial neoplasia (PIN), and prostate cancer. Cancer Causes and Control. 2011;22(3):417–426. doi: 10.1007/s10552-010-9713-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rickles AS, Iannuzzi JC, Mironov O, et al. Visceral obesity and colorectal cancer: are we missing the boat with BMI? Journal of Gastrointestinal Surgery. 2013;17(1):133–143. doi: 10.1007/s11605-012-2045-9. [DOI] [PubMed] [Google Scholar]
- 11.Hammarsten J, Peeker R. Urological aspects of the metabolic syndrome. Nature Reviews Urology. 2011;8(9):483–494. doi: 10.1038/nrurol.2011.112. [DOI] [PubMed] [Google Scholar]
- 12.Porter MP, Stanford JL. Obesity and the risk of prostate cancer. The Prostate. 2005;62(4):316–321. doi: 10.1002/pros.20121. [DOI] [PubMed] [Google Scholar]
- 13.Carmichael AR. Obesity as a risk factor for development and poor prognosis of breast cancer. BJOG: An International Journal of Obstetrics and Gynaecology. 2006;113(10):1160–1166. doi: 10.1111/j.1471-0528.2006.01021.x. [DOI] [PubMed] [Google Scholar]
- 14.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. Adults. The New England Journal of Medicine. 2003;348(17):1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 15.Ma J, Li H, Giovannucci E, et al. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. The Lancet Oncology. 2008;9(11):1039–1047. doi: 10.1016/S1470-2045(08)70235-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fowke JH, Matthews CM, Buchowski MS, et al. Association between prostate-specific antigen and leptin, adiponectin, HbA1c or C-peptide among African-American and Caucasian men. Prostate Cancer and Prostatic Diseases. 2008;11(3):264–269. doi: 10.1038/sj.pcan.4501022. [DOI] [PubMed] [Google Scholar]
- 17.Jayachandran J, Aronson WJ, Terris MK, et al. Obesity and positive surgical margins by anatomic location after radical prostatectomy: results from the shared equal access regional cancer hospital database. BJU International. 2008;102(8):964–968. doi: 10.1111/j.1464-410X.2008.07881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez C, Patel AV, Calle EE, Jacobs EJ, Chao A, Thun MJ. Body mass index, height, and prostate cancer mortality in two large cohorts of adult men in the United States. Cancer Epidemiology Biomarkers and Prevention. 2001;10(4):345–353. [PubMed] [Google Scholar]
- 19.Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annual Review of Medicine. 2013;64:45–57. doi: 10.1146/annurev-med-121211-091527. [DOI] [PubMed] [Google Scholar]
- 20.Hursting SD, Berger NA. Energy balance, host-related factors, and cancer progression. Journal of Clinical Oncology. 2010;28(26):4058–4065. doi: 10.1200/JCO.2010.27.9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iyer A, Fairlie DP, Prins JB, Hammock BD, Brown L. Inflammatory lipid mediators in adipocyte function and obesity. Nature Reviews Endocrinology. 2010;6(2):71–82. doi: 10.1038/nrendo.2009.264. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. Journal of Clinical Investigation. 2004;114(12):1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Luca C, Olefsky JM. Inflammation and insulin resistance. The FEBS Letters. 2008;582(1):97–105. doi: 10.1016/j.febslet.2007.11.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das UN. Essential fatty acids enhance free radical generation and lipid peroxidation to induce apoptosis of tumor cells. Clinical Lipidology. 2011;6(4):463–489. [Google Scholar]
- 25.Nomura DK, Lombardi DP, Chang JW, et al. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chemistry and Biology. 2011;18(7):846–856. doi: 10.1016/j.chembiol.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cathcart M-C, Lysaght J, Pidgeon GP. Eicosanoid signalling pathways in the development and progression of colorectal cancer: novel approaches for prevention/intervention. Cancer and Metastasis Reviews. 2011;30(3-4):363–385. doi: 10.1007/s10555-011-9324-x. [DOI] [PubMed] [Google Scholar]
- 27.Locke JA, Tomlinson Guns ES, Lehman ML, et al. Arachidonic acid activation of intratumoral steroid synthesis during prostate cancer progression to castration resistance. The Prostate. 2010;70(3):239–251. doi: 10.1002/pros.21057. [DOI] [PubMed] [Google Scholar]
- 28.Ahima R, Osei S. Adipokines in obesity. Frontiers of Hormone Research. 2007;36:182–197. doi: 10.1159/000115365. [DOI] [PubMed] [Google Scholar]
- 29.Hursting SD, DiGiovanni J, Dannenberg AJ, et al. Obesity, energy balance, and cancer: new opportunities for prevention. Cancer Prevention Research. 2012;5(11):1260–1272. doi: 10.1158/1940-6207.CAPR-12-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arditi JD, Venihaki M, Karalis KP, Chrousos GP. Antiproliferative effect of adiponectin on MCF7 breast cancer cells: a potential hormonal link between obesity and cancer. Hormone and Metabolic Research. 2007;39(1):9–13. doi: 10.1055/s-2007-956518. [DOI] [PubMed] [Google Scholar]
- 31.Cox ME, Gleave ME, Zakikhani M, et al. Insulin receptor expression by human prostate cancers. The Prostate. 2009;69(1):33–40. doi: 10.1002/pros.20852. [DOI] [PubMed] [Google Scholar]
- 32.Isbarn H, Boccon-Gibod L, Carroll PR, et al. Androgen deprivation therapy for the treatment of prostate cancer: consider both benefits and risks. European Urology. 2009;55(1):62–75. doi: 10.1016/j.eururo.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahra IB, Laurent K, Loubat A, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27(25):3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 34.Smith MR, Lee H, Nathan DM. Insulin sensitivity during combined androgen blockade for prostate cancer. Journal of Clinical Endocrinology and Metabolism. 2006;91(4):1305–1308. doi: 10.1210/jc.2005-2507. [DOI] [PubMed] [Google Scholar]
- 35.Hammarsten J, Högstedt B. Hyperinsulinaemia: a prospective risk factor for lethal clinical prostate cancer. European Journal of Cancer. 2005;41(18):2887–2895. doi: 10.1016/j.ejca.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Neuhouser ML, Till C, Kristal A, et al. Finasteride modifies the relation between serum C-peptide and prostate cancer risk: results from the prostate cancer prevention trial. Cancer Prevention Research. 2010;3(3):279–289. doi: 10.1158/1940-6207.CAPR-09-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulanet DB, Ludwig DL, Kahn CR, Hanahan D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(24):10791–10798. doi: 10.1073/pnas.0914076107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H, Fagan DH, Zeng X, Freeman KT, Sachdev D, Yee D. Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene. 2010;29(17):2517–2527. doi: 10.1038/onc.2010.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seim I, Lubik AL, Lehman M, et al. Cloning of a novel insulin-regulated ghrelin transcript in prostate cancer. Journal of Molecular Endocrinology. 2012 doi: 10.1530/JME-12-0150. [DOI] [PubMed] [Google Scholar]
- 40.Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocrine Reviews. 2009;30(6):586–623. doi: 10.1210/er.2008-0047. [DOI] [PubMed] [Google Scholar]
- 41.del Barco S, Vazquez-Martin A, Cufí S, et al. Metformin: multi-faceted protection against cancer. Oncotarget. 2011;2(12):896–917. doi: 10.18632/oncotarget.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heni M, Hennenlotter J, Scharpf M, et al. Insulin receptor isoforms A and B as well as insulin receptor substrates-1 and -2 are differentially expressed in prostate cancer. PLoS ONE. 2012;7(12) doi: 10.1371/journal.pone.0050953.e50953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Call R, Grimsley M, Cadwallader L, et al. Insulin-carcinogen or mitogen? Preclinical and clinical evidence from prostate, breast, pancreatic, and colorectal cancer research. Postgraduate Medicine. 2010;122(3):158–165. doi: 10.3810/pgm.2010.05.2153. [DOI] [PubMed] [Google Scholar]
- 44.Stein KB, Snyder CF, Barone BB, et al. Colorectal cancer outcomes, recurrence, and complications in persons with and without diabetes mellitus: a systematic review and meta-analysis. Digestive Diseases and Sciences. 2010;55(7):1839–1851. doi: 10.1007/s10620-009-0944-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simon D, Balkau B. Diabetes mellitus, hyperglycaemia and cancer. Diabetes and Metabolism. 2010;36(3):182–191. doi: 10.1016/j.diabet.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Fukui M, Tanaka M, Kadono M, et al. Serum prostate-specific antigen levels in men with type 2 diabetes. Diabetes Care. 2008;31(5):930–931. doi: 10.2337/dc07-1962. [DOI] [PubMed] [Google Scholar]
- 47.Pierce BL, Plymate S, Ostrander EA, Stanford JL. Diabetes mellitus and prostate cancer risk. The Prostate. 2008;68(10):1126–1132. doi: 10.1002/pros.20777. [DOI] [PubMed] [Google Scholar]
- 48.Abdollah F, Briganti A, Suardi N, et al. Does diabetes mellitus increase the risk of high-grade prostate cancer in patients undergoing radical prostatectomy. Prostate Cancer and Prostatic Diseases. 2011;14(1):74–78. doi: 10.1038/pcan.2010.41. [DOI] [PubMed] [Google Scholar]
- 49.Wallner LP, Morgenstern H, McGree ME, et al. The effects of type 2 diabetes and hypertension on changes in serum prostate specific antigen levels: results from the olmsted county study. Urology. 2011;77(1):137–141. doi: 10.1016/j.urology.2010.07.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snyder CF, Stein KB, Barone BB, et al. Does pre-existing diabetes affect prostate cancer prognosis A systematic review. Prostate Cancer and Prostatic Diseases. 2010;13(1):58–64. doi: 10.1038/pcan.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kasper JS, Giovannucci E. A meta-analysis of diabetes mellitus and the risk of prostate cancer. Cancer Epidemiology Biomarkers and Prevention. 2006;15(11):2056–2062. doi: 10.1158/1055-9965.EPI-06-0410. [DOI] [PubMed] [Google Scholar]
- 52.Daousi C, Casson IF, Gill GV, MacFarlane IA, Wilding JPH, Pinkney JH. Prevalence of obesity in type 2 diabetes in secondary care: association with cardiovascular risk factors. Postgraduate Medical Journal. 2006;82(966):280–284. doi: 10.1136/pmj.2005.039032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Currie CJ, Poole CD, Gale EAM. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52(9):1766–1777. doi: 10.1007/s00125-009-1440-6. [DOI] [PubMed] [Google Scholar]
- 54.Jonasson JM, Ljung R, Talbäck M, Haglund B, Gudbjörnsdòttir S, Steineck G. Insulin glargine use and short-term incidence of malignancies-a population-based follow-up study in Sweden. Diabetologia. 2009;52(9):1745–1754. doi: 10.1007/s00125-009-1444-2. [DOI] [PubMed] [Google Scholar]
- 55.Hemkens LG, Grouven U, Bender R, et al. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: a cohort study. Diabetologia. 2009;52(9):1732–1744. doi: 10.1007/s00125-009-1418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29(2):254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 57.Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. The British Medical Journal. 2005;330(7503):1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Landman GWD, Kleefstra N, van Hateren KJJ, Groenier KH, Gans ROB, Bilo HJG. Metformin associated with lower cancer mortality in type 2 diabetes: zodiac-16. Diabetes Care. 2010;33(2):322–326. doi: 10.2337/dc09-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He XX, Tu SM, Lee MH, Yeung SCJ. Thiazolidinediones and metformin associated with improved survival of diabetic prostate cancer patients. Annals of Oncology. 2011;22(12):2640–2645. doi: 10.1093/annonc/mdr020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cauley JA, McTiernan A, Rodabough RJ, et al. Statin use and breast cancer: prospective results from the women's health initiative. Journal of the National Cancer Institute. 2006;98(10):700–707. doi: 10.1093/jnci/djj188. [DOI] [PubMed] [Google Scholar]
- 61.Karp I, Behlouli H, LeLorier J, Pilote L. Statins and cancer risk. The American Journal of Medicine. 2008;121(4):302–309. doi: 10.1016/j.amjmed.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 62.Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocrine-Related Cancer. 2010;17(2):351–360. doi: 10.1677/ERC-09-0252. [DOI] [PubMed] [Google Scholar]
- 63.DeCensi A, Puntoni M, Goodwin P, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prevention Research. 2010;3(11):1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 64.Wright JL, Stanford JL. Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes and Control. 2009;20(9):1617–1622. doi: 10.1007/s10552-009-9407-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dowling RJO, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Research. 2007;67(22):10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 66.Green AS, Chapuis N, Maciel TT, et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood. 2010;116(20):4262–4273. doi: 10.1182/blood-2010-02-269837. [DOI] [PubMed] [Google Scholar]
- 67.Tao R, Gong J, Luo X, et al. AMPK exerts dual regulatory effects on the PI3K pathway. Journal of Molecular Signaling. 2010;5(1, article 1) doi: 10.1186/1750-2187-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Larsson O, Morita M, Topisirovic I, et al. Distinct perturbation of the translatome by the antidiabetic drug metformin. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(23):8977–8982. doi: 10.1073/pnas.1201689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oliveras-Ferraros C, Vazquez-Martin A, Menendez JA. Genome-wide inhibitory impact of the AMPK activator metformin on [kinesins, tubulins, histones, auroras and polo-like kinases] M-phase cell cycle genes in human breast cancer cells. Cell Cycle. 2009;8(10):1633–1636. doi: 10.4161/cc.8.10.8406. [DOI] [PubMed] [Google Scholar]
- 70.Swinnen JV, Heemers H, van de Sande T, et al. Androgens, lipogenesis and prostate cancer. Journal of Steroid Biochemistry and Molecular Biology. 2004;92(4):273–279. doi: 10.1016/j.jsbmb.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 71.Samarajeewa NU, Ham S, Yang F, Simpson ER, Brown KA. Promoter-specific effects of metformin on aromatase transcript expression. Steroids. 2011;76(8):768–771. doi: 10.1016/j.steroids.2011.02.041. [DOI] [PubMed] [Google Scholar]
- 72.Wu B, Li S, Sheng L, et al. Metformin inhibits the development and metastasis of ovarian cancer. Oncology Reports. 2012;28(3):903–908. doi: 10.3892/or.2012.1890. [DOI] [PubMed] [Google Scholar]
- 73.Li W, Yuan Y, Huang L, Qiao M, Zhang Y. Metformin alters the expression profiles of microRNAs in human pancreatic cancer cells. Diabetes Research and Clinical Practice. 2012;96(2):187–195. doi: 10.1016/j.diabres.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 74.Bao B, Wang Z, Ali S, et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prevention Research. 2012;5(3):355–364. doi: 10.1158/1940-6207.CAPR-11-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hadad SM, Fleming S, Thompson AM. Targeting AMPK: a new therapeutic opportunity in breast cancer. Critical Reviews in Oncology/Hematology. 2008;67(1):1–7. doi: 10.1016/j.critrevonc.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 76.Pollak M. Metformin and other biguanides in oncology: advancing the research agenda. Cancer Prevention Research. 2010;3(9):1060–1065. doi: 10.1158/1940-6207.CAPR-10-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Derosa G, Cicero AFG, D’Angelo A, Fogari E, Maffioli P. Effects of 1-year orlistat treatment compared to placebo on insulin resistance parameters in patients with type 2 diabetes. Journal of Clinical Pharmacy and Therapeutics. 2012;37(2):187–195. doi: 10.1111/j.1365-2710.2011.01280.x. [DOI] [PubMed] [Google Scholar]
- 78.Oliveras G, Blancafort A, Urruticoechea A, et al. Novel anti-fatty acid synthase compounds with anti-cancer activity in HER2+ breast cancer. Annals of the New York Academy of Sciences. 2010;1210:86–92. doi: 10.1111/j.1749-6632.2010.05777.x. [DOI] [PubMed] [Google Scholar]
- 79.Tomek K, Wagner R, Varga F, Singer CF, Karlic H, Grunt TW. Blockade of fatty acid synthase induces ubiquitination and degradation of phosphoinositide-3-kinase signaling proteins in ovarian cancer. Molecular Cancer Research. 2011;9(12):1767–1779. doi: 10.1158/1541-7786.MCR-10-0467. [DOI] [PubMed] [Google Scholar]
- 80.Mansour M, Schwartz D, Judd R, et al. Thiazolidinediones/PPARγ agonists and fatty acid synthase inhibitors as an experimental combination therapy for prostate cancer. International Journal of Oncology. 2011;38(2):537–546. doi: 10.3892/ijo.2010.877. [DOI] [PubMed] [Google Scholar]
- 81.Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Research. 2004;64(6):2070–2075. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- 82.Knowles LM, Yang C, Osterman A, Smith JW. Inhibition of fatty-acid synthase induces caspase-8-mediated tumor cell apoptosis by up-regulating DDIT4. The Journal of Biological Chemistry. 2008;283(46):31378–31384. doi: 10.1074/jbc.M803384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fujiwara J, Sowa Y, Horinaka M, et al. The anti-obesity drug orlistat promotes sensitivity to TRAIL by two different pathways in hormone-refractory prostate cancer cells. International Journal of Oncology. 2012;40(5):1483–1491. doi: 10.3892/ijo.2012.1353. [DOI] [PubMed] [Google Scholar]
- 84.Little JL, Wheeler FB, Fels DR, Koumenis C, Kridel SJ. Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer Research. 2007;67(3):1262–1269. doi: 10.1158/0008-5472.CAN-06-1794. [DOI] [PubMed] [Google Scholar]
- 85.Browning DRL, Martin RM. Statins and risk of cancer: a systematic review and metaanalysis. International Journal of Cancer. 2007;120(4):833–843. doi: 10.1002/ijc.22366. [DOI] [PubMed] [Google Scholar]
- 86.Hamilton RJ, Banez LL, Aronson WJ, et al. Statin medication use and the risk of biochemical recurrence after radical prostatectomy: results from the shared equal access regional cancer hospital (SEARCH) database. Cancer. 2010;116(14):3389–3398. doi: 10.1002/cncr.25308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shafique K, McLoone P, Qureshi K, Leung H, Hart C, Morrison DS. Cholesterol and the risk of grade-specific prostate cancer incidence: evidence from two large prospective cohort studies with up to 37 years' follow up. BMC Cancer. 2012;12(article 25) doi: 10.1186/1471-2407-12-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Loeb S, Kan D, Helfand BT, Nadler RB, Catalona WJ. Is statin use associated with prostate cancer aggressiveness? BJU International. 2010;105(9):1222–1225. doi: 10.1111/j.1464-410X.2009.09007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mostaghel EA, Solomon KR, Pelton K, Freeman MR, Montgomery RB. Impact of circulating cholesterol levels on growth and intratumoral androgen concentration of prostate tumors. PLoS ONE. 2012;7(1) doi: 10.1371/journal.pone.0030062.e30062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu O, Boudreau DM, Buist DSM, Miglioretti DL. Statin use and female reproductive organ cancer risk in a large population-based setting. Cancer Causes and Control. 2009;20(5):609–616. doi: 10.1007/s10552-008-9271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Segawa Y, Yoshimura R, Hase T, et al. Expression of peroxisome proliferator-activated receptor (PPAR) in human prostate cancer. The Prostate. 2002;51(2):108–116. doi: 10.1002/pros.10058. [DOI] [PubMed] [Google Scholar]
- 92.Du Q, Wang Y-J, Yang S, Wu B, Han P, Zhao Y-Y. A systematic review and meta-analysis of randomized controlled trials comparing pioglitazone versus metformin in the treatment of polycystic ovary syndrome. Current Medical Research and Opinion. 2012;28(5):723–730. doi: 10.1185/03007995.2012.681636. [DOI] [PubMed] [Google Scholar]
- 93.Colmers IN, Bowker SL, Johnson JA. Thiazolidinedione use and cancer incidence in type 2 diabetes: a systematic review and meta-analysis. Diabetes and Metabolism. 2012;38(6):475–484. doi: 10.1016/j.diabet.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 94.Bolden A, Bernard L, Jones D, Akinyeke T, Stewart LV. The PPAR γ agonist troglitazone regulates Erk 1/2 phosphorylation via a PPARγ-independent, MEK-dependent pathway in human prostate cancer cells. PPAR Research. 2012;2012:9 pages. doi: 10.1155/2012/929052.929052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang M, Fernandez S, Jerome WG, et al. Disruption of PPARγ signaling results in mouse prostatic intraepithelial neoplasia involving active autophagy. Cell Death and Differentiation. 2010;17(3):469–481. doi: 10.1038/cdd.2009.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Al-Alem L, Southard RC, Kilgore MW, Curry TE. Specific thiazolidinediones inhibit ovarian cancer cell line proliferation and cause cell cycle arrest in a PPARγ independent manner. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016179.e16179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim S, Lee JJ, Heo DS. PPARγ ligands induce growth inhibition and apoptosis through p63 and p73 in human ovarian cancer cells. Biochemical and Biophysical Research Communications. 2011;406(3):389–395. doi: 10.1016/j.bbrc.2011.02.052. [DOI] [PubMed] [Google Scholar]
- 98.Lyles BE, Akinyeke TO, Moss PE, Stewart LV. Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARγ-dependent and PPARγ-independent pathways. Cell Cycle. 2009;8(2):268–277. doi: 10.4161/cc.8.2.7584. [DOI] [PubMed] [Google Scholar]
- 99.Tan XH, Dagher H, Hutton CA, Bourke JE. Effects of PPARγ ligands on TGF-β1-induced epithelial-mesenchymal transition in alveolar epithelial cells. Respiratory Research. 2010;11(article 21) doi: 10.1186/1465-9921-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kurtzhals P, Schäffer L, Sørensen A, et al. Correlations of receptor binding and metabolic and mitogenic potencies of insulin analogs designed for clinical use. Diabetes. 2000;49(6):999–1005. doi: 10.2337/diabetes.49.6.999. [DOI] [PubMed] [Google Scholar]
- 101.Hansen BF, Danielsen GM, Drejer K, et al. Sustained signalling from the insulin receptor after stimulation with insulin analogues exhibiting increased mitogenic potency. Biochemical Journal. 1996;315(1):271–279. doi: 10.1042/bj3150271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hansen BF, Kurtzhals P, Jensen AB, Dejgaard A, Russell-Jones D. Insulin X10 revisited: a super-mitogenic insulin analogue. Diabetologia. 2011;54(9):2226–2231. doi: 10.1007/s00125-011-2203-8. [DOI] [PubMed] [Google Scholar]
- 103.Registries, A.I.o.H.a.W.A.A.o.C. Cancer in Australia: An Overview, 2010. Vol. 60. Canberra, Australia: AIHW; 2010. (Australian Institute of Health and Welfare’s Cancer series). [Google Scholar]
- 104.Tewari A, Raman JD, Chang P, Rao S, Divine G, Menon M. Long-term survival probability in men with clinically localized prostate cancer treated either conservatively or with definitive treatment (radiotherapy or radical prostatectomy) Urology. 2006;68(6):1268–1274. doi: 10.1016/j.urology.2006.08.1059. [DOI] [PubMed] [Google Scholar]
- 105.Huggins C. Effect of orchiectomy and irradiation on cancer of the prostate. Annals of Surgery. 1942;115(6):1192–1200. doi: 10.1097/00000658-194206000-00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tolis G, Koutsilieris M, Stellos A. Metastatic prostate cancer: hormone dependency and therapeutic efficacy of GnRH analogues. Acta Endocrinologica, Supplementum. 1984;107(265):22–23. doi: 10.1530/acta.0.107s0022. [DOI] [PubMed] [Google Scholar]
- 107.Hellerstedt BA, Pienta KJ. The current state of hormonal therapy for prostate cancer. CA: A Cancer Journal for Clinicians. 2002;52(3):154–179. doi: 10.3322/canjclin.52.3.154. [DOI] [PubMed] [Google Scholar]
- 108.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. The New England Journal of Medicine. 2004;351(15):1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 109.Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. Journal of Clinical Oncology. 2008;26(2):242–245. doi: 10.1200/JCO.2007.12.4008. [DOI] [PubMed] [Google Scholar]
- 110.de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. The Lancet. 2010;376(9747):1147–1154. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 111.Small EJ, Srinivas S. The antiandrogen withdrawal syndrome: experience in a large cohort of unselected patients with advanced prostate cancer. Cancer. 1995;76(8):1428–1434. doi: 10.1002/1097-0142(19951015)76:8<1428::aid-cncr2820760820>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 112.Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: a phase III trial (CALGB 9583) Journal of Clinical Oncology. 2004;22(6):1025–1033. doi: 10.1200/JCO.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 113.Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Research. 2008;68(11):4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. The New England Journal of Medicine. 1995;332(21):1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 115.Buchanan G, Yang M, Cheong A, et al. Structural and functional consequences of glutamine tract variation in the androgen receptor. Human Molecular Genetics. 2004;13(16):1677–1692. doi: 10.1093/hmg/ddh181. [DOI] [PubMed] [Google Scholar]
- 116.Veldscholte J, Ris-Stalpers C, Kuiper GGJM, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochemical and Biophysical Research Communications. 1990;173(2):534–540. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 117.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Research. 2008;68(13):5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nature Medicine. 2004;10(1):33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 119.Dehm SM, Tindall DJ. Androgen receptor structural and functional elements: role and regulation in prostate cancer. Molecular Endocrinology. 2007;21(12):2855–2863. doi: 10.1210/me.2007-0223. [DOI] [PubMed] [Google Scholar]
- 120.Holzbeierlein J, Lal P, LaTulippe E, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. American Journal of Pathology. 2004;164(1):217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Linja MJ, Savinainen KJ, Saramäki OR, Tammela TLJ, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Research. 2001;61(9):3550–3555. [PubMed] [Google Scholar]
- 122.Mostaghel EA, Page ST, Lin DW, et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: therapeutic implications for castration-resistant prostate cancer. Cancer Research. 2007;67(10):5033–5041. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 123.Geller J, Albert J, Nachtsheim D, Loza D, Lippman S. Steroid levels in cancer of the prostate—markers of tumor differentiation and adequacy of anti-androgen therapy. Progress in Clinical and Biological Research. 1979;33:103–111. [PubMed] [Google Scholar]
- 124.Geller J, de la Vega DJ, Albert JD, Nachtsheim DA. Tissue dihydrotestosterone levels and clinical response to hormonal therapy in patients with advanced prostate cancer. Journal of Clinical Endocrinology and Metabolism. 1984;58(1):36–40. doi: 10.1210/jcem-58-1-36. [DOI] [PubMed] [Google Scholar]
- 125.Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clinical Cancer Research. 2004;10(21):7121–7126. doi: 10.1158/1078-0432.CCR-04-0913. [DOI] [PubMed] [Google Scholar]
- 126.Locke JA, Guns ES, Lubik AA, et al. Androgen Levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Research. 2008;68(15):6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 127.Leon CG, Locke JA, Adomat HH, et al. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. The Prostate. 2010;70(4):390–400. doi: 10.1002/pros.21072. [DOI] [PubMed] [Google Scholar]
- 128.Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Watson PA, Chen YF, Balbas MD, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(39):16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Research. 2012;72(6):1494–1503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Attard G, Reid AHM, A’Hern R, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. Journal of Clinical Oncology. 2009;27(23):3742–3748. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Potter GA, Elaine Barrie S, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017α (17α-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. Journal of Medicinal Chemistry. 1995;38(13):2463–2471. doi: 10.1021/jm00013a022. [DOI] [PubMed] [Google Scholar]
- 133.Barrie SE, Potter GA, Goddard PM, Haynes BP, Dowsett M, Jarman M. Pharmacology of novel steroidal inhibitors of cytochrome p450(17α) (17α-hydroxylase/C17-20 lyase) Journal of Steroid Biochemistry and Molecular Biology. 1994;50(5-6):267–273. doi: 10.1016/0960-0760(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 134.Fizazi K, Scher HI, Molina A, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study (vol 13, pg 983, 2012) The Lancet Oncology. 2012;13(11):E464–E464. doi: 10.1016/S1470-2045(12)70379-0. [DOI] [PubMed] [Google Scholar]
- 135.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. The New England Journal of Medicine. 2013;368(2):138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vasaitis T, Belosay A, Schayowitz A, et al. Androgen receptor inactivation contributes to antitumor efficacy of 17α-hydroxylase/17,20-lyase inhibitor 3β-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Molecular Cancer Therapeutics. 2008;7(8):2348–2357. doi: 10.1158/1535-7163.MCT-08-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Attard G, Reid AHM, Yap TA, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. Journal of Clinical Oncology. 2008;26(28):4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 138.Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. The New England Journal of Medicine. 2012;367(13):1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 139.Basaria S, Muller DC, Carducci MA, Egan J, Dobs AS. Hyperglycemia and insulin resistance in men with prostate carcinoma who receive androgen-deprivation therapy. Cancer. 2006;106(3):581–588. doi: 10.1002/cncr.21642. [DOI] [PubMed] [Google Scholar]
- 140.Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C. Definition of metabolic syndrome: report of the national heart, lung, and blood institute/American heart association conference on scientific issues related to definition. Circulation. 2004;109(3):433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 141.Yialamas MA, Dwyer AA, Hanley E, Lee H, Pitteloud N, Hayes FJ. Acute sex steroid withdrawal reduces insulin sensitivity in healthy men with idiopathic hypogonadotropic hypogonadism. Journal of Clinical Endocrinology and Metabolism. 2007;92(11):4254–4259. doi: 10.1210/jc.2007-0454. [DOI] [PubMed] [Google Scholar]
- 142.Yannucci J, Manola J, Garnick MB, Bhat G, Bubley GJ. The effect of androgen deprivation therapy on fasting serum lipid and glucose parameters. Journal of Urology. 2006;176(2):520–525. doi: 10.1016/j.juro.2006.03.057. [DOI] [PubMed] [Google Scholar]
- 143.Flanagan J, Gray PK, Hahn N, et al. Presence of the metabolic syndrome is associated with shorter time to castration-resistant prostate cancer. Annals of Oncology. 2011;22(4):801–807. doi: 10.1093/annonc/mdq443. [DOI] [PubMed] [Google Scholar]
- 144.Giganti MG, Minella D, Testa B, et al. A pilot study on the transcriptional response of androgen- and insulin-related genes in peripheral blood mononuclear cells induced by testosterone administration in hypogonadal men. Journal of Biological Regulators and Homeostatic Agents. 2011;25(2):291–294. [PubMed] [Google Scholar]
- 145.Rohrmann S, Shiels MS, Lopez DS, et al. Body fatness and sex steroid hormone concentrations in US men: results from NHANES III. Cancer Causes and Control. 2011;22(8):1141–1151. doi: 10.1007/s10552-011-9790-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Haffner SM, Valdez RA, Mykkanen L, Stern MP, Katz MS. Decreased testosterone and dehydroepiandrosterone sulfate concentrations are associated with increased insulin and glucose concentrations in nondiabetic men. Metabolism. 1994;43(5):599–603. doi: 10.1016/0026-0495(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 147.Moriarty-Kelsey M, Harwood JEF, Travers SH, Zeitler PS, Nadeau KJ. Testosterone, obesity and insulin resistance in young males: evidence for an association between gonadal dysfunction and insulin resistance during puberty. Journal of Pediatric Endocrinology and Metabolism. 2010;23(12):1281–1287. doi: 10.1515/jpem.2010.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang C, Feng R, Sun D, Li Y, Bi X, Sun C. Metabolic profiling of urine in young obese men using ultra performance liquid chromatography and Q-TOF mass spectrometry (UPLC/Q-TOF MS) Journal of Chromatography B. 2011;879(27):2871–2876. doi: 10.1016/j.jchromb.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 149.Simon D, Preziosi P, Barrett-Connor E, et al. Interrelation between plasma testosterone and plasma insulin in healthy adult men: the telecom study. Diabetologia. 1992;35(2):173–177. doi: 10.1007/BF00402551. [DOI] [PubMed] [Google Scholar]
- 150.Pitteloud N, Mootha VK, Dwyer AA, et al. Relationship between testosterone levels, insulin sensitivity, and mitochondrial function in men. Diabetes Care. 2005;28(7):1636–1642. doi: 10.2337/diacare.28.7.1636. [DOI] [PubMed] [Google Scholar]
- 151.Rubinow KB, Snyder CN, Amory JK, Hoofnagle AN, Page ST. Acute testosterone deprivation reduces insulin sensitivity in men. Clinical Endocrinology. 2012;76(2):281–288. doi: 10.1111/j.1365-2265.2011.04189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Smith MR, Lee H, Fallon MA, Nathan DM. Adipocytokines, obesity, and insulin resistance during combined androgen blockade for prostate cancer. Urology. 2008;71(2):318–322. doi: 10.1016/j.urology.2007.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Quinn DI, Henshall SM, Sutherland RL. Molecular markers of prostate cancer outcome. European Journal of Cancer. 2005;41(6):858–887. doi: 10.1016/j.ejca.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 154.Wang Y, Zhu Y, Zhang L, et al. Insulin promotes proliferation, survival, and invasion in endometrial carcinoma by activating the MEK/ERK pathway. Cancer Letters. 2012;322(2):223–231. doi: 10.1016/j.canlet.2012.03.026. [DOI] [PubMed] [Google Scholar]
- 155.Lubik AA, Gunter JH, Hendy SC, et al. Insulin increases de novo steroidogenesis in prostate cancer cells. Cancer Research. 2011;71(17):5754–5764. doi: 10.1158/0008-5472.CAN-10-2470. [DOI] [PubMed] [Google Scholar]
- 156.Eichholz A, Ferraldeschi R, Attard G, de Bono JS. Putting the brakes on continued androgen receptor signaling in castration-resistant prostate cancer. Molecular and Cellular Endocrinology. 2012;360(1-2):68–75. doi: 10.1016/j.mce.2011.09.038. [DOI] [PubMed] [Google Scholar]
- 157.Reis LO. Old issues and new perspectives on prostate cancer hormonal therapy: the molecular substratum. Medical Oncology. 2012;29(3):1948–1955. doi: 10.1007/s12032-011-9991-z. [DOI] [PubMed] [Google Scholar]
- 158.Cohen BL, Gomez P, Omori Y, et al. Cyclooxygenase-2 (cox-2) expression is an independent predictor of prostate cancer recurrence. International Journal of Cancer. 2006;119(5):1082–1087. doi: 10.1002/ijc.21749. [DOI] [PubMed] [Google Scholar]
- 159.Cao ZX, Liu LZ, Dixon DA, Zheng JZ, Chandran B, Jiang BH. Insulin-like growth factor-I induces cyclooxygenase-2 expression via PI3K, MAPK and PKC signaling pathways in human ovarian cancer cells. Cellular Signalling. 2007;19(7):1542–1553. doi: 10.1016/j.cellsig.2007.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stoeltzing O, Liu W, Fan F, et al. Regulation of cyclooxygenase-2 (COX-2) expression in human pancreatic carcinoma cells by the insulin-like growth factor-I receptor (IGF-IR) system. Cancer Letters. 2007;258(2):291–300. doi: 10.1016/j.canlet.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zoubeidi A, Zardan A, Beraldi E, et al. Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Research. 2007;67(21):10455–10465. doi: 10.1158/0008-5472.CAN-07-2057. [DOI] [PubMed] [Google Scholar]
- 162.Zoubeidi A, Zardan A, Wiedmann RM, et al. Hsp27 promotes insulin-like growth factor-I survival signaling in prostate cancer via p90Rsk-dependent phosphorylation and inactivation of BAD. Cancer Research. 2010;70(6):2307–2317. doi: 10.1158/0008-5472.CAN-09-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang J, Lei T, Chen X, et al. Resistin up-regulates COX-2 expression via TAK1-IKK-NF-κB signaling pathway. Inflammation. 2010;33(1):25–33. doi: 10.1007/s10753-009-9155-x. [DOI] [PubMed] [Google Scholar]
- 164.Mullen E, O’Reilly E, Ohlendieck K. Skeletal muscle tissue from the Goto-Kakizaki rat model of type-2 diabetes exhibits increased levels of the small heat shock protein Hsp27. Molecular Medicine Reports. 2011;4(2):229–236. doi: 10.3892/mmr.2011.437. [DOI] [PubMed] [Google Scholar]
- 165.Burut DFP, Borai A, Livingstone C, Ferns G. Serum heat shock protein 27 antigen and antibody levels appear to be related to the macrovascular complications associated with insulin resistance: a pilot study. Cell Stress and Chaperones. 2010;15(4):379–386. doi: 10.1007/s12192-009-0152-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Price RS, Cavazos DA, de Angel RE, Hursting SD, DeGraffenried LA. Obesity-related systemic factors promote an invasive phenotype in prostate cancer cells. Prostate Cancer and Prostatic Diseases. 2012;15(2):135–143. doi: 10.1038/pcan.2011.54. [DOI] [PubMed] [Google Scholar]
- 167.Gunter JH, Lubik AA, McKenzie I, Pollak M, Nelson CC. The interactions between insulin and androgens in progression to castrate-resistant prostate cancer. Advances in Urology. 2012;2012:11 pages. doi: 10.1155/2012/248607.248607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate Ccancer. Cancer Cell. 2011;19(5):575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kaarbø M, Mikkelsen OL, Malerød L, et al. PI3K-AKT-mTOR pathway is dominant over androgen receptor signaling in prostate cancer cells. Cellular Oncology. 2010;32(1-2):11–27. doi: 10.3233/CLO-2009-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mulholland DJ, Tran LM, Li Y, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19(6):792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, Witte ON. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(20):7789–7794. doi: 10.1073/pnas.0602567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Malinowska K, Neuwirt H, Cavarretta IT, et al. Interleukin-6 stimulation of growth of prostate cancer in vitro and in vivo through activation of the androgen receptor. Endocrine-Related Cancer. 2009;16(1):155–169. doi: 10.1677/ERC-08-0174. [DOI] [PubMed] [Google Scholar]
- 174.Robinson DR, Zylstra CR, Williams BO. Wnt signaling and prostate cancer. Current Drug Targets. 2008;9(7):571–580. doi: 10.2174/138945008784911831. [DOI] [PubMed] [Google Scholar]
- 175.Seaton A, Scullin P, Maxwell PJ, et al. Interleukin-8 signaling promotes androgen-independent proliferation of prostate cancer cells via induction of androgen receptor expression and activation. Carcinogenesis. 2008;29(6):1148–1156. doi: 10.1093/carcin/bgn109. [DOI] [PubMed] [Google Scholar]
- 176.Frigo DE, Howe MK, Wittmann BM, et al. CaM kinase kinase β-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Research. 2011;71(2):528–537. doi: 10.1158/0008-5472.CAN-10-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Massie CE, Lynch A, Ramos-Montoya A, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. The EMBO Journal. 2011;30(13):2719–2733. doi: 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6(1):91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 179.Kodaman PH, Duleba AJ. HMG-CoA reductase inhibitors: do they have potential in the treatment of polycystic ovary syndrome? Drugs. 2008;68(13):1771–1785. doi: 10.2165/00003495-200868130-00001. [DOI] [PubMed] [Google Scholar]
- 180.Zheng X, Cui XX, Gao Z, et al. Atorvastatin and celecoxib in combination inhibits the progression of androgen-dependent LNCaP xenograft prostate tumors to androgen independence. Cancer Prevention Research. 2010;3(1):114–124. doi: 10.1158/1940-6207.CAPR-09-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Swinnen JV, Roskams T, Joniau S, et al. Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. International Journal of Cancer. 2002;98(1):19–22. doi: 10.1002/ijc.10127. [DOI] [PubMed] [Google Scholar]
- 182.Rashid MH, Chaudhary UB. Intermittent androgen deprivation therapy for prostate cancer. Oncologist. 2004;9(3):295–301. doi: 10.1634/theoncologist.9-3-295. [DOI] [PubMed] [Google Scholar]
- 183.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. Journal of Clinical Oncology. 2005;23(32):8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 184.So A, Gleave M, Hurtado-Col A, Nelson C. Mechanisms of the development of androgen independence in prostate cancer. World Journal of Urology. 2005;23(1):1–9. doi: 10.1007/s00345-004-0473-1. [DOI] [PubMed] [Google Scholar]
- 185.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Research. 2006;66(5):2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 186.Redig AJ, Munshi HG. Care of the cancer survivor: metabolic syndrome after hormone-modifying therapy. The American Journal of Medicine. 2010;123(1):87.e1–87.e6. doi: 10.1016/j.amjmed.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]