

The tryparedoxin peroxidase from L. braziliensis was crystallized and diffraction data were collected to a maximum resolution of 3.3 Å.
Keywords: tryparedoxin peroxidase, Leishmania braziliensis
Abstract
Tryparedoxin peroxidase (TXNPx) is an essential constituent of the main enzymatic scavenger system for reactive oxygen species (ROS) in trypanosomatids. Genetic studies have demonstrated the importance of this system for the development and virulence of these parasites, representing a potential target for the discovery of new trypanocidal drugs. In this work, the mitochondrial TXNPx from Leishmania braziliensis was cloned, overexpressed, purified and crystallized. The crystals diffracted to 3.3 Å resolution and belonged to space group P42212, with unit-cell parameters a = b = 131.8, c = 44.4 Å. These studies will contribute to a better understanding of the molecular mechanisms involved in ROS detoxification by trypanosomatids.
1. Introduction
Leishmania braziliensis is the causative agent of cutaneous and muco-cutaneous leishmaniasis. It is widely distributed from Central to South America. The World Health Organization (WHO) estimates that the worldwide prevalence of leishmaniasis is approximately 12 million cases, with an annual mortality of about 60 000 people. The size of the population at risk is around 350 million (World Health Organization, 2010 ▶). Cutaneous leishmaniasis is endemic in more than 70 countries, and 90% of cases occur in Afghanistan, Algeria, Brazil, Pakistan, Peru, Saudi Arabia and Syria (Desjeux, 2004 ▶; Reithinger et al., 2007 ▶).
Leishmania parasites present two well defined forms in their life cycle: the promastigote in insects and the intracellular amastigote in the mammalian host (Wheeler et al., 2011 ▶). At the infection stage, a large amount of reactive oxygen or nitrogen intermediates are generated by the host cell to create unfavourable conditions for parasite development (Eslami et al., 2011 ▶). These compounds are toxic to parasites and their ability to combat the pro-oxidants will determine the success and persistence of the infection (Piñeyro et al., 2011 ▶).
In trypanosomatids, peroxides are eliminated through an enzymatic cascade that involves trypanothione reductase (TR), tryparedoxin (TXN) and tryparedoxin peroxidase (TXNPx) with the thiol trypanothione [N1,N8-bis(glutathionyl)spermidine] serving as a mediator for transfer of reducing equivalents (Nogoceke et al., 1997 ▶). The relevance of this redox cascade to the vitality and virulence of trypanosomatids was demonstrated by inverse genetics in Trypanosoma brucei, in which the absence of TR causes the inhibition of proliferation, increase of sensitivity to H2O2 and abrogation of virulence (Tovar et al., 1998 ▶; Krieger et al., 2000 ▶). Moreover, cells overexpressing the Leishmania TXNPx had a higher resistance to oxidative stress, higher growth and lower division rates (Iyer, 2008 ▶). These studies suggest that this enzyme is a promising molecular target for the development of new trypanocidal drugs.
The present work describes the cloning, overexpression in Escherichia coli cells, purification to homogeneity, crystallization and preliminary X-ray diffraction studies of the mitochondrial TXNPx from L. braziliensis (LbTXNPx).
2. Materials and methods
2.1. Molecular cloning
The tryparedoxin peroxidase coding sequence (GenBank accession number: FR798998) was amplified by polymerase chain reaction (PCR) using Pfx Taq polymerase, total DNA from L. braziliensis and the oligonucleotide primers LbTXNPx-F (5′-CATATGCGAATTTTTTGAGAAGAAT-3′) and LbTXNPx-R (5′-GTCGACTTAATTCTTCTCAAAAAATTCG-3′). The amplified gene was inserted into the pGEM-T vector (Promega) and then sequenced to check for PCR-induced errors. The coding region of LbTXNPx, cloned in the pGEM-T vector, was digested with NdeI and SalI enzymes, and the resulting fragment was inserted into the pET-28a expression vector (Invitrogen) previously digested with the same enzymes. Positive clones were selected by PCR, confirmed by DNA sequencing and transformed in E. coli BL21(DE3)ΔSLyD strain.
2.2. Protein expression
BL21(DE3)DSlyD cells, harbouring the LbTXNPx+pET-28a vector, were grown in Luria Broth (LB) medium containing kanamycin (100 µg ml−1) and chloramphenicol (34 µg ml−1) at 310 K and 200 rev min−1. Cells were grown to an A 600 of ∼0.8 and expression was induced by adding isopropyl β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM at 303 K. After 4 h, cultured cells were harvested by centrifugation for 10 min at 2600g and suspended in a buffer consisting of 25 mM Tris–HCl pH 7.5, 4 mM phenylmethylsulfonyl fluoride (PMSF). The cell suspension was sonicated and the supernatant was separated by centrifugation (20 000g, 15 min).
2.3. Purification
The His6-tagged protein (HHHHHHSSGLVPRGSRTATVRD, the tag sequence corresponds to the residues given in bold) was initially purified by nickel-affinity chromatography using a 5 ml HiTrap Chelating column (GE Healthcare Life Sciences) pre-equilibrated with 25 mM Tris–HCl pH 7.5 (buffer A). The bound proteins were eluted using a 0–1 M gradient of imidazole in buffer A. Fractions corresponding to LbTXNPx were identified by SDS–PAGE and dialysed against buffer A to remove imidazole. Further, the dialysed sample was submitted to cation-exchange chromatography (CEC) in a HiTrap SP column (GE Healthcare Life Sciences). The elution was performed using a 0–500 mM non-linear gradient of NaCl in buffer A. CEC was found to be an essential purification step for the crystallization process, yielding single crystals adequate for X-ray diffraction experiments. Previous crystallization attempts with a sample from a purification protocol consisting of nickel-affinity chromatography followed by molecular-exclusion chromatography were ineffective in obtaining crystals. We speculate that CEC was efficient in removing oxidized forms of protein, since after this step a secondary minor band at 45 kDa was not observed (data not shown).
2.4. Mass spectrometric analysis (LC-MS/MS)
The protein band was excized, reduced, alkylated and submitted to in-gel digestion with trypsin. An aliquot (4.5 µl) of the resulting peptide mixture was separated in a C18 column coupled with a Q-Tof Ultima mass spectrometer (Waters) with a nano-electrospray source at 0.6 µl min−1. The gradient was 2–90%(v/v) acetonitrile in 0.1%(v/v) formic acid over 45 min. The instrument was operated in the ‘top three’ mode, in which one MS spectrum is acquired followed by MS/MS of the top three most-intense peaks detected. The spectra were collected using the software MassLynx v.4.1 (Waters Company) and the raw data files were converted to a peak list format (mgf) by the software Mascot Distiller v.2.3.2.0 2009 (Matrix Science Ltd); we searched against a non-redundant protein database using the engine Mascot v.2.3 (Matrix Science Ltd), with carbamidomethylation as fixed modification, oxidation of methionine as variable modification, one trypsin missed cleavage and a tolerance of 0.1 Da for both precursor and fragment ions.
2.5. Dynamic light scattering
Dynamic light-scattering (DLS) experiments were carried out using a DynaPro MS/X (Wyatt Technology Corporation) device equipped with a Peltier temperature controller. The wavelength of the laser light and the output power were set to 830 nm and 30 mW, respectively. Around 20 measurements were made at 15 s intervals for each run at 291 K. The protein concentration was adjusted to 2.0 mg ml−1 to verify aggregate formation. Hydrodynamic parameters were determined using the DYNAMICS v.6.3.40 software (Wyatt Technology Corporation). The hydrodynamic radius (Rh) was extrapolated from the translational diffusion coefficient (Dt) using the Stokes–Einstein equation.
2.6. Crystallization
His6-tagged LbTXNPx was concentrated to 6.5 mg ml−1 in buffer A containing 5 mM EDTA using an Amicon Ultra 10 K centrifugal filter device (Millipore). Crystallization trials were carried out by the sitting-drop vapour-diffusion method, mixing 0.2 µl of the protein sample with an equal volume of screening solution and equilibrated over 80 µl of the latter in the reservoir. Initial screening was performed using 536 conditions from the commercially available kits Crystal Screen and Crystal Screen 2 (Hampton Research), Wizard I and II (Emerald BioSystems), JCSG-plus (Emerald BioSystems), Precipitant Synergy (Emerald BioSystems), PACT (Qiagen) and Salt Rx (Hampton Research).
Small and imperfect crystals appeared in the following conditions: PACT (Qiagen), condition No. 73 [20%(w/v) PEG 3350, 0.2 M sodium fluoride, 0.1 M bis-tris propane pH 7.5]; PACT (Qiagen), condition No. 52 [25%(w/v) PEG 3350, 0.2 M potassium thiocyanate]; and JCSG-plus (Emerald BioSystems), condition No. 17 [0.1 M sodium cacodylate pH 6.5, 40%(w/v) 2-methyl-2,4-pentanediol and 5%(w/v) PEG 8000]. Only in condition No. 17 [0.10 M Tris–HCl pH 8.5, 30%(w/v) PEG 4000, 0.2 M lithium sulfate] from Crystal Screen HT (Hampton Research) did we obtain well formed crystals but with small dimensions for X-ray diffraction experiments at our available beamlines. In order to obtain larger crystals, we refined the initial condition by varying the precipitant concentration within an established range [30–20%(w/v) of PEG 4000]. In addition, we increased the drop volume from 0.2 to 0.5 µl. The reservoir volume was also changed to 200 µl, because of the larger drop volume. The best single crystals were obtained from a solution consisting of 27%(w/v) PEG 4000, 0.1 M Tris–HCl pH 8.5, 0.2 M lithium sulfate.
2.7. Data collection, processing and structure determination
X-ray diffraction data were collected on the W01B-MX2 beamline at the Brazilian Synchrotron Light Laboratory, Campinas, Brazil (Guimarães et al., 2009 ▶). A full data set was collected using a crystal-to-detector distance of 167.2 mm with 1° oscillation and 60 s exposure per image. Data were indexed, integrated, merged and scaled using the HKL-2000 package (Otwinowski & Minor, 1997 ▶). The molecular-replacement method implemented in the MOLREP program (Vagin & Teplyakov, 1998 ▶) from the CCP4 suite (Winn et al., 2011 ▶) was employed for phasing. The atomic coordinates of a mammalian 2-Cys peroxiredoxin (PDB entry 1qq2, Hirotsu et al., 1999 ▶), which shares 60% sequence identity with LbTXNPx, was used as the search model.
3. Results and discussion
The E. coli BL21(DE3)ΔSlyD strain containing the pET-28a+LbTXNPx vector showed a high level of LbTXNPx expression in the soluble extract. A crystal-grade sample was obtained by two purification steps involving nickel-affinity (Fig. 1 ▶ a) and cation-exchange (Fig. 1 ▶ b) chromatographies. Mass spectrometric analysis of the trypsin-digested sample confirmed that the purified protein corresponded to LbTXNPx with the identification of three major peptides and sequence coverage of 80% (Fig. 1 ▶ c).
Figure 1.

SDS–PAGE analysis of the purification steps: (a) fractions from Ni–NTA affinity chromatography and (b) fractions from CEC. Lane M, protein molecular-weight marker (Fermentas; labelled in kDa); lane FT, flow-through; LbTXPNx, fractions corresponding to the purified protein. (c) Sequence of LbTXNPx with peptides identified by LC-MS/MS analysis shaded in grey.
The structural homogeneity of the purified protein was evaluated by DLS assays indicating a monodispersive sample (19.8% polydispersity) with an extrapolated hydrodynamic radius of 2.31 nm, which corresponds to a molecular weight of about 42 kDa (dimer), using a calibration curve based on the diffusion coefficients of globular proteins.
Prior to crystallization experiments, the purified His6-tagged protein was dialysed against a 25 mM Tris–HCl pH 7.5 buffer containing 5 mM EDTA. The presence of the chelating agent was shown to be efficient in preventing protein precipitation and allowed a higher protein concentration to be reached (6.5 mg ml−1). In the absence of EDTA, the maximum protein concentration was 2–3 mg ml−1.
A single crystal with dimensions of 175 × 105 µm (Fig. 2 ▶ a) was used for X-ray diffraction experiments and data were collected to 3.3 Å resolution (Fig. 2 ▶ b) under cryogenic conditions (100 K). The reflections were indexed in the tetragonal crystal system with unit-cell parameters a = b = 131.8, c = 44.4 Å. An examination of the systematic absences indicated that the crystal belonged to space group P42212. Calculation of the Matthews coefficient (Matthews, 1968 ▶) based on the molecular weight of 22 028 Da (monomer plus His tag) resulted in a V M of 1.9 Å3 Da−1 and a solvent content of 35.40%, which corresponds to the presence of two molecules per asymmetric unit. The statistics of the data processing are summarized in Table 1 ▶.
Figure 2.

(a) Photomicrograph of LbTXNPx crystals. (b) X-ray diffraction pattern of an LbTXNPx crystal. The edge of the diffraction frame is at 3.0 Å resolution.
Table 1. Data-collection statistics for the LbTXNPx crystal.
Values in parentheses are for the outermost resolution shell.
| Beamline | W01B-MX2, LNLS |
| Wavelength (Å) | 1.459 |
| Space group | P42212 |
| Unit-cell dimensions (Å) | a = b = 131.8, c = 44.4 |
| Resolution range (Å) | 40.0–3.30 (3.42–3.30) |
| No. of unique reflections | 6176 (578) |
| Multiplicity | 5.8 (4.2) |
| Completeness (%) | 97.6 (92.9) |
| 〈I/σ(I)〉 | 18.6 (2.7) |
| R merge † (%) | 13.2 (56.9) |
| Matthews coefficient (Å3 Da−1) | 1.9 |
| Corresponding solvent content (%) | 35.9 |
| Molecules per asymmetric unit | 2 |
R
merge = 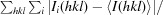
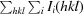 , where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average over all observations of reflection hkl.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average over all observations of reflection hkl.
Molecular-replacement calculations, using the atomic coordinates of mammalian 2-Cys peroxiredoxin, Hbp23 (PDB entry 1qq2, Hirotsu et al., 1999 ▶) as a template, confirmed the presence of two molecules in the asymmetric unit and the initial model after rigid-body refinement resulted in an R factor/R free of 39/44%. The structure refinement is currently in progress by intercalated steps of manual building and isotropic restrained refinement with COOT (Emsley & Cowtan, 2004 ▶) and REFMAC5 (Murshudov et al., 1997 ▶), respectively. In parallel with crystallographic studies, functional and biophysical experiments are being carried out in order to shed light on the correlations between oligomeric state and catalytic activity. A better understanding of the molecular mechanisms governing the reactive oxygen species detoxification in trypanosomatids is of great importance for the development of new trypanocidal drugs.
Acknowledgments
This research was supported by grants from Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES).
References
- Desjeux, P. (2004). Nature Rev. Microbiol. 2, 692. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Eslami, G., Frikha, F., Salehi, R., Khamesipour, A., Hejazi, H. & Nilforoushzadeh, M. A. (2011). Mol. Biol. Rep. 38, 3765–3776. [DOI] [PubMed]
- Guimarães, B. G., Sanfelici, L., Neuenschwander, R. T., Rodrigues, F. & Grizolli, W. C. (2009). J. Synchrotron Radiat. 16, 69–75. [DOI] [PubMed]
- Hirotsu, S., Abe, Y., Okada, K., Nagahara, N., Hori, H., Nishino, T. & Hakoshima, T. (1999). Proc. Natl Acad. Sci. USA, 96, 12333–12338. [DOI] [PMC free article] [PubMed]
- Iyer, J. P. (2008). Mol. Microbiol. 2, 372–391. [DOI] [PubMed]
- Krieger, S., Schwarz, W., Ariyanayagam, M. R., Fairlamb, A. H., Krauth-Siegel, R. L. & Clayton, C. (2000). Mol. Microbiol. 35, 542–552. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Nogoceke, E., Gommel, D. U., Kiess, M., Kalisz, H. M. & Flohé, L. (1997). Biol. Chem. 378, 827–836. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Piñeyro, M. D., Arcari, T., Robello, C., Radi, R. & Trujillo, M. (2011). Arch. Biochem. Biophys. 507, 287–295. [DOI] [PubMed]
- Reithinger, R., Dujardin, J. C., Louzir, H., Pirmez, C., Alexander, B. & Brooker, S. (2007). Lancet Infect. Dis. 7, 581–596. [DOI] [PubMed]
- Tovar, J., Cunningham, M. L., Smith, A. C., Croft, S. L. & Fairlamb, A. H. (1998). Proc. Natl Acad. Sci. USA, 95, 5311–5316. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (1998). Acta Cryst. D54, 400–402. [DOI] [PubMed]
- Wheeler, R. J., Gluenz, E. & Gull, K. (2011). Mol. Microbiol. 79, 647–662. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- World Health Organization (2010). World Health Organ. Tech. Rep. Ser. 949, 1–186. [PubMed]