

Proliferating cell nuclear antigen (PCNA) plays essential roles in DNA replication, DNA repair, cell-cycle regulation and chromatin metabolism. The PCNA from Drosophila melanogaster (DmPCNA) has been purified and crystallized.
Keywords: PCNA, homotrimer, DNA replication and repair, cell-cycle regulation
Abstract
Proliferating cell nuclear antigen (PCNA) plays essential roles in DNA replication, DNA repair, cell-cycle regulation and chromatin metabolism. The PCNA from Drosophila melanogaster (DmPCNA) was purified and crystallized. The crystal of DmPCNA diffracted to 2.0 Å resolution and belonged to space group H3, with unit-cell parameters a = b = 151.16, c = 38.28 Å. The structure of DmPCNA was determined by molecular replacement. DmPCNA forms a symmetric homotrimer in a head-to-tail manner. An interdomain connector loop (IDCL) links the N- and C-terminal domains. Additionally, the N-terminal and C-terminal domains contact each other through hydrophobic associations. Compared with human PCNA, the IDCL of DmPCNA has conformational changes, which may explain their difference in function. This work provides a structural basis for further functional and evolutionary studies of PCNA.
1. Introduction
Proliferating cellular nuclear antigen (PCNA) was initially identified by Miyachi and coworkers as an autoantigen in the serum of patients with systemic lupus erythematosus (Kelman, 1997 ▶; Miyachi et al., 1978 ▶; Strzalka & Ziemienowicz, 2011 ▶). PCNA was subsequently identified universally in the proliferating cells of a variety of organisms. PCNA is a member of the sliding-clamp family (β clamps; Krishna, Kong et al., 1994 ▶). The functions of PCNA are conserved in different species and are involved in many cellular aspects such as DNA replication, DNA repair, cell-cycle control, chromatin assembly and remodelling, sister chromatin cohesion and survival (Moldovan et al., 2007 ▶; Strzalka & Ziemienowicz, 2011 ▶).
In the process of DNA replication in eukaryotic cells, replication factor C (RFC) loads PCNA to the primase–polymerase α (Polα) complex (Bowman et al., 2004 ▶). PCNA acts as a processivity factor for DNA polymerases δ and ∊ (Polδ and Pol∊; Moldovan et al., 2007 ▶). As a sliding clamp, PCNA functions by forming a trimeric ring, with homotrimers in eukaryotes and T4 bacteriophage and heterotrimers in archaea. Duplex DNA can cross and slide freely through PCNA in both directions. PCNA provides a docking station for association of many other proteins, such as p21, FEN-1, Cdt1, Rad6, Rad18, EXO1 and so on, which are involved in many different cellular processes (Arias & Walter, 2006 ▶; Bruning & Shamoo, 2004 ▶; Gulbis et al., 1996 ▶; Hoege et al., 2002 ▶; Nielsen et al., 2004 ▶; Watanabe et al., 2004 ▶). Partner proteins interact with PCNA via a conserved motif termed the PCNA-interacting protein box (PIP-box; Williams et al., 2006 ▶). The PIP-box anchors into a hydrophobic pocket under the PCNA interdomain connector loop (IDCL; Gulbis et al., 1996 ▶).
In Drosophila melanogaster, DmPCNA is encoded by mutagen-sensitive 209 (Mus209). Mutation of DmPCNA exhibits a complex pleiotropy of temperature-sensitive lethality, hypersensitivity to DNA-damaging agents, suppression of position-effect variegation and female sterility, suggesting that DmPCNA functions in DNA repair, chromatin assembly and modification (Henderson, 1999 ▶; Henderson et al., 1994 ▶, 2000 ▶). In vitro binding assays showed that the cyclin-dependent kinase inhibitor Dacapo, which is a Drosophila homologue of p21, and Pogo transposase interact with DmPCNA (Warbrick et al., 1998 ▶). However, the precise function and mechanism of DmPCNA has not been fully studied.
Here, we report the crystal structure of DmPCNA, which forms a symmetric homotrimer in a head-to-tail manner. The N-terminal and C-terminal domains are bridged by the IDCL and contact each other through hydrophobic associations.
2. Materials and methods
2.1. Gene cloning and expression
Full-length DmPCNA was obtained by PCR with a D. melanogaster whole cDNA template and was inserted into BamHI and XhoI sites in HT-pET28a, which was modified from pET28a (Novagen) and encodes an N-terminal 6×His tag. The recombinant plasmid (HT-pET28a-DmPCNA) was validated by sequencing.
The HT-pET28a-DmPCNA plasmid was transformed into Escherichia coli BL21(DE3) CodonPlus competent cells. The cells were incubated in 750 ml Terrific Broth (TB) medium with 30 µg ml−1 kanamycin at 310 K until the absorbance at 600 nm reached 1.0. The temperature was then lowered to 289 K, isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM and the culture was incubated for a further 18 h at 289 K.
2.2. Protein purification
The following procedures were carried out at 277 K. The E. coli cells were collected at 6300 rev min−1 for 8 min and suspended in five times the volume of lysis buffer consisting of 20 mM HEPES pH 7.5, 500 mM NaCl, 20 mM imidazole, 5% glycerol, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride (PMSF). The cells were then broken using a High Pressure Homogenizer (JNBIO) at 130 MPa. The debris was removed by centrifugation at 20 000g for 40 min at 277 K. The supernatant was mixed with pre-equilibrated Ni Sepharose (GE Healthcare) for 1 h and the beads were then washed with lysis buffer without PMSF. The proteins were eluted with 300 mM imidazole in lysis buffer. The sample was desalted to 20 mM HEPES pH 7.5, 250 mM NaCl, 5% glycerol, 1 mM DTT, 10 mM imidazole. The 6×His-tagged DmPCNA was digested with TEV protease. The 6×His tag and TEV protease were then removed using Ni Sepharose. The protein solution was concentrated using a 10 kDa cutoff Amicon Ultra-15 (Millipore) and was then applied onto a HiLoad 16/60 Superdex 75 column (GE Healthcare) equilibrated with 20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM DTT. The purity of the protein was monitored by SDS–PAGE (Figs. 1 ▶ a and 1 ▶ b). Purified DmPCNA was concentrated to 13 mg ml−1, aliquoted and stored at 193 K.
Figure 1.

(a) Elution profile of the purification of DmPCNA using a HiLoad 16/60 Superdex 75 column. (b) SDS–PAGE of the DmPCNA which was used in the crystallization experiment. The left lane contains molecular-mass markers (labelled in kDa). (c) Crystals of DmPCNA were grown from 0.1 M HEPES pH 7.5, 1.2 M sodium citrate tribasic dehydrate using sitting-drop vapour diffusion.
2.3. Crystallization
Crystallization trials were carried out at 289 K by the sitting-drop vapour-diffusion method. The 2 µl sitting drops consisted of 1 µl protein solution and 1 µl reservoir solution and were equilibrated against 100 µl reservoir solution. The crystals were optimized using sitting-drop and hanging-drop vapour diffusion and were grown in reservoir solution consisting of 0.1 M HEPES pH 7.5, 1.2 M sodium citrate tribasic dehydrate (Fig. 1 ▶ c). The crystals were soaked in a cryoprotection solution consisting of 0.1 M HEPES pH 7.5, 1.2 M sodium citrate tribasic dehydrate, 20% glycerol and flash-cooled in liquid nitrogen.
2.4. Data collection, structure determination and refinement
Diffraction data were collected on beamline BL17U at Shanghai Synchrotron Radiation Facility (SSRF) and processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). The structure of DmPCNA was solved by molecular replacement with the program Phaser (McCoy et al., 2007 ▶) in the CCP4 package (Winn et al., 2011 ▶) using human PCNA (hPCNA; PDB entry 1u7b; Bruning & Shamoo, 2004 ▶) as the search model. A solution with one DmPCNA molecule in the asymmetric unit was found. The structure was refined using phenix.refine (Afonine et al., 2005 ▶) and model building was performed in Coot (Emsley et al., 2010 ▶).
2.5. Structure deposition
The coordinate file and structure factors for the DmPCNA crystal structure have been deposited in the RCSB Protein Data Bank under accession code 4hk1.
3. Results and discussion
3.1. Determination of the DmPCNA structure
We purified full-length DmPCNA using nickel-affinity and gel-filtration chromatography (Figs. 1 ▶ a and 1 ▶ b). DmPCNA eluted as a trimer from the gel-filtration chromatography, which was consistent with its homologues from other species (Krishna, Fenyö et al., 1994 ▶). Crystallization trials were set up using the sitting-drop and hanging-drop vapour-diffusion methods. The best crystal was grown in a condition consisting of 0.1 M HEPES pH 7.5, 1.2 M sodium citrate tribasic dehydrate using sitting-drop vapour diffusion (Fig. 1 ▶ c). Diffraction data were obtained to a resolution of 2.0 Å and the crystal belonged to space group H3, with unit-cell parameters a = b = 151.16, c = 38.28 Å. The structure of DmPCNA was solved by molecular replacement using human PCNA (hPCNA; PDB entry 1u7b; Bruning & Shamoo, 2004 ▶) as a search model. Details of the refinement statistics are summarized in Table 1 ▶.
Table 1. Data-collection and refinement statistics for DmPCNA.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | H3 |
| Unit-cell parameters (Å) | a = b = 151.16, c = 38.28 |
| Wavelength (Å) | 0.97915 |
| Resolution range (Å) | 50.0–2.00 (2.03–2.00) |
| Total reflections | 20100 |
| Unique reflections | 1026 |
| Completeness (%) | 91.6 (95.5) |
| Multiplicity | 2.8 (2.9) |
| R merge † | 0.070 (0.282) |
| 〈I/σ(I)〉 | 28.9 (4.2) |
| Mosaicity (°) | 0.300 |
| Matthews coefficient (Å3 Da−1) | 3.94 |
| Refinement | |
| Resolution (Å) | 28.57–2.00 |
| No. of reflections | 19958 (1028) |
| R work/R free ‡ | 0.228 (0.278) |
| No. of atoms | |
| Protein | 1845 |
| Water | 116 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.006 |
| Bond angles (°) | 1.156 |
| Ramachandran favoured (%) | 95.63 |
| Ramachandran outliers (%) | 0.87 |
| Average B factors (Å2) | 34.35 |
R
merge = 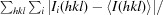
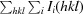 .
.
R
work = 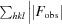 −
− 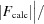
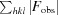 . R
free was computed identically except that all reflections belonged to a test set consisting of a randomly selected 5% of the data.
. R
free was computed identically except that all reflections belonged to a test set consisting of a randomly selected 5% of the data.
3.2. Overall structure of DmPCNA
DmPCNA and hPCNA are highly conserved in primary sequence, with 70% shared identity (Fig. 2 ▶). DmPCNA consists of an N-terminal domain (amino acids 1–118), a flexible IDCL (amino acids 119–135) and a C-terminal domain (amino acids 136–260). The DmPCNA monomer belongs to the α/β protein family (Fig. 3 ▶). The structure of DmPCNA contains four α-helices and a twisted β-sheet which is composed of 18 antiparallel β-strands. α-Helices α1 and α2 and β-strands β1–β9 constitute the N-terminal domain and α-helices α3 and α4 and β-strands β10–β18 make up the C-terminal domain. These two domains share a similar overall structure. DmPCNA forms a symmetric homotrimer in the crystal structure, which is consistent with the results of gel-filtration chromatography. The conformations of the three protomers are identical and form a ring-like structure. The diameter of the internal void of this ring is about 28 Å. In the process of eukaryotic DNA replication, DNA binds to and runs through this ring.
Figure 2.

Sequence alignment of DmPCNA and hPCNA was performed with ClustalW2 (Goujon et al., 2010 ▶; Larkin et al., 2007 ▶) and ESPript (Gouet et al., 1999 ▶). The secondary structure is shown according to the DmPCNA structure. Residues that are indentical in the two species are highlighted with a red background and highly conserved residues are coloured red; all of these residues are boxed.
Figure 3.

DmPCNA forms a symmetric homodimer. Three monomers of DmPCNA are shown in cartoon and surface representation and are coloured blue, green and pink.
3.3. Interface of the DmPCNA homotrimer
DmPCNA forms a homotrimer in a head-to-tail manner. The C-terminal domain of monomer A contacts the N-terminal domain of monomer B (Fig. 4 ▶). The buried area is up to 716 Å2. Strand β13 of monomer A forms an antiparallel β-sheet with strand β9 of monomer B. On the outside surface, residue Asn179 in strand β13 of monomer A interacts with Asp113 in strand β9 of monomer B by electrostatic interactions. Monomers A and B form a hydrophobic core in which Leu151, Phe154, Ala178 and Ile180 from helix α3 and strand β13 form hydrophobic interactions with Ile78 and Tyr114 from strand β13 of monomer B. Additionally, the side chains of Glu143 and Arg146 in helix α3 of monomer A interact with the side chain of Lys110 in strand β9 and the main chains of Lys80 and Ala82 in helix α2 and the adjacent loop by electrostatic interactions. The hydrophobic and electrostatic interactions maintain the ring-shaped conformation of DmPCNA.
Figure 4.

The hydrophobic and electrostatic interactions maintain the ring-shaped conformation of DmPCNA.
3.4. The N-terminal and C-terminal domains of DmPCNA are bridged by the IDCL and hydrophobic interactions
The IDCL of PCNA is important for binding PIP-box-containing proteins. In human PCNA, mutations of Val125, Leu126, Gly127 and Ile128 in the IDCL affect DNA synthesis by inhibiting the stimulation of Polδ and its binding to hPCNA (Zhang et al., 1998 ▶). These sites are also important for the binding of p21 and Fen1, implying that the loop is important for DNA replication and cell-cycle regulation (Jónsson et al., 1998 ▶). The N-terminal and C-terminal domains of DmPCNA are linked by the IDCL (Fig. 5 ▶ a). The residues in the tail of the IDCL are disordered owing to its flexibility. The binding of partners may stabilize the conformation of the IDCL. In particular, residues Leu126–Pro129 of IDCL are highly conserved, indicating their functional importance (Fig. 2 ▶). These residues are hydrophobic and contact Met40 in strand β3, Leu47 in strand β4, Val236 in strand β17 and Tyr250 in strand β18 by hydrophobic interactions.
Figure 5.

(a) The IDLC links the N- and C-terminal domains of DmPCNA. The IDCL is coloured cyan. (b) The N- and C-terminal domains of DmPCNA contact each other by hydrophobic interactions. The N- and C-terminal domains are coloured yellow and wheat, respectively.
An antiparallel β-sheet forms between strand β4 in the N-terminal domain and the last strand β18 in the C-terminal domain of DmPCNA (Fig. 5 ▶ b). Besides the IDCL, a wide hydrophobic network connects the N-terminal and C-terminal domains. Leu6 in strand β1, Ile11 and Ile15 in helix α1, Leu22 in the α1–β2 loop, Val48, Leu50 and Leu52 in strand β4 and Leu57 in the β4–β5 loop in the N-terminal domain interact with residues Phe215 and Leu221 in helix α4, Ile241 in strand β17 and Leu244, Ile247 and Tyr249 in strand β18 of the C-terminal domain. Therefore, combined with the analysis above, it appears that hydrophobic interactions play a dominant role during the folding of dPCNA into a ring-shaped structure.
3.5. Structural comparison of DmPCNA and hPCNA
Previously, structures of hPCNA have been determined by Gulbis et al. (1996 ▶) and Kontopidis et al. (2005 ▶). DmPCNA also forms a symmetric trimer. The overall structures of DmPCNA and hPCNA (PDB entry 1vym; Kontopidis et al., 2005 ▶) are highly similar (Fig. 6 ▶), with an r.m.s.d. value of 0.534 Å on superimposing the main-chain atoms of the two monomers. However, the IDCL of DmPCNA shows conformational changes compared with hPCNA. Although most of the amino acids in this loop are highly conserved between DmPCNA and hPCNA (Fig. 2 ▶), there are certain variations in the sequences of the IDCLs, with residues Asn120, Gln123, His125, Thr131, Asp132 and Phe133 in DmPCNA corresponding to Asp, Val, Gln, Gln, Glu and Tyr, respectively, in hPCNA. These differences may result in structural changes and may explain their different functions.
Figure 6.

DmPCNA and hPCNA (PDB entry 1vym) are coloured blue and wheat, respectively. Left, comparison of the overall structures; right, comparison of the details of the IDCLs.
In previous studies, purified calf thymus Polδ was dramatically stimulated by hPCNA but not by wild-type DmPCNA (Mozzherin et al., 2004 ▶). Mozzherin and coworkers reported that mutations of DmPCNA involving both the substitution of a single residue (Q123V) of DmPCNA and the replacement of Met119–Phe133 by the corresponding human amino acids substantially increased the stimulation of Polδ. This different ability to bind Polδ may be caused by the structural distinctiveness of these two PCNAs. The function of DmPCNA is still not well understood, and DmPCNA may have other unique functions that are distinct from those of hPCNA. Our structural research provides a basis for further functional and evolutionary studies of PCNA.
Supplementary Material
PDB reference: PCNA, 4hk1
Acknowledgments
We would like to thank Dr Zhaocai Zhou at Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences for helpful advice and discussion. We also thank the staff of beamline BL17U at Shanghai Synchrotron Radiation Facility (SSRF) for help with data collection. This work was supported by the Natural Science Foundation of Shandong Province of China (Y2006D05), and the Science and Technology Project of Shandong Provincial Education Department of China (J07YJI9-1).
References
- Afonine, P. V., Grosse-Kunstleve, R. W. & Adams, P. D. (2005). CCP4 Newsl. Protein Crystallogr. 42, contribution 8.
- Arias, E. E. & Walter, J. C. (2006). Nature Cell Biol. 8, 84–90. [DOI] [PubMed]
- Bowman, G. D., O’Donnell, M. & Kuriyan, J. (2004). Nature (London), 429, 724–730. [DOI] [PubMed]
- Bruning, J. B. & Shamoo, Y. (2004). Structure, 12, 2209–2219. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Goujon, M., McWilliam, H., Li, W., Valentin, F., Squizzato, S., Paern, J. & Lopez, R. (2010). Nucleic Acids Res. 38, W695–W699. [DOI] [PMC free article] [PubMed]
- Gulbis, J. M., Kelman, Z., Hurwitz, J., O’Donnell, M. & Kuriyan, J. (1996). Cell, 87, 297–306. [DOI] [PubMed]
- Henderson, D. S. (1999). Methods, 18, 377–400. [DOI] [PubMed]
- Henderson, D. S., Banga, S. S., Grigliatti, T. A. & Boyd, J. B. (1994). EMBO J. 13, 1450–1459. [DOI] [PMC free article] [PubMed]
- Henderson, D. S., Wiegand, U. K., Norman, D. G. & Glover, D. M. (2000). Genetics, 154, 1721–1733. [DOI] [PMC free article] [PubMed]
- Hoege, C., Pfander, B., Moldovan, G. L., Pyrowolakis, G. & Jentsch, S. (2002). Nature (London), 419, 135–141. [DOI] [PubMed]
- Jónsson, Z. O., Hindges, R. & Hübscher, U. (1998). EMBO J. 17, 2412–2425. [DOI] [PMC free article] [PubMed]
- Kelman, Z. (1997). Oncogene, 14, 629–640. [DOI] [PubMed]
- Kontopidis, G., Wu, S.-Y., Zheleva, D. I., Taylor, P., McInnes, C., Lane, D. P., Fischer, P. M. & Walkinshaw, M. D. (2005). Proc. Natl Acad. Sci. USA, 102, 1871–1876. [DOI] [PMC free article] [PubMed]
- Krishna, T. S. R., Fenyö, D., Kong, X.-P., Gary, S., Chait, B. T., Burgers, P. & Kuriyan, J. (1994). J. Mol. Biol. 241, 265–268. [DOI] [PubMed]
- Krishna, T. S. R., Kong, X.-P., Gary, S., Burgers, P. M. & Kuriyan, J. (1994). Cell, 79, 1233–1243. [DOI] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Miyachi, K., Fritzler, M. J. & Tan, E. M. (1978). J. Immunol. 121, 2228–2234. [PubMed]
- Moldovan, G. L., Pfander, B. & Jentsch, S. (2007). Cell, 129, 665–679. [DOI] [PubMed]
- Mozzherin, D. J., McConnell, M., Miller, H. & Fisher, P. A. (2004). BMC Biochem. 5, 13. [DOI] [PMC free article] [PubMed]
- Nielsen, F. C., Jäger, A. C., Lützen, A., Bundgaard, J. R. & Rasmussen, L. J. (2004). Oncogene, 23, 1457–1468. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Strzalka, W. & Ziemienowicz, A. (2011). Ann. Bot. 107, 1127–1140. [DOI] [PMC free article] [PubMed]
- Warbrick, E., Heatherington, W., Lane, D. P. & Glover, D. M. (1998). Nucleic Acids Res. 26, 3925–3932. [DOI] [PMC free article] [PubMed]
- Watanabe, K., Tateishi, S., Kawasuji, M., Tsurimoto, T., Inoue, H. & Yamaizumi, M. (2004). EMBO J. 23, 3886–3896. [DOI] [PMC free article] [PubMed]
- Williams, G. J., Johnson, K., Rudolf, J., McMahon, S. A., Carter, L., Oke, M., Liu, H., Taylor, G. L., White, M. F. & Naismith, J. H. (2006). Acta Cryst. F62, 944–948. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Zhang, P., Sun, Y., Hsu, H., Zhang, L., Zhang, Y. & Lee, M. Y. W. T. (1998). J. Biol. Chem. 273, 713–719. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: PCNA, 4hk1