

FeoB is a transmembrane protein involved in ferrous iron uptake in prokaryotic organisms. Here, the structure of the cytoplasmic domain of FeoB from Gallionella capsiferriformans is reported.
Keywords: FeoB, domain-swapped dimer, NFeoB, Gallionella capsiferriformans
Abstract
FeoB is a transmembrane protein involved in ferrous iron uptake in prokaryotic organisms. FeoB comprises a cytoplasmic soluble domain termed NFeoB and a C-terminal polytopic transmembrane domain. Recent structures of NFeoB have revealed two structural subdomains: a canonical GTPase domain and a five-helix helical domain. The GTPase domain hydrolyses GTP to GDP through a well characterized mechanism, a process which is required for Fe2+ transport. In contrast, the precise role of the helical domain has not yet been fully determined. Here, the structure of the cytoplasmic domain of FeoB from Gallionella capsiferriformans is reported. Unlike recent structures of NFeoB, the G. capsiferriformans NFeoB structure is highly unusual in that it does not contain a helical domain. The crystal structures of both apo and GDP-bound protein forms a domain-swapped dimer.
1. Introduction
The ability to acquire sufficient quantities of iron from the environment is vital for almost all prokaryotes (Cartron et al., 2006 ▶; Emerson et al., 2010 ▶; Kammler et al., 1993 ▶). In many bacterial species, the major route of ferrous iron uptake is through the anaerobically induced FeoABC (ferrous iron-transport) system (Cartron et al., 2006 ▶; Kammler et al., 1993 ▶). The major component of the FeoABC system is FeoB, a transmembrane protein, which is most likely to act as a permease through which ferrous iron is transported into the cell. The structural organization of FeoB is divided into three domains: two N-terminal cytoplasmic domains and a C-terminal hydrophobic membrane-spanning domain. The two soluble domains are collectively termed NFeoB and comprise a GTPase domain (termed the G-domain) followed by a five-helix helical bundle designated the helical domain. Owing to this unprecedented occurrence of a polytopic membrane protein covalently fused to an intracellular G-protein, FeoB is often considered to be a missing link in the evolution of G-protein-coupled membrane processes in higher organisms (Guilfoyle et al., 2009 ▶; Hattori et al., 2009 ▶).
The structure of the NFeoB portion of FeoB has recently been reported from a number of different organisms, all of which revealed the G-domain to be closely associated with the helical domain (Ash et al., 2011a ▶,b ▶, 2012 ▶; Guilfoyle et al., 2009 ▶; Hattori et al., 2009 ▶; Hung et al., 2010 ▶, 2012 ▶; Petermann et al., 2010 ▶). The structure of the G-domain revealed a classical G-protein fold, which is distinguished by the presence of five conserved sequence motifs (G1–G5) critical for nucleotide recognition and hydrolysis (Ash et al., 2011a ▶; Guilfoyle et al., 2009 ▶; Wittinghofer & Vetter, 2011 ▶). Like other small G-proteins, the G-domain of FeoB also possesses two Switch regions (Switch I and Switch II) that undergo conformational changes in response to nucleotide binding and hydrolysis.
While the G-domain functions as a molecular switch regulating or providing the power stroke for iron transport, the presence of the helical domain has been shown to increase the affinity of the G-domain for GDP by up to 12-fold, with little effect on GTP affinity (Eng et al., 2008 ▶). As such, the helical domain has been postulated to function as a GDP-dissociation inhibitor (GDI) domain that stabilizes the GDP-bound state. Another proposed role is in communicating structural changes between the G-domain and the membrane domain (Petermann et al., 2010 ▶; Hattori et al., 2009 ▶), although large structural changes between the two domains have not been observed in the published structures. FeoB harbouring mutations in the interface between the two domains has been shown to be unable to import iron in vivo, whilst GTP-hydrolysis rates in vitro remain unaffected (Eng et al., 2008 ▶). Thus, while it appears clear that the helical domain has an important function, the precise role of this domain has so far remained elusive.
Here, we report the X-ray crystal structure of the cytoplasmic region of FeoB from Gallionella capsiferriformans, an iron-oxidizing Gram-negative bacterium (Emerson et al., 2010 ▶; Weiss et al., 2007 ▶). The three-dimensional structure was determined in two states: (i) the apo state at 2.15 Å resolution and (ii) with bound GDP at 1.9 Å resolution. Compared with the previously determined structures of NFeoB, the structure illustrates a high degree of structural conservation of the GTPase core domain. However, it differs by the conspicuous absence of a large portion of the helical domain, an absence that facilitates the formation of a putative domain-swapped dimer.
2. Methods and materials
2.1. Cloning, expression and purification
A DNA fragment encoding the soluble cytoplasmic portion of FeoB (NFeoB; corresponding to residues 1–202 of UniProtKB entry D9SIP4) was PCR-amplified from genomic G. capsiferriformans ES-2 (NCMA B102) DNA using the primers 5′-TATATAGAATTCGAAAACCTGTACTTCCAGGGTCAATTCAAACGCATCGCGTTAC-3′ and 5′-TTTTCTCGAGTTATTCAGGCAGGCGGGC-3′. The resulting PCR product, incorporating a TEV protease cleavage site as well as EcoRI and XhoI restriction sites, was ligated into pGEX-4T-1 vector (GE Healthcare Life Sciences) to create an N-terminally GST-tagged recombinant clone. The resulting vector was amplified in Escherichia coli TOP10 cells (Invitrogen) and a sequence-verified clone was then transformed into E. coli BL21(DE3) cells for protein expression.
The cells were grown under aerobic conditions at 310 K in Luria–Bertani broth (4 × 1 l) and induced with 0.2 mM IPTG upon reaching an OD600 of 0.6. The cells were grown at 298 K for 6 h post-induction, after which they were harvested by centrifugation and resuspended in 50 ml buffer A (20 mM Tris pH 8, 100 mM NaCl) containing 10 mM EDTA and stored at 253 K.
For protein purification, cells were thawed at room temperature and lysed by three passes through a cooled EmulsiFlex-C3 homogenizer (Avestin). The resulting cell lysate was centrifuged (81 000g at 279 K) to remove unbroken cells and cell debris. The supernatant was then incubated for 1 h at 277 K with Glutathione Sepharose resin (GE Healthcare Life Sciences) which had been pre-equilibrated with buffer A containing 1 mM EDTA. The protein-bound resin was loaded onto a Poly-Prep gravity-flow column (Bio-Rad) and the resin was subsequently washed with five column volumes of buffer A supplemented with 1 mM EDTA, followed by a second wash with five column volumes of buffer consisting of 20 mM Tris pH 8, 300 mM NaCl, 1 mM EDTA. The GST moiety was cleaved using TEV protease under dialysis with 20 mM Tris pH 8, 300 mM NaCl (overnight at 277 K). The untagged protein was eluted and concentrated to 1.8 ml before loading onto a Superdex 75 16/600 size-exclusion column (GE Healthcare Life Sciences) pre-equilibrated with buffer A. The eluted protein of ∼23 kDa was concentrated to ∼5 mg ml−1 as determined by the BCA assay method (Thermo Fisher Scientific) and stored at 277 K.
2.2. Crystallization and X-ray data collection
Crystallization conditions for NFeoB were screened by the hanging-drop vapour-diffusion method in 96-well plates (Greiner Bio-One) using a Mosquito nanolitre liquid-handling robot (TTP LabTech). Protein (250 nl) was mixed with commercially available crystallization screens (The JCSG+, PACT and Classics Suites; Qiagen) in a 1:1 or 2:1 ratio and incubated at 289 K. To obtain nucleotide-bound crystal forms, protein solution was mixed with MgCl2 (10 mM) and GDP (3 mM) or GMPPNP (2 mM) prior to crystallization. Native multi-faceted pyramid-shaped crystals (Fig. 1 ▶) of NFeoB grew to dimensions of ∼0.1 × 0.1 × 0.05 mm within a week in condition H2 of the The JCSG+ Suite [1 M ammonium sulfate, 0.1 M bis-tris pH 5.5, 1%(w/v) PEG 3350]. Cocrystallization trials with NFeoB and GMPPNP (2 mM) resulted in thin plate-like crystals of dimensions ∼0.2 × 0.05 × 0.01 mm comprised of GDP-bound protein (from the slow intrinsic hydrolysis of GMPPNP) in condition H7 of The JCSG+ Suite [0.2 M ammonium sulfate, 0.1 M bis-tris pH 5.5, 25%(w/v) PEG 3350] after two weeks. Prior to flash-cooling in liquid nitrogen, the crystals were cryoprotected in mother liquor containing 25% glycerol.
Figure 1.

Crystals of G. capsiferriformans NFeoB. (a) Crystals of the apo form grown using condition H2 of The JCSG+ Suite [1 M ammonium sulfate, 0.1 M bis-tris pH 5.5, 1%(w/v) PEG 3350]. (b) Crystals of the GDP-bound form obtained using condition H7 of The JCSG+ Suite [0.2 M ammonium sulfate, 0.1 M bis-tris pH 5.5, 25%(w/v) PEG 3350].
In-house diffraction data (100 K) were initially collected from the native NFeoB crystals to 2.9 Å resolution on a MAR 345 image-plate detector (MAR Research) using Cu Kα X-rays. Diffraction data (100 K) were collected from a second native crystal from the same drop to a resolution of 2 Å using an ADSC Quantum 315r detector on beamline MX2 of the Australian Synchrotron at a wavelength of 0.9 Å. 1.9 Å resolution diffraction data (100 K) were collected from the GDP-bound crystal form on the 23-ID-D beamline of the Advanced Photon Source (APS; Argonne National Laboratory) at a wavelength of 1.0 Å using a MAR 300 CCD detector.
2.3. Structure solution and refinement
X-ray diffraction data for the apo NFeoB crystals were processed with MOSFLM (Leslie, 2006 ▶) and scaled together with SCALA from the CCP4 suite (Winn et al., 2011 ▶). The data-processing statistics are summarized in Table 1 ▶. Structure solution was initially achieved using the 2.9 Å resolution in-house data. Briefly, the program CHAINSAW (Stein, 2008 ▶) was used to create a search model for molecular replacement (MR) using the structure of the FeoB G-domain from Legionella pneumophila (PDB entry 3iby, chain A; Petermann et al., 2010) and its sequence alignment with G. capsiferriformans NFeoB. A unique solution (TFZ = 11.7, LLG = 194) was found by Phaser (McCoy, 2007 ▶) in the orthorhombic space group C2221 with two molecules in the asymmetric unit. The resulting model underwent rigid-body refinement in REFMAC5 (Murshudov et al., 2011 ▶) followed by restrained refinement with simulated annealing using phenix.refine (Afonine et al., 2005 ▶) within PHENIX (Zwart et al., 2008 ▶). Higher resolution synchrotron data were subsequently collected to 2.15 Å resolution, allowing a more complete model to be automatically built using AutoBuild (as implemented in PHENIX; Terwilliger et al., 2008 ▶). The resulting model was manually corrected within Coot (Emsley & Cowtan, 2004 ▶) interspersed with iterative rounds of refinement in REFMAC5 and phenix.refine. Water molecules were included automatically using phenix.refine and checked manually in Coot to retain only those that made appropriate hydrogen-bonding contacts and had spherical densities above 1σ in the 2F o − F c map. Three tetrahedral-shaped electron-density peaks were modelled as sulfate ions derived from the crystallant. Refinement converged with residuals R work = 0.221 and R free = 0.261.
Table 1. Data-processing and refinement statistics for G. capsiferriformans NFeoB.
Values in parentheses are for the highest resolution shell.
| Apo | GDP-bound | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.9 | 1.0 |
| Space group | C2221 | P212121 |
| Unit-cell parameters (Å, °) | a = 100.7, b = 112.5, c = 97.9, α = β = γ = 90 | a = 45.8, b = 84.0, c = 95.5, α = β = γ = 90 |
| Resolution (Å) | 75.03–2.15 (2.27–2.15) | 63.08–1.93 (2.04–1.93) |
| Total reflections | 213515 (31462) | 200435 (28347) |
| Unique reflections | 30386 (4371) | 28390 (4084) |
| Completeness (%) | 99.5 (99.2) | 100 (100) |
| Multiplicity | 7.0 (7.2) | 7.1 (6.9) |
| Mean I/σ(I) | 14.8 (2.7) | 9.0 (2.6) |
| R merge † | 0.083 (0.717) | 0.151 (0.796) |
| R pim ‡ | 0.034 (0.284) | 0.061 (0.323) |
| Refinement | ||
| Resolution | 75.03–2.15 (2.20–2.15) | 63.08–1.93 (1.98–1.93) |
| Unique reflections | 28887 (2220) | 26903 (2065) |
| Completeness (%) | 99.2 (98.9) | 100 (100) |
| R work § | 0.221 | 0.191 |
| R free | 0.261 | 0.231 |
| 〈Protein B factor〉 (Å2) | 29.7 | 17.7 |
| No. of protein molecules per asymmetric unit | 2 | 2 |
| R.m.s.d., bonds (Å) | 0.009 | 0.012 |
| R.m.s.d., angles (°) | 1.097 | 1.306 |
| Ramachandran plot statistics¶ | ||
| Favoured (%) | 98.7 | 98.7 |
| Allowed (%) | 100 | 100 |
| PDB code | 3w5i | 3w5j |
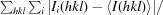
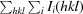
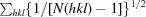
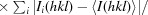
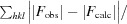
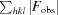
The diffraction data for the GDP-bound crystals were indexed, integrated and scaled using the HKL-2000 suite of programs (Otwinowski, 1993 ▶). MR was carried out with Phaser using the apo structure as a search model with residues 26–50 (including Switch I) omitted. This gave a clear solution in the orthorhombic space group P212121 with two molecules in the asymmetric unit (TFZ = 21.2, LLG = 343). The resulting model was refined in a similar fashion as the apo structure, converging to give refinement residuals of R work = 0.191 and R free = 0.231. In chain A clear difference density was visible for one molecule of GDP, which was included in the model with full occupancy in the later stages of refinement. No electron density was apparent at the corresponding position in chain B to indicate that another GDP molecule binds in this subunit. However, the electron density does indicate that a sulfate ion derived from the mother liquor is bound. An additional electron-density peak corresponding to glycerol (which was used as cryoprotectant) was also modelled.
The quality of the structures was assessed regularly during the refinement process using MolProbity (Chen et al., 2010 ▶), which was used to validate the geometry of the final models (Table 1 ▶). Subunit interactions and assemblies were analyzed using PISA (Krissinel & Henrick, 2007 ▶). All structural figures were produced using PyMOL (DeLano, 2002 ▶). Refined atomic coordinates and experimental structure factors have been deposited in the Protein Data Bank (PDB entries 3w5i and 3w5j). Data-collection and refinement statistics are given in Table 1 ▶.
3. Results and discussion
3.1. Overall structure of G. capsiferriformans NFeoB
The crystal structure of NFeoB from G. capsiferriformans in the nucleotide-free state reveals two chains per asymmetric unit in space group C2221. Within the asymmetric unit, the main chain was traced for residues 1–197 of chain A and residues 1–200 of chain B. In both chains residues 144–146 could not be located in the electron density and could not be modelled. Overall, the three-dimensional fold of the G-domain (residues 1–162) is well conserved and aligns with the G-domain of E. coli NFeoB (residues 1–166; PDB entry 3hyt; Guilfoyle et al., 2009 ▶) with an r.m.s.d. of 1.20 Å (145 Cα positions). The G-domain comprises a seven-stranded β-sheet (β1–β7) flanked by six α-helices (α1–α6) (Fig. 2 ▶), which includes the five signature sequence motifs (G1–G5) involved in GTP and Mg2+ binding in all G proteins (Wittinghofer & Vetter, 2011 ▶). The G5 motif, which is located in the loop between strand β7 and helix α6, was disordered in both the apo and the GDP-bound structures and could not be confidently included in the final model, strongly indicating flexibility in this region of the molecule.
Figure 2.

Overall structure of the G. capsiferriformans GDP-bound NFeoB dimer. (a) Ribbon representation of the GDP-bound dimer viewed from the side. Individual protomers are coloured purple (chain A) and blue (chain B). The switch regions are shaded in orange. Loop regions of poor density connecting β7 to α6 (residues 144–147) are represented by dotted lines. A bound molecule of GDP and a sulfate ion occupying the nucleotide-binding site in chain A and chain B, respectively, are represented by sticks and coloured by atom type (C, yellow; O, red; N, blue; P, yellow). (b) Rotated view of (a).
The GDP-bound form of NFeoB crystallized in space group P212121 with two molecules in the asymmetric unit (Fig. 2 ▶), which overlay with an r.m.s.d. of 0.9 Å. Within the asymmetric unit, the main chain was traced for residues 1–197 of both chains A and B. A GDP molecule occupies the nucleotide-binding site in chain A, whilst in chain B a sulfate ion, presumably derived from the crystallization condition, occupies a position similar to that of the β-phosphate of GDP. Hydrogen-bonding interactions are observed between the GDP molecule and a number of residues in the binding site, including Asn11, Thr12, Gly13, Lys14, Ser15, Thr16 and Glu118 (Fig. 3 ▶). As previously observed in NFeoB from Streptococcus thermophilus (Ash et al., 2010 ▶), the Switch I loop (residues 22–41) is oriented away from the GDP-binding site and does not interact with the GDP molecule.
Figure 3.

A 2F o − F c map around the GDP molecule (ball-and-stick representation coloured by atom type as in Fig. 2 ▶) contoured at 1σ. The protein backbone is shown as grey ribbons. The electron density is shown in light blue. The residues forming hydrogen-bonding interactions with the GDP molecule are shown in stick representation (grey) and are coloured red for oxygen and blue for nitrogen. Hydrogen bonds are shown as black dashed lines, and green spheres represent the waters in the GDP-binding network.
3.2. Structural comparison
In comparison to other NFeoB structures, a distinct difference in G. capsiferriformans NFeoB is the absence of the C-terminal helical domain (Fig. 4 ▶). Prior to our structure of G. capsiferriformans NFeoB, all determined structures of NFeoB consisted of the G-domain followed by a helical domain comprised of a four-helix bundle and a C-terminal extended helix (hd5; Ash et al., 2011a ▶; Guilfoyle et al., 2009 ▶; Hattori et al., 2009 ▶). In these structures, the C-terminal extended helix is positioned in a groove formed between helices α3 and α5 (Fig. 4 ▶). In contrast, NFeoB from G. capsiferriformans only contains the G-domain and the C-terminal extended helix (α7; residues 168–194). Here, the absence of the four-helix bundle of the helical domain renders the extended C-terminal helix unable to be located in the same position owing to geometrical and distance restraints. The C-terminal helix instead positions itself in the same groove (i.e. between α3 and α5) but in the opposing protomer in the asymmetric unit (Figs. 2 ▶ and 4 ▶). PISA (Krissinel & Henrick, 2007 ▶) analysis points to a stable dimeric interface with buried surface areas of 1535.9 and 1541.7 Å2 for the apo and the GDP-bound form, respectively.
Figure 4.

Amino-acid sequence alignment and structural comparison of G. capsiferriformans NFeoB and E. coli NFeoB. (a) Sequence alignment of E. coli NFeoB and G. capsiferriformans NFeoB, with the conserved residues shaded in grey. (b) Structural superimposition of G. capsiferriformans NFeoB chain A (coloured as in Fig. 2 ▶; PDB entry 3w5j) with E. coli NFeoB (yellow; PDB entry 3hyt), illustrating the absence of a large portion of the helical domain in the former. (c) and (d) show structural superimpositions of the G. capsiferriformans NFeoB dimer (views and colours as in Fig. 2 ▶) with E. coli NFeoB, illustrating the analogous positioning of the C-terminal extended helix in the two structures.
The majority of the inter-protomeric interactions are contributed by hydrogen-bonding associations between helix α7′ and α3, α5 and the α4–α5 loop of the other monomer, as well as between α2 and α3′ and between α3′ and the β4–α2 loop. Two salt bridges (Arg94′–Asp66 and Glu185′–Lys103) further stabilize the dimer. G. capsiferriformans NFeoB elutes at a volume consistent with its monomeric molecular weight in size-exclusion chromatography (data not shown). However, this does not necessarily exclude a biological dimer. Given that the protein is expressed as a subclone of a much larger protein, the potential contact surface between protomers is drastically reduced. In addition, the protein is initially fused to a large GST moiety, which again could potentially limit the ability of the protein to form a correct oligomeric structure. However, when purified and highly concentrated, such as in the crystallization drop, the likelihood of forming an oligomer increases as small changes in protein composition or environment can often tip the balance from one state to the next (Ali & Imperiali, 2005 ▶; Tanaka et al., 2011 ▶; Sakurai et al., 2001 ▶; Yang et al., 2005 ▶).
The domain-swapped dimer structure that is presented here is intriguing and re-opens the debate on the oligomeric state of FeoB. Although a high degree of care should be taken when examining oligomeric structures from crystallographic data alone, the structure of G. capsiferriformans NFeoB indicates it to be highly unlikely that the C-terminal extended helix folds back onto its own protomer; rather, it crosses over to the neighbouring protomer. This raises the question of whether this domain-swapped dimer is indeed the correct oligomeric state and, if so, are the previous structures adopting a structural architecture that is solely facilitated by the added flexibility conferred by the four-helix bundle of the helical domain which allows the C-terminal helix to fold back on its own protomer? Additional experiments are required to address these questions.
4. Conclusions
We have reported here the crystal structure of NFeoB from G. capsiferriformans in both apo and GDP-bound states. While the structures illustrate a G-domain that is structurally very similar to other recently determined NFeoB structures, a marked difference is the absence of a large part of the helical domain. The helical domain has previously been implicated in having a GDI-like activity; however, given the absence of this domain in our NFeoB structure the precise role of the helical domain requires additional studies. In addition, the absence of the helical domain appears to prevent the G. capsiferriformans protein from forming the same intra-protomeric architecture as the previously determined structures; rather, it seems to facilitate the formation of a putative domain-swapped dimer. Further studies are required to corroborate the biological implications of this and to determine the precise oligomeric state of FeoB.
Supplementary Material
PDB reference: NFeoB, 3w5i
PDB reference: GDP-bound, 3w5j
Acknowledgments
This study was supported by the National Health and Medical Research Council (grants 632703 and 1035693), the GM/CA-CAT beamline 23-ID at the Advanced Photon Source (which is supported by National Cancer Institute grant Y1-CO-1020 and National Institute of General Medical Sciences grant Y1-GM-1104), and by beamline MX-2 at the Australian Synchrotron. We acknowledge travel funding provided by the International Synchrotron Access Program (ISAP) managed by the Australian Synchrotron and funded by the Australian Government. CD is supported by a National Breast Cancer Foundation Postdoctoral Fellowship. APM is supported by an NHMRC CJ Martin Postdoctoral Research Fellowship. MJM is supported by a LIMS Senior Research Fellowship.
References
- Afonine, P. V., Grosse-Kunstleve, R. W. & Adams, P. D. (2005). CCP4 Newsl. Protein Crystallogr. 42, contribution 8.
- Ali, M. H. & Imperiali, B. (2005). Bioorg. Med. Chem. 13, 5013–5020. [DOI] [PubMed]
- Ash, M. R., Guilfoyle, A., Clarke, R. J., Guss, J. M., Maher, M. J. & Jormakka, M. (2010). J. Biol. Chem. 285, 14594–14602. [DOI] [PMC free article] [PubMed]
- Ash, M.-R., Maher, M. J., Guss, J. M. & Jormakka, M. (2011a). Acta Cryst. D67, 973–980. [DOI] [PubMed]
- Ash, M.-R., Maher, M. J., Guss, J. M. & Jormakka, M. (2011b). Acta Cryst. F67, 1511–1515. [DOI] [PMC free article] [PubMed]
- Ash, M.-R., Maher, M. J., Guss, J. M. & Jormakka, M. (2012). FEBS Lett. 586, 2218–2224. [DOI] [PubMed]
- Cartron, M. L., Maddocks, S., Gillingham, P., Craven, C. J. & Andrews, S. C. (2006). Biometals, 19, 143–157. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Emerson, D., Fleming, E. J. & McBeth, J. M. (2010). Annu. Rev. Microbiol. 64, 561–583. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Eng, E. T., Jalilian, A. R., Spasov, K. A. & Unger, V. M. (2008). J. Mol. Biol. 375, 1086–1097. [DOI] [PMC free article] [PubMed]
- Guilfoyle, A., Maher, M. J., Rapp, M., Clarke, R., Harrop, S. & Jormakka, M. (2009). EMBO J. 28, 2677–2685. [DOI] [PMC free article] [PubMed]
- Hattori, M., Jin, Y., Nishimasu, H., Tanaka, Y., Mochizuki, M., Uchiumi, T., Ishitani, R., Ito, K. & Nureki, O. (2009). Structure, 17, 1345–1355. [DOI] [PubMed]
- Hung, K.-W., Chang, Y.-W., Eng, E. T., Chen, J.-H., Chen, Y.-C., Sun, Y.-J., Hsiao, C.-D., Dong, G., Spasov, K. A., Unger, V. M. & Huang, T.-H. (2010). J. Struct. Biol. 170, 501–512. [DOI] [PMC free article] [PubMed]
- Hung, K.-W., Tsai, J.-Y., Juan, T.-H., Hsu, Y.-L., Hsiao, C.-D. & Huang, T.-H. (2012). J. Bacteriol. 194, 6518–6526. [DOI] [PMC free article] [PubMed]
- Kammler, M., Schön, C. & Hantke, K. (1993). J. Bacteriol. 175, 6212–6219. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 56–62. Warrington: Daresbury Laboratory.
- Petermann, N., Hansen, G., Schmidt, C. L. & Hilgenfeld, R. (2010). FEBS Lett. 584, 733–738. [DOI] [PubMed]
- Sakurai, K., Oobatake, M. & Goto, Y. (2001). Protein Sci. 10, 2325–2335. [DOI] [PMC free article] [PubMed]
- Stein, N. (2008). J. Appl. Cryst. 41, 641–643.
- Tanaka, Y., Hirano, N., Kaneko, J., Kamio, Y., Yao, M. & Tanaka, I. (2011). Protein Sci. 20, 448–456. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Moriarty, N. W., Zwart, P. H., Hung, L.-W., Read, R. J. & Adams, P. D. (2008). Acta Cryst. D64, 61–69. [DOI] [PMC free article] [PubMed]
- Weiss, M. S. (2001). J. Appl. Cryst. 34, 130–135.
- Weiss, J. V., Rentz, J. A., Plaia, T., Neubauer, S. C., Merrill-Floyd, M., Lilburn, T., Bradburne, C., Megonigal, J. P. & Emerson, D. (2007). Geomicrobiol. J. 24, 559–570.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wittinghofer, A. & Vetter, I. R. (2011). Annu. Rev. Biochem. 80, 943–971. [DOI] [PubMed]
- Yang, S., Levine, H. & Onuchic, J. N. (2005). J. Mol. Biol. 352, 202–211. [DOI] [PubMed]
- Zwart, P. H., Afonine, P. V., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., McKee, E., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K., Storoni, L. C., Terwilliger, T. C. & Adams, P. D. (2008). Methods Mol. Biol. 426, 419–435. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: NFeoB, 3w5i
PDB reference: GDP-bound, 3w5j