

The structural study of a bacterial anhydrolase/dipeptidase closely related to human prolidase is reported.
Keywords: organophosphorus acid anhydrolase, prolidases, bifunctional
Abstract
The bacterial enzyme organophosphorus acid anhydrolase (OPAA) is able to catalyze the hydrolysis of both proline dipeptides (Xaa-Pro) and several types of organophosphate (OP) compounds. The full three-dimensional structure of the manganese-dependent OPAA enzyme is presented for the first time. This enzyme, which was originally isolated from the marine bacterium Alteromonas macleodii, was prepared recombinantly in Escherichia coli. The crystal structure was determined at 1.8 Å resolution in space group C2, with unit-cell parameters a = 133.8, b = 49.2, c = 97.3 Å, β = 125.0°. The enzyme forms dimers and their existence in solution was confirmed by dynamic light scattering and size-exclusion chromatography. The enzyme shares the pita-bread fold of its C-terminal domain with related prolidases. The binuclear manganese centre is located in the active site within the pita-bread domain. Moreover, an Ni2+ ion from purification was localized according to anomalous signal. This study presents the full structure of this enzyme with complete surroundings of the active site and provides a critical analysis of its relationship to prolidases.
1. Introduction
Organophosphorus acid anhydrolase (OPAA; EC 3.1.8.2) is an enzyme capable of the hydrolysis of both toxic organic phosphorus-containing compounds (organophosphates; OPs; DeFrank & Cheng, 1991 ▶) and dipeptides with C-terminal proline (Xaa-Pro; Cheng et al., 1997 ▶). OPAA was firstly discovered by Mazur (1946 ▶) as a rabbit tissue enzyme which hydrolyzes diisopropyl fluorophosphate (DFP). A similar enzyme was later localized in squid axons but had slightly different properties. The squid-type OPAA with molecular mass 30 kDa required Ca2+ ions for activity, in contrast to the Mazur-type OPAA with molecular mass 45–90 kDa, which was stimulated by Mn2+ ions (DeFrank, 1991 ▶). The squid-type OPAA hydrolyzes DFP more rapidly than O-pinacolyl methylphosphonofluoridate (soman), unlike the Mazur-type OPAA, which hydrolyzes soman more rapidly than DFP (Scharff et al., 2001 ▶). The issue of the degradation of dangerous OPs is of interest because of their negative effects on living beings. Harmful OPs also include soman, sarin, tabun, VX and other chemical warfare agents. Many OPs inhibit the enzyme acetylcholinesterase (AChE; EC 3.1.1.7) and cause neuromuscular paralysis (Herkenhoff et al., 2004 ▶). AChE catalyzes the rapid hydrolysis of the neurotransmitter acetylcholine to acetyl and choline and is responsible for termination of synapse transmission. The exposure of animals or humans to organophosphates results in an instant and irreversible interaction between OP and AChE followed by the accumulation of acetylcholine, causing neuromuscular paralysis which can also have lethal consequences (Harel et al., 2000 ▶). After the discovery of this effect of toxic OPs on living beings they became widely used in agriculture and by the military. They are used as insecticides (malathion, parathion and chlorpyrifos), nerve agents (soman, sarin and VX), antihelminthics and herbicides.
The severity of intoxication depends on several factors such as the dose, the route of entry, the type of agent and also its stereoisomer. In general, the V-agents are tenfold more poisonous than sarin (GB). Nerve gases exist as optical stereoisomers owing to the potential attachment of up to four various chiral groups to the P atom. The individual stereoisomers inhibit acetylcholine esterase at different levels and as a consequence evoke varied intoxication effects.
Thousands of tons of these organophosphates have been accumulated around the world (Mansee et al., 2005 ▶). The enzymatic biodegradation of organophosphate pesticide waste and demilitarization of nerve agents is more effective, economical and safer than other methods (e.g. incineration; Mansee et al., 2005 ▶). There are several types of enzymes that are capable of the hydrolysis of toxic OPs, but most of them have poor activity, are unstable or are scarcely accessible. Organophosphorus acid hydrolase (OPH; EC 3.1.8.1) and organophosphorus acid anhydrolase (OPAA; EC 3.1.8.2) are important enzymes for the biodegradation of OPs. In contrast to OPH, the natural substrate and role of which are still unknown, OPAA has been identified as a proline dipeptidase (Cheng et al., 1997 ▶), which means that it can cleave the P—F bond in G-type nerve agents as well as dipeptides containing a C-terminal proline residue (Xaa-Pro). OPAA from Alteromonas species (aOPAA) hydrolyses DFP at a high rate (V max = 2.70 mV s−1 obtained using a pH electrode; K m = 0.54 mM with DFP as substrate; Zheng et al., 2005 ▶). aOPAA is able to cleave the P—F bond in OPs, while its catalytic activity towards P—O, P—S and P—CN bonds is minimal (Zheng et al., 2005 ▶). Activity assays of aOPAAs towards DFP and several types of G-agents have been described previously (Cheng et al., 1997 ▶). All aOPAAs displayed the highest activity towards soman as a substrate, which is in agreement with the definition of a Mazur-type OPAA. It was also confirmed that their catalytic activity towards phosphate and phosphonate esters is stereoisomer-dependent. aOPAAs are optimally active at pH values between 7.5 and 8.5 and at temperatures between 313 and 328 K, and Mn2+ ions increase their activity and stability (DeFrank & Cheng, 1991 ▶). They are inhibited by the DFP analogue mipafox (N,N′-diisopropyl phosphorodiamidofluoridate; Vyas et al., 2010 ▶).
Cheng et al. (1997 ▶) tested the prolidase and aminopeptidase activities of aOPAAs and demonstrated their ability to hydrolyze dipeptides but not tripeptides. Activity was not detectable towards tested dipeptide substrates with Pro at the N-terminus (Pro-Xaa; Cheng et al., 1997 ▶). The mechanism of the peptide-bond cleavage is the same as in Escherichia coli aminopeptidase (eAMPP; Lowther & Matthews, 2000 ▶). A water molecule activated by the two metal ions in the active site plays the role of the nucleophile during hydrolysis (Lowther & Matthews, 2000 ▶).
The structural studies proved that aOPAA shares the fold of its C-terminal domain with prolidases (EC 3.4.13.9), aminopeptidases (AMPPs; EC 3.4.11.9) and methionine aminopeptidases (MetAPs; EC 3.4.11.18). This fold was first described for E. coli methionine aminopeptidase (eMetAP) and is named the pita-bread domain (Roderick & Matthews, 1993 ▶). The architecture of the active site and the location of the binuclear metal centre within this domain are similar for all enzymes based on the pita-bread fold, including aOPAA.
A sequence alignment of aOPAA from A. macleodii (amOPAA) with human prolidase (PDB entry 2iw2; U. Mueller, F. H. Niesen, Y. Roske, F. Goetz, J. Behlke, K. Buessow & U. Heinemann, unpublished work) shows 27% identity (Fig. 1 ▶). Owing to the high similarity in sequence, structure and function, it has been speculated that aOPAA is a prolidase (Cheng et al., 1997 ▶) or that the human prolidase is a type of OPAA (Wang et al., 2006 ▶). No sequence homology was found between OPAA and OPH, which is also capable of the hydrolysis of OPs. OPH cleaves P—O, P—F, P—S and P—CN bonds via an SN2 mechanism (Zheng et al., 2005 ▶).
Figure 1.

Sequence alignment of amOPAA and human prolidase (PDB entry 2iw2). Sequence identity is highlighted by a black background; the N-domain is highlighted by a red box in the amOPAA sequence and the C-domain by a yellow box. The magenta lines show the positions of the finger loops responsible for dimerization. Active-site residues suggested to be involved in ligand binding and residues binding nickel and manganese are marked.
Here, we present the three-dimensional structure of an aOPAA from the marine halophilic bacterium A. macleodii. The Mn2+-dependent amOPAA belongs to the Mazur-type OPAAs, with a molecular mass of 51 kDa and 445 amino acids. The recently reported aOPAA from Alteromonas sp. JD6.5 (aOPAA-JD6.5) is composed of 517 amino acids and, according to the authors, forms functional tetramers (Vyas et al., 2010 ▶).
2. Materials and methods
2.1. Expression and purification
The strain A. macleodii (NCIMN1963) was grown on Marine agar 2216 at 299 K. An overnight culture was performed in Marine Broth at 299 K and shaken at 250 rev min−1. The pellet was resuspended in 100 µl TEL buffer (Tris–EDTA buffer supplemented with 50 mg ml−1 lysozyme). The genomic DNA was isolated according to Pitcher et al. (1989 ▶). The resulting genomic DNA was resuspended in 50 µl H2O and used as a template for PCR amplification of the opaa structural genes from A. macleodii (GenBank JF922916). To facilitate the cloning of the PCR product, the primers were designed with appropriate restriction sites added to the 5′-end. The sequences of the forward and reverse primers were 5′-GGAATTCCATATGTCGCAACATAAAGCAACCTAC-3′ and 5′-CCGCTCGAGATTTAAGTCCAAATCTCGTGTCATG-3′, respectively. PCR amplification was performed using the Expand Long Template PCR System (Boehringer Mannheim) under the following conditions: 367 K for 2 min followed by 30 cycles of 367 K for 30 s, 323 K for 1 min and 341 K for 2 min. The resulting PCR fragment was purified and digested with the relevant restriction enzymes. The resulting fragment was cloned into NdeI- and XhoI-digested E. coli vector pET-30a(+) using standard ligation techniques. The resulting plasmid was transformed into the expression host E. coli BL21. The plasmid construct was isolated and verified by DNA sequencing.
Recombinant amOPAA was produced in E. coli BL21. The cells were grown at 310 K in LB medium supplemented with 100 mg l−1 kanamycin to an OD600 of 0.6; expression was then induced by the addition of 0.4 mM isopropyl β-d-1-thiogalactopyranoside and growth was continued for approximately 3 h. Cells were harvested by centrifugation at 10 000g for 10 min and the obtained pellet was resuspended in binding buffer (0.5 M NaCl, 20 mM Tris–HCl, 5 mM imidazole pH 7.9). The cells were subjected to sonication and the resulting lysate was clarified by centrifugation for 30 min at 15 000g and 277 K. The cleared lysate was loaded onto a column of His·Bind Resin (Novagen) pre-equilibrated in the binding buffer. The column was washed with six column volumes of wash buffer (0.5 M NaCl, 60 mM imidazole, 20 mM Tris–HCl pH 7.9) to remove unbound proteins. The bound protein was eluted with six column volumes of elution buffer (1 M imidazole, 0.5 M NaCl, 20 mM Tris–HCl pH 7.9). The fractions containing pure amOPAA, as determined by SDS–PAGE, were pooled, washed and concentrated using Amicon centrifugal filters.
2.2. Gel-filtration chromatography
Samples with a concentration of about 10 mg ml−1 were loaded onto a Tricorn 10/300 column packed with Superdex 200 (GE Healthcare) equilibrated with 50 mM NaH2PO4/Na2HPO4 pH 7.5, 100 mM NaCl, 1 mM NaN3 at a flow rate of 1 ml min−1. The elution of amOPAA was monitored at 280 nm. According to the column calibration with protein standards, the position of the amOPAA peak corresponded to a molecular weight of approximately 101 kDa.
2.3. Dynamic light scattering and hydrodynamic radius calculation
Filtered samples of amOPAA diluted to a concentration of 1.3 mg ml−1 in 50 mM NaH2PO4/Na2HPO4 buffer pH 7.5 were loaded into a 45 µl quartz cuvette and DLS measurements were performed at a temperature of 291 K using a Malvern Instruments ZEN3600 operated using the DYNAMICS software. The hydrodynamic radii of monomers and dimers of the enzyme were estimated by the program HYDROPRO v.7c (García de la Torre et al., 2000 ▶) using standard procedures. The calculated Stokes radii were compared with the DLS results.
2.4. Crystallization and X-ray data collection
The amOPAA enzyme was crystallized by the vapour-diffusion method, mostly in hanging drops. The hanging drops consisted of 0.5 µl precipitant solution and 0.5 µl protein solution. The protein solution consisted of 50 mM NaH2PO4/Na2HPO4 pH 7.5, 1 mM NaN3 and 13.4 mg ml−1 protein. For the initial screening of crystallization conditions, several commercially available sets of crystallization solutions were tested at a temperature of 291 K: Crystal Screen (Hampton Research; Jancarik & Kim, 1991 ▶), Crystal Screen 2 (Hampton Research; Cudney et al., 1994 ▶), Index (Hampton Research; D’Arcy et al., 2003 ▶), PEG/Ion (Hampton Research; McPherson, 2001 ▶) and Poly A and Poly B (Skálová et al., 2010 ▶). Crystals of different shapes, sizes and quality were formed in several screening conditions within a few days.
A plate-like crystal of amOPAA was grown in 0.056 M NaH2PO4, 1.344 M K2HPO4 pH 8.2 (Index condition No. 19; Hampton Research; D’Arcy et al., 2003 ▶) as a precipitant solution at 291 K in 2 d. CoCl2 was added to the crystallization drop to a final concentration of 5 mM to increase the crystal size.
Prior to data collection, a crystal with a longest dimension of about 300 µm was soaked in mother liquor with 25%(v/v) ethylene glycol as a cryoprotectant and was stored in a nylon cryoloop under liquid nitrogen.
All crystals were cooled in a stream of N2 gas (100 K) during data collection. X-ray diffraction data were collected by the oscillation method on the PX14.1 beamline (λ = 0.91841 Å) at the BESSY II synchrotron-radiation source, Helmholtz-Zentrum Berlin using a MAR Mosaic 225 CCD detector (Mueller et al., 2012 ▶).
2.5. Structure determination and refinement
Data processing and scaling were carried out using the HKL-2000 software package (Otwinowski & Minor, 1997 ▶). The data were processed to a resolution limit of 1.8 Å (R merge = 0.088) in space group C2, with unit-cell parameters a = 134.8, b = 49.2, c = 97.2 Å, β = 125.0° and unit-cell volume 524 459 Å3. The phase problem was solved by molecular replacement using MOLREP (Vagin & Teplyakov, 2010 ▶) in the CCP4 suite (Winn et al., 2011 ▶) with one chain of aOPAA-JD6.5 (PDB entry 3l24; space group I222; resolution 2.3 Å; Vyas et al., 2010 ▶) as a template. One monomer was located in the asymmetric unit with an initial R factor of 0.36. Computation of the Matthews coefficient suggested that the asymmetric unit contains one monomer, with a crystal solvent content of 52.5%.
Subsequent refinement involved iterative cycles of model building in Coot (Emsley & Cowtan, 2004 ▶) according to F o − F c and 2F o − F c difference electron-density maps and restrained refinement in REFMAC5 v. 5.6.0111 (Murshudov et al., 2011 ▶) in CCP4 (Winn et al., 2011 ▶) without any I/σ(I) cutoff for the reflections used. The process of structure building and refinement was monitored using the R free statistic calculated for 5% of reflections.
Water molecules were automatically added by REFMAC5 (Murshudov et al., 2011 ▶) in peaks of the 2F o − F c Fourier with a minimum level of 2σ and with distances in the range 2.2–3.5 Å from a hydrogen-bond donor or acceptor.
Analysis and validation of the structure were carried out with the assistance of the program PROCHECK (Laskowski et al., 1993 ▶). 99.8% of the amOPAA residues lie in the allowed or preferred regions of the Ramachandran plot and one residue is in the disallowed region but is supported by a very good fit to the electron density (see §3).
The amOPAA coordinates and structure factors have been deposited in the PDB (Berman et al., 2000 ▶) as entry 3rva.
The figures were created with PyMOL (DeLano, 2002 ▶) and CCP4mg (McNicholas et al., 2011 ▶) and the sequence alignment was created with ClustalW (Thompson et al., 1994 ▶) and ESPript (Gouet et al., 1999 ▶). Data-collection and refinement statistics are given in Table 1 ▶.
Table 1. Data and structure statistics for the structure of OPAA from A. macleodii .
Values in parentheses are for the highest resolution shell.
| X-ray source and beamline | BESSY II, PX14.1 |
| X-ray beam shape and size | Elliptical, major axis 70 µm |
| Oscillation range ω (°) | 180 |
| Δω (°) | 0.5 |
| Resolution range (Å) | 40.0–1.8 (1.83–1.80) |
| Crystal system | Monoclinic |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 134.8, b = 49.2, c = 97.3, α = γ = 90, β = 125.0 |
| 〈I/σ(I)〉 | 16.5 (2.0) |
| R merge † | 0.088 (0.698) |
| Data completeness (%) | 100 (100) |
| Average multiplicity | 4.4 (4.4) |
| No. of measured unique reflections | 48066 (2405) |
| Criterion for observed reflection | I/σ(I) > −3.7 |
| No. of observed reflections | 48066 (2405) |
| No. of observed reflections used in refinement | 47975 |
| No. of non-H atoms | 8670 |
| No. of monomers per asymmetric unit | 1 |
| No. of molecules in unit cell (Z) | 4 |
| Matthews coefficient V M (Å3 Da−1) | 2.5 |
| Localized residues | 2–445 + 1 His |
| No. of water molecules | 464 |
| Other solvent molecules | 1 PO4 3− |
| Localized metal ions | 2 Mn2+, 1 Ni2+ |
| Average ADP (Å2) | |
| All atoms | 23.8 |
| Protein | 22.5 |
| Water | 33.8 |
| Ligands/ions | 58.6 |
| R work ‡ | 0.1573 |
| R free ‡ | 0.2047 |
| No. of reflections for R free | 2371 [5%] |
| R.m.s.d. from ideal, bond lengths (Å) | 0.013 |
| R.m.s.d. from ideal, bond angles (°) | 1.323 |
R
merge = 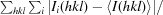
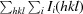 , where I
i(hkl) is the intensity of individual observations of reflections.
, where I
i(hkl) is the intensity of individual observations of reflections.
R
work = 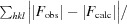
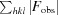 , where F
obs and F
calc are the observed and calculated structure factors, respectively, and the statistic is calculated for all reflections except for the test set. R
free is calculated accordingly for reflections excluded from refinement (the test set).
, where F
obs and F
calc are the observed and calculated structure factors, respectively, and the statistic is calculated for all reflections except for the test set. R
free is calculated accordingly for reflections excluded from refinement (the test set).
3. Results and discussion
3.1. Structure of OPAA from A. macleodii
Wild-type amOPAA modified by a six-histidine tag at the C-terminus was expressed in E. coli. The enzyme crystallized in 0.056 M NaH2PO4, 1.344 M K2HPO4 pH 8.2 at 291 K in the form of plates shredded at one side. The Hampton Research Additive Screen (Trakhanov & Quiocho, 1995 ▶) was tested for its influence on the shape and size of the crystals. The addition of 5 mM barium chloride dehydrate, cadmium chloride hydrate, calcium chloride dehydrate, cobalt(II) chloride hexahydrate, copper(II) chloride dehydrate and magnesium chloride hexahydrate improved the shape and quality of the crystals: larger thick single crystals were grown. The structure-solution process started with attempts to solve the phase problem by multiple-wavelength anomalous dispersion (MAD) of the natively present manganese ions. Even when the presence of manganese anomalous signal in the MAD data set was confirmed, it was not sufficient for successful structure determination. In the course of further phasing efforts, the structure of aOPAA-JD6.5 (Vyas et al., 2010 ▶) became available. The structure of amOPAA was determined by the molecular-replacement method using aOPAA-JD6.5 (PDB entry 3l24; Vyas et al., 2010 ▶) as the model. The refined model contained residues 2–445. The asymmetric unit contained one monomer, whereas the functional enzyme is composed of two chains. The existence of dimers was also confirmed in solution by dynamic light scattering and size-exclusion chromatography experiments.
3.1.1. Monomer of amOPAA
The amOPAA monomer is composed of two domains (Fig. 2 ▶). The smaller N-terminal domain is composed of residues 1–160 and is also called the N-domain. The larger C-terminal domain or C-domain comprises residues 161–445. The arrangement of the domains resembles the letter V, with the chain ‘bent’ between the two domains. The overall fold of amOPAA is the same as in the case of aOPAA-JD6.5 (PDB entries 3l24 and 3l7g; Vyas et al., 2010 ▶). The N-domain is composed of four α-helices (α1, residues 4–26; α2, 54–57; α3, 133–139; α4, 147–157), two 310-helices (η1, residues 101–103; η2, 112–118) and five β-strands (β1, residues 30–34; β2, 69–72; β3, 79–83; β4, 105–109; β5, 126–129). Interestingly, four of the β-strands run parallel to each other, with only the middle β2 being antiparallel. The C-domain is composed of nine α-helices (α5, residues 162–187; α6, 192–203; α7, 268–288; α8, 295–312; α9, 320–325; α10, 329–332; α11, 391–398; α12, 408–414; α13, 436–439), two 310-helices (η3, residues 220–223; η4, 401–405) and six β-strands (β6, residues 215–218; β7, 240–249; β8, 252–261; β9, 380–388; β10, 419–428; β11, 433–435). In contrast to the N-domain β-sheet, all of the β-strands in the C-domain are antiparallel and form the ‘core’ of the C-domain surrounded by the α-helical scaffold. The β-strands are long and strongly curved, evoking pita bread (as first described for eMetAP; Roderick & Matthews, 1993 ▶). The four longest α-helices run parallel to the β-strands and most of the other shorter helices form a cluster.
Figure 2.

Structure of the monomer of organophosphorus acid anhydrolase from A. macleodii. The monomer is coloured by secondary structure: α-helices, cyan; 310-helices, blue; β-strands, purple; Mn2+ ions, light green.
The position of the first residue (Met1) is slightly uncertain owing to unclear electron density. The rest of the 444 residues are well supported by electron density (Fig. 3 ▶); additionally, one histidine of the C-terminal His tag can be localized. All residues were modelled in the trans conformation. Ten residues contain atoms modelled in alternative positions. Owing to radiation damage, several side-chain carboxyl groups were observed to be partially decarboxylated and were modelled with an occupancy of 0.5 for the terminal groups.
Figure 3.

The active site of OPAA from A. macleodii. The binuclear metal centre and the coordinating residues are shown as spheres and sticks colour-coded by atom type; the contours of the 2F o − F c Fourier are at the 1σ level.
3.1.2. Active site
The active site is located in an oval pocket in the β-sheet part of the C-domain. The active site is built around the manganese-binding residues Asp244, Asp255, His339, Glu384 and Glu423. Two metal ions are coordinated by these residues and their presence was confirmed by anomalous difference peaks (peak heights of 5.5σ and 11.1σ for MN1 and MN2, respectively).
At the time of the first MAD experiment, an X-ray fluorescence spectrum was measured from a crystal on a synchrotron beamline (PX14.1 at BESSY II; E = 13.5 keV) and a strong nonbackground peak corresponded to the Mn Kα1,2 line. After the structure of the enzyme had been solved by molecular replacement, the structure in a different space group originally used to obtain the MAD data set was refined. The two strongest maxima of the Fourier calculated for anomalous differences at the peak incident energy (1.894 Å) belonged to these metal ions (22σ and 6σ levels for the MN1 and MN2 sites, respectively). Based on the X-ray fluorescence data and the presence of anomalous signal in all cases, we concluded that two Mn2+ ions were present in the active site of amOPAA, one with decreased occupancy. MN1 was refined with an occupancy of 0.7 according to the anomalous contribution as implemented in REFMAC5 (Murshudov et al., 2011 ▶). MN2 was refined with full occupancy. The distance between MN1 and MN2 is 3.28 Å.
MN1 is coordinated in a slightly distorted octahedron by six atoms: four protein O atoms (from Asp244, Asp255 and Glu423) and two water molecules (460 and 857). Its coordination closely resembles those observed in other homologous enzymes; for example, aOPAA-JD6.5 (Vyas et al., 2010 ▶). MN2 is coordinated only by five atoms, four of which come from protein and form a distorted coordination square (Asp255, His339, Glu384 and Glu423). The fifth ligand is water 460, which bridges the two Mn2+ ions. A similar situation is found in homologous enzymes, making the fifth position for a ligand or activated water a strong binding site. The details of the coordination of the manganese ions are summarized in Table 2 ▶ and are illustrated in Figs. 3 ▶ and 4 ▶.
Table 2. Coordination of the two manganese ions by residues and water in the active site with indicated interatomic distances in Å.
| Residue | Atom | MN1 | MN2 |
|---|---|---|---|
| Asp244 | Oδ1 | 2.4 | — |
| Oδ2 | 2.4 | — | |
| Asp255 | Oδ1 | 2.2 | — |
| Oδ2 | — | 2.2 | |
| Glu423 | O∊1 | 2.3 | — |
| O∊2 | — | 2.0 | |
| His339 | N∊2 | — | 2.2 |
| Glu384 | O∊2 | — | 2.6 |
| W460 | O | 2.3 | 2.2 |
| W857 | O | 2.5 | — |
Figure 4.

A detailed view of the active site. The binuclear metal centre coordinating residues are colour-coded by atom type (yellow C atoms); residues proposed to be involved in substrate binding are colour-coded by atom type (grey C atoms), Mn2+ ions are shown in light green. Contact distances are shown in Å for the manganese cluster.
Significant nonproteinaceous electron density was observed in the active site apart from the metal sites. This density did not allow any reasonable ligands or constituents of the used solutions to be fitted and therefore several water molecules were modelled to satisfy the hydrogen-bonding interaction criteria. Water 460 bridging the metal ions (distances of 2.3 and 2.2 Å) is in the position of the expected nucleophile necessary for catalysis in eMetAP (Lowther & Matthews, 2000 ▶). Water molecules 460 and 459 were built in close contact with an occupancy of 0.5 to partially model the unidentified ligand in the active site.
The substrate-binding site resembles a deep pocket and is determined by nine residues as summarized in Table 3 ▶ and Fig. 4 ▶. The C-terminal end of the pocket (from the point of view of a peptide substrate) makes interactions with a loop (finger) of the second molecule (residues 36–54). Therefore, enzyme dimerization is very likely to influence its activity.
Table 3. Comparison of the amino-acid residues involved in ligand binding in amOPAA and human prolidase.
Prediction of the residues involved in contacts with organophosphate substrates is based on a superposition of the active sites of amOPAA and of aOPAA-JD6.5 with mipafox. Residues involved in Mn2+ coordination are determined from the X-ray structures of amOPAA and human prolidase. The roles of residues in peptide substrate binding are predicted based on superpositions of the structures of amOPAA, E. coli aminopeptidase (e.g. PDB entry 2v3x; Graham & Guss, 2008 ▶) and human prolidase. For a peptide substrate of type Xaa-Pro the interacting part and the prolidase subpocket are presented.
| amOPAA | Organophosphate substrate | Mn2+ sites | Peptide substrate | Human prolidase |
|---|---|---|---|---|
| Tyr212 | CH3 (C2) | Xaa, S1 | Tyr242 | |
| His226 | Pro, S1′ | His256 | ||
| Arg370 | Pro, S1′ | Arg399 | ||
| Asp244 | NH | MN1 | Asp277 | |
| Asp255 | MN1 and MN2 | Asp288 | ||
| Glu423 | MN1 and MN2 | Glu45 | ||
| Glu384 | NH | MN2 and activated water ligand | Glu413 | |
| His339 | MN2 | His371 | ||
| His346 | Phosphate O2 | Xaa, S1 | His378 | |
| Val345 | CH3 (C2) | Xaa, S1 | Val377 | |
| Arg421 | CH3 (C5) | Pro, S1′ | Arg451 | |
| His335 | CH3 (C6) | His367 |
3.1.3. Dimer of amOPAA
The overall shape of the dimer is shown in Fig. 5 ▶ viewed along the crystallographic twofold axis. The dimer arrangement resembles a handshake of two hands without thumbs. The N-domains in Fig. 5 ▶ are closer to the observer and the C-domains are in the background. The chains interact in two main areas: via the bridging loops in the small N-domains (residues 36–54 and 82–99) and by contacts between residues in the α6 α-helices. The contacts are realised as protein–protein hydrogen bonds, solvent-mediated hydrogen bonds or hydrophobic interactions. According to the PISA server (Krissinel & Henrick, 2007 ▶), 64 residues of each monomer participate in the contacts and 10.8% of the solvent-accessible surface area (2196 Å2) of each monomer is buried in the dimerization interfaces. The bridging loops of the N-domain project towards the other monomer and penetrate deep into the opening of the V shape. The longer loop (residues 36–54) of monomer A practically fills the ‘valley’ of monomer B. Both loops reach the surroundings of the active site of the second monomer and thus are very likely to participate in the full completion of the functional state of the enzyme.
Figure 5.

The dimer of OPAA from A. macleodii consists of two chains denoted A (coloured chain) and B (grey chain). The N-domains are highlighted by saturated colours (the N-domain of monomer A is in red and the N-domain of chain B is in black) and the C-domains are shown in light colours (the C-domain of monomer A is in orange and the C-domain of monomer B is in grey). The two fingers responsible for dimerization are coloured magenta in monomer A. Manganese ions are shown as spheres in both active sites. Chain B is also represented by a semitransparent surface.
3.1.4. Localized solvent
Apart from the two expected manganese ions (MN1 and MN2) in the active site, a nickel ion was most likely found binding to the N-terminus. The presence of a metal was confirmed by an intensive peak in the anomalous difference Fourier (9.1σ) and by a strong 2F o − F c maximum. The metal appeared to be coordinated by four N atoms of surrounding amino acids with short contact distances typical of metals of the fourth period (Fig. 6 ▶). Nickel and cobalt were the only metals of such type which were in contact with the enzyme at significant concentrations to our knowledge; nickel during the affinity-chromatography purification step and cobalt during crystallization. Nevertheless, specific searches for known cases of metal–protein interactions in the MESPEUS database (Hsin et al., 2008 ▶) showed that such coordination and distances to N atoms in proteins have to date never been observed for Co2+ ions. However, 11 structures available from the PDB contained a total of 55 cases with nickel ions in a very similar situation to ours (PDB entries 1jvn, 1ox4, 1ox5, 1ox6, 1q0f, 1q0g, 1q0k, 1q0m, 1hbk, 1oao and 1xmk). In several cases an almost identical situation (square-planar coordination, repeating set of distances) occurred and the element was assigned as Ni2+. Chaudhuri et al. (2001 ▶) confirmed the presence of nickel in PDB entry 1jvn in such a coordination by X-ray fluorescence. The metal-binding site in our structure was therefore modelled as an Ni2+ ion with 100% occupancy.
Figure 6.

The nickel-binding site of OPAA from A. macleodii. The metal centre and the coordinating residues are shown as a sphere and as sticks colour-coded by atom type, respectively. Contours of the 2F o − F c Fourier are shown at 1σ (cyan) and 6σ (coral) levels.
The structure contains 464 localized water molecules and one phosphate anion as other solvent molecules.
3.2. Dimeric form of amOPAA in solution
The protein was stable in solution throughout all experiments and no aggregation was observed. According to the DLS results, the enzyme in solution at a concentration of 1.3 mg ml−1 in 50 mM NaH2PO4/Na2HPO4 buffer pH 7.5 is monodisperse, with an average particle size of 7.9 nm.
This measured value was compared with the calculated diameters for globular proteins of molecular weight 50 623 Da (amOPAA monomer) and 101 246 Da (dimer) and it was suggested that the measured value corresponds very well to the value for a dimer (8.03 nm). Calculations were performed with the program HYDROPRO v.c7 (García de la Torre et al., 2000 ▶).
The oligomerization state of amOPAA was also confirmed by size-exclusion chromatography. The position of the peak in the chromatogram corresponded to a molecular mass of ∼101 kDa, which is in excellent agreement with the expected value for an amOPAA dimer (2 × 50.6 = 101.2 kDa).
3.3. A. macleodii OPAA and other OPAA enzymes
Structures of both of the two types of OPAA are known: squid-type OPAA (e.g. PDB entry 1pjx; Koepke et al., 2003 ▶) and Mazur-type OPAA (PDB entry 3l24; Vyas et al., 2010 ▶). amOPAA and the published squid-type OPAAs are completely dissimilar in both structure and sequence.
The sequences of amOPAA and of previously published aOPAA-JD6.5 (Vyas et al., 2010 ▶) show 61.7% identity. The surface residues vary more significantly, while the residues that are essential for enzymatic function are completely conserved. Comparison of the structures of amOPAA and aOPAA-JD6.5 (Vyas et al., 2010 ▶) shows a high level of similarity, with an overall r.m.s.d. of 0.8 Å for 421 aligned Cα atoms. Monomers of the two enzymes share overall topology and shape. They both consist of a small N-domain and a larger C-domain with a pita-bread fold. However, the currently presented structure provides the first complete description of the three-dimensional structure of this type of OPAA. Part of the protein chain in aOPAA-JD6.5 in the residue range 346–368 is missing from the model (PDB entries 3l24 and 3l7g; Vyas et al., 2010 ▶); recalculated electron density based on the deposited data and coordinates does not allow the localization of any of the missing amino acids. In the reported structure of amOPAA this region is very well defined and all amino acids were modelled without ambiguity. Residues Phe368 and Arg370 of this part of the protein chain lie very close to the active site and participate in formation of the substrate-binding pocket, especially Arg370. This residue is in direct contact with the water network in the active site and its guanidinium group stabilizes the side chain of His346 by a stacking interaction. His346 plays an important role in ligand binding in the active site, as observed for example in the complex with mipafox (Vyas et al., 2010 ▶). According to comparison of the active site of amOPAA and enzymes with prolidase and aminopeptidase activity (AMPP and prolidase), the same binding pattern of peptide substrates can be expected and therefore it can be assumed that the Arg370 side chain is also likely to participate in contacts with the C-terminal carboxylate group.
Other structural differences follow from sequence variation. There is an insertion in amOPAA with respect to aOPAA-JD6.5 (Vyas et al., 2010 ▶; residues 265–267 in amOPAA numbering). The segment between Phe262 and Phe268 of aOPAA-JD6.5 is shorter by about three residues in amOPAA and is structured differently.
Surprisingly, our data are in clear contrast to previously published results on the oligomerization state of aOPAA-JD6.5. In the original paper the enzyme was presented as a monomer in solution and three independent molecules were present in the asymmetric unit of the crystal. However, chains generated by crystal symmetry complete dimers of aOPAA-JD6.5 which are very similar to the dimers of amOPAA. Originally, the authors of the publication did not notice the formation of aOPAA-JD6.5 dimers. In additional errata to the original work the dimers were mentioned and the occurrence of tetramers of aOPAA-JD6.5 in solution (Vyas et al., 2010 ▶) was referred to as an experimentally confirmed fact. Apart from the enzyme dimerization, there is no other significant similarity in the packing of the molecules in the crystals of amOPAA and aOPAA-JD6.5.
According to our analysis, the amOPAA enzyme observed in the crystal structure naturally forms dimers (see the discussion of dimerization contacts via extended loops in §3.1.3). The existence of dimers of amOPAA was also confirmed in this work by two independent methods. From our results, it follows that the formation of dimers is most likely to be a prerequisite for enzymatic function, as the two bridging loops of the small N-domain are not only important in dimerization; both of them are also directly involved in contacts with the residues that make up the substrate-binding pocket of the second protein chain.
In comparison to the aOPAA-JD6.5 structure the architecture of the active site is entirely homologous and the types of residues surrounding the active site are preserved in both enzymes. The Mn2+ cluster coordination and the MN1–MN2 distances are also similar (3.27 and 3.26 Å in amOPAA and aOPAA-JD6.5, respectively).
3.4. Structural comparison with related peptidases
The amOPAA enzyme was compared with other groups of enzymes with a pita-bread fold (MetAMP, AMPP and prolidases). All of them are metal-dependent and have a bimetallic cluster located in the active site. Despite the different activities and substrate specificities, the amino acids coordinating the binuclear metal centre are conserved in all four enzyme groups.
3.4.1. Structural comparison with prolidases
As amOPAA can hydrolyze the same substrates as prolidases and there are indications that prolidases hydrolyze several types of OPs (Park et al., 2004 ▶), a closer comparison of these two types of enzymes is necessary. Prolidase is a ubiquitous enzyme in mammals (Endo et al., 1989 ▶), bacteria (Park et al., 2004 ▶) and archaea (Ghosh et al., 1998 ▶; Maher et al., 2004 ▶); in humans, prolidase is responsible for the final step of the degradation of endogenous and dietary proteins and is involved in collagen catabolism. Its deficiency causes an autosomal recessive disorder characterized by skin lesions, mental retardation and recurrent infections. The natural role of this enzyme in bacteria and archaea is still unclear (Maher et al., 2004 ▶). The PDB contains seven structures of prolidases. Here, we compare all of them (bacterial, archeal and human prolidases) with amOPAA.
3.4.2. Structural comparison with bacterial prolidase
Only the N-terminal domain has been deposited in the PDB in the cases of the prolidases from Streptococcus pyogenes M1 GAS (PDB entries 3ovk and 3o5v; Midwest Center for Structural Genomics, unpublished work) and S. pneumoniae (PDB entry 3pn9; Midwest Center for Structural Genomics, unpublished work). In contrast to amOPAA, these N-domains lack the extended loops.
The structure of the prolidase enzyme from Bacillus anthracis is very similar to amOPAA. The N-domain of B. anthracis prolidase has no extended loops, in contrast to amOPAA. Also, the C-domain of B. anthracis prolidase is shorter compared with amOPAA, but the pita-bread fold is retained. The types and architecture of the amino-acid residues coordinating the manganese ions are also conserved. However, parts of these two enzymes that are more distant from the binuclear metal cluster differ. In the case of amOPAA, Arg370 and the corresponding part of the protein chain over the active site are present and are likely to be involved in substrate binding. Prolidase from B. anthracis entirely lacks this part of the protein in this region. Therefore, a wide opening is formed over the active site in this case.
As discussed above, it is not possible to compare this active-site entrance between amOPAA and aOPAA-JD6.5 (published as a ‘bacterial prolidase’ as well as the Mazur-type OPAA) owing to this missing part of the protein chain in aOPAA-JD6.5.
3.4.3. Structural comparison with archeal prolidase
Both crystal structures of prolidases from archaea, those from Pyrococcus furiosus (PDB entry 1pv9; Maher et al., 2004 ▶) and P. horikoshii OT3 (PDB entry 1wy2; RIKEN Structural Genomics/Proteomics Initiative, unpublished work), are very similar to amOPAA, with the exception of the extended loops in the N-terminal domain which are missing in the archaeal prolidases. The bimetallic cluster is coordinated in the same way in all cases. The active-site entrance (‘above the site’) differs slightly: Arg370 in amOPAA discussed above has an equivalent in the archaeal prolidase (Arg295 in the 1pv9 numbering) but the position of the side chain of this residue is somewhat different and also the whole relevant part of the main chain runs differently.
The sequence identity between amOPAA and the enzyme from P. horikoshii (PDB entry 1wy2) is 24% and that between amOPAA and the enzyme from P. furiosus (PDB entry 1pv9) is 23%.
3.4.4. Structural comparison with human prolidase
Overall, human prolidase and amOPAA are very similar. Interesting differences can be found in the regions of loops 148–161 and 264–257 (human prolidase numbering), which run differently in human prolidase in the close vicinity of the active site and cause some important structural differences in the surroundings of the active sites when the two dimers are compared (Fig. 7 ▶). The active sites of these enzymes can be approached via two openings. As the dimer is assembled an arc is formed by the second molecule above the active site (loop 82–99 in amOPAA), under which a second narrower tunnel occurs. Ligands can access the active site via a widely open tunnel along His226 (256 in human prolidase) and Arg370 (399) or through this narrower tunnel along Gln229 and Thr213. Relatively large compounds including dipeptides and small organophosphates could certainly access the active site via this smaller opening in amOPAA. In contrast, in human prolidase the chain Gly258–Asn264 protrudes into this channel, making it much tighter and making passage of the assumed ligands likely to be impossible (a cross-section of the tunnels is shown in Supplementary Fig. S11). Also, the presence of the 149–160 loop in human prolidase at the main route into the active site as well as partial occlusion of the opening by Glu54 makes the only access route to the active site that is left more restricted when compared with the bacterial enzyme. The bacterial enzyme is probably more versatile and accessible to various types of ligands, with two possible routes of entry or release, while the human enzyme has limited access to the substrate-binding pocket, possibly to enhance its specificity. These differences in ligand ‘logistics’ may have consequences for enzymatic rates and substrate affinities.
Figure 7.

Comparison of the surroundings of the active sites of amOPAA and human prolidase. Protein loops are shown as coils coloured according to the protein chain of the dimer and the organism: light and dark blue, amOPAA chains A and B, respectively; yellow and orange, the corresponding chains in human prolidase. The two longest loops form the ‘fingers’ contributing to dimer formation. Ligand access to the active site through the tunnel between the molecules of human prolidase is more restricted than in amOPAA (see §3.4.4 for details). The active-site residues of amOPAA are represented as sticks with colour-coding according to atom type; Mn2+ ions are shown as green spheres.
4. Conclusions
The reported structure of OPAA from the marine bacterium A. macleodii clearly identifies organophosphorous acid anhydrolases with dipeptidases. It also completes the details of the active site, as several amino-acid residues had not been localized previously in the structure of aOPAA-JD6.5. Arg370 of this enzyme part is suggested to play an important role in ligand binding. The ligand-binding site is largely formed owing to enzyme dimerization. amOPAA dimerization was confirmed in the crystal and in solution. Loops 36–54 and 82–99 participate in completion of the environment around the active site. Therefore, we conclude that the dimer is the biologically relevant oligomeric form of amOPAA and that interference with dimerization would significantly affect its activity towards both dipeptides and organophosphates.
Human prolidase has been tested positive for activity towards nerve gases in at least one study (Wang et al., 2006 ▶). Given the structural results for the complex of aOPAA-JD6.5 with mipafox, the amino-acid residues critical for decontamination of fluorine-containing OPs are also conserved in amOPAA and human prolidase. It should be concluded that apart from differences in the close vicinity of the active site the two enzymes represent a variation on the same topic: one in mammals and the other in bacteria. If proline recycling remains the key reason for the production of this type of enzyme in both humans and bacteria then their shared ability to degrade OPs is a consequence of the conserved construction of the active sites, even though the sequence identity between these two representatives is only 24%.
Supplementary Material
PDB reference: organophosphorus acid anhydrolase, 3rva
Supplementary material file. DOI: 10.1107/S1744309113002674/hv5230sup1.pdf
Acknowledgments
Support from the Czech Science Foundation (project No. 310/09/1407 and 305/07/1073 grant), the European Commission (project No. 226716; user access for projects 2010.2.100364 and 2010.2.100242) and the Ministry of Education, Youth and Sports of the Czech Republic (grant No. CZ.1.07/2.3.00/30.0029) is gratefully acknowledged. The authors wish to thank Dr U. Müller of Helmholtz-Zentrum Berlin for support at the beamline.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: HV5230).
References
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed]
- Chaudhuri, B. N., Lange, S. C., Myers, R. S., Chittur, S. V., Davisson, V. J. & Smith, J. L. (2001). Structure, 9, 987–997. [PubMed]
- Cheng, T., Liu, L., Wang, B., Wu, J., DeFrank, J. J., Anderson, D. M., Rastogi, V. K. & Hamilton, A. B. (1997). J. Ind. Microbiol. Biotechnol. 18, 49–55. [DOI] [PubMed]
- Cudney, R., Patel, S., Weisgraber, K., Newhouse, Y. & McPherson, A. (1994). Acta Cryst. D50, 414–423. [DOI] [PubMed]
- D’Arcy, A., Mac Sweeney, A., Stihle, M. & Haber, A. (2003). Acta Cryst. D59, 396–399. [DOI] [PubMed]
- DeFrank, J. J. (1991). Applications of Enzyme Biotechnology, edited by J. W. Kelly & T. O. Baldwin, pp. 165–188. New York: Plenum Press.
- DeFrank, J. J. & Cheng, T.-C. (1991). J. Bacteriol. 173, 1938–1943. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Endo, F., Tanoue, A., Nakai, H., Hata, A., Indo, Y., Titani, K. & Matsuda, I. (1989). J. Biol. Chem. 264, 4476–4481. [PubMed]
- García de la Torre, J., Huertas, M. L. & Carrasco, B. (2000). Biophys. J. 78, 719–730. [DOI] [PMC free article] [PubMed]
- Ghosh, M., Grunden, A. M., Dunn, D. M., Weiss, R. & Adams, M. W. W. (1998). J. Bacteriol. 180, 4781–4789. [DOI] [PMC free article] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Graham, S. C. & Guss, J. M. (2008). Arch. Biochem. Biophys. 469, 200–208. [DOI] [PubMed]
- Harel, M., Kryger, G., Rosenberry, T. L., Mallender, W. D., Lewis, T., Fletcher, R. J., Guss, J., Silman, I. & Sussman, J. L. (2000). Protein Sci. 9, 1063–1072. [DOI] [PMC free article] [PubMed]
- Herkenhoff, S., Szinicz, L., Rastogi, V. K., Cheng, T.-C., DeFrank, J. J. & Worek, F. (2004). Arch. Toxicol. 78, 338–343. [DOI] [PubMed]
- Hsin, K., Sheng, Y., Harding, M. M., Taylor, P. & Walkinshaw, M. D. (2008). J. Appl. Cryst. 41, 963–968.
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409–411.
- Koepke, J., Scharff, E. I., Lücke, C., Rüterjans, H. & Fritzsch, G. (2003). Acta Cryst. D59, 1744–1754. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lowther, W. T. & Matthews, B. W. (2000). Biochim. Biophys. Acta, 1477, 157–167. [DOI] [PubMed]
- Maher, M. J., Ghosh, M., Grunden, A. M., Menon, A. L., Adams, M. W., Freeman, H. C. & Guss, J. M. (2004). Biochemistry, 43, 2771–2783. [DOI] [PubMed]
- Mansee, A. H., Chen, W. & Mulchandani, A. (2005). J. Ind. Microbiol. Biotechnol. 32, 554–560. [DOI] [PubMed]
- Mazur, A. (1946). J. Biol. Chem. 164, 271–289. [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- McPherson, A. (2001). Protein Sci. 10, 418–422. [DOI] [PMC free article] [PubMed]
- Mueller, U., Darowski, N., Fuchs, M. R., Förster, R., Hellmig, M., Paithankar, K. S., Pühringer, S., Steffien, M., Zocher, G. & Weiss, M. S. (2012). J. Synchrotron Rad. 19, 442–449. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Park, M. S., Hill, C. M., Li, Y., Hardy, R. K., Khanna, H., Khang, Y.-H. & Raushel, F. M. (2004). Arch. Biochem. Biophys. 429, 224–230. [DOI] [PubMed]
- Pitcher, D., Saunders, N. & Owen, R. (1989). Lett. Appl. Microbiol. 8, 151–156.
- Roderick, S. L. & Matthews, B. W. (1993). Biochemistry, 32, 3907–3912. [DOI] [PubMed]
- Scharff, E. I., Koepke, J., Fritzsch, G., Lücke, C. & Rüterjans, H. (2001). Structure, 9, 493–502. [DOI] [PubMed]
- Skálová, T., Dušková, J., Hašek, J., Kolenko, P., Štěpánková, A. & Dohnálek, J. (2010). J. Appl. Cryst. 43, 737–742.
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Trakhanov, S. & Quiocho, F. A. (1995). Protein Sci. 4, 1914–1919. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Vyas, N. K., Nickitenko, A., Rastogi, V. K., Shah, S. S. & Quiocho, F. A. (2010). Biochemistry, 49, 547–559. [DOI] [PubMed]
- Wang, S. H., Zhi, Q. W. & Sun, M. J. (2006). Toxicol. In Vitro, 20, 71–77. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zheng, J., Constantine, C. A., Zhao, L., Rastogi, V. K., Cheng, T.-C., DeFrank, J. J. & Leblanc, R. M. (2005). Biomacromolecules, 6, 1555–1560. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: organophosphorus acid anhydrolase, 3rva
Supplementary material file. DOI: 10.1107/S1744309113002674/hv5230sup1.pdf