

The serine acetylxylan esterase from G. stearothermophilus (Axe2) has been crystallized in the tetragonal space group I422. Complete diffraction data sets have been measured for the selenomethionine derivative (SAD data, 1.70 Å resolution) and the wild-type enzyme (1.85 Å resolution) to be used for a full three-dimensional structural analysis of the Axe2 protein.
Keywords: Geobacillus stearothermophilus, xylan esterases, serine esterases, selenium absorption edge, SAD data collection
Abstract
Acetylxylan esterases are part of the hemi-cellulolytic system of many microorganisms which utilize plant biomass for growth. Xylans, which are polymeric sugars that constitute a significant part of the plant biomass, are usually substituted with acetyl side groups attached at position 2 or 3 of the xylose backbone units. Acetylxylan esterases hydrolyse the ester linkages of the xylan acetyl groups and thus improve the ability of main-chain hydrolysing enzymes to break down the sugar backbone units. As such, these enzymes play an important part in the hemi-cellulolytic utilization system of many microorganisms that use plant biomass for growth. Interest in the biochemical characterization and structural analysis of these enzymes stems from their numerous potential biotechnological applications. An acetylxylan esterase (Axe2) of this type from Geobacillus stearothermophilus T-6 has recently been cloned, overexpressed, purified, biochemically characterized and crystallized. One of the crystal forms obtained (RB1) belonged to the tetragonal space group I422, with unit-cell parameters a = b = 110.2, c = 213.1 Å. A full diffraction data set was collected to 1.85 Å resolution from flash-cooled crystals of the wild-type enzyme at 100 K using synchrotron radiation. A selenomethionine derivative of Axe2 has also been prepared and crystallized for single-wavelength anomalous diffraction experiments. The crystals of the selenomethionine-derivatized Axe2 appeared to be isomorphous to those of the wild-type enzyme and enabled the measurement of a full 1.85 Å resolution diffraction data set at the selenium absorption edge and a full 1.70 Å resolution data set at a remote wavelength. These data are currently being used for three-dimensional structure determination of the Axe2 protein.
1. Introduction
Acetylxylan esterases are part of the hemi-cellulolytic system of many microorganisms which utilize plant biomass for growth. Xylans are in many cases decorated with acetyl side groups attached at position 2 or 3 of the xylose backbone units. For example, 70% of the xylose units in 4-O-methyl-d-glucuronoxylan, the main hardwood hemi-cellulose, are acetylated (Sjöström, 1993 ▶). Acetylxylan esterases hydrolyse the ester linkages of acetyl groups in xylan and thus improve the ability of the main-chain hydrolysing enzymes to break down the sugar backbone units (Biely et al., 2011 ▶). Many of the acetylxylan esterases studied and reported to date have been classified in the CAZy database and are found in eight of the 16 carbohydrate esterase (CE) families characterized so far (Cantarel et al., 2009 ▶). Families CE3 (acetylxylan esterases) and CE12 (pectin acetylesterases, rhamnogalacturonan acetylesterases and acetylxylan esterases) are also classified as lipase GDSL family proteins (Pfam accession number PF00657). Geobacillus stearothermophilus T-6 is a Gram-positive thermophilic bacterium that possesses an extensive hemi-cellulolytic system, with 40 genes involved in the utilization of hemi-cellulose (Shulami et al., 1999 ▶, 2007 ▶; Tabachnikov & Shoham, 2013 ▶). For the utilization of xylan, the bacterium initially secretes an extracellular xylanase (Gat et al., 1994 ▶; Khasin et al., 1993 ▶; Lapidot et al., 1996 ▶; Bar et al., 2004 ▶), which partially degrades xylan to decorated xylo-oligomers that are transported into the cell via a specific ATP-binding cassette (ABC) transport system (Shulami et al., 2007 ▶). Inside the cell, the decorated xylo-oligomers are hydrolysed by side-chain-cleaving enzymes, including arabinofuranosidases (Shallom, Belakhov, Solomon, Gilead-Gropper et al., 2002 ▶; Shallom, Belakhov, Solomon, Shoham et al., 2002 ▶; Hövel et al., 2003 ▶) and glucuronidase (Zaide et al., 2001 ▶; Golan et al., 2004 ▶; Shallom et al., 2004 ▶), and finally by intracellular xylanase (Solomon et al., 2007 ▶) and xylosidases (Bravman et al., 2003 ▶; Ben-David et al., 2007 ▶).
We have recently identified in G. stearothermophilus the axe2 gene (GenBank accession number ABI49953.1) as part of a three-gene operon, which also includes xynB3 (encoding β-xylosidase) (Brüx et al., 2006 ▶; Shallom et al., 2005 ▶) and an uncharacterized gene, xynX (GenBank accession number DQ868502.2). According to sequence similarities, the axe2 gene product, Axe2, is an intracellular serine hydrolase belonging to the lipase GDSL family (UniProtKB/TrEMBL accession number Q09LX1) and is made up of 219 amino acids with a calculated molecular mass of 24 770 Da. The lipase GDSL family is one of four families that make up the SGNH hydrolase superfamily (Pfam clan accession number CL0264) (Akoh et al., 2004 ▶; Finn et al., 2010 ▶). Axe2 was characterized biochemically, and the purified protein completely deacetylates xylobiose peracetate (fully acetylated) and hydrolyses the synthetic substrates 2-naphthyl acetate, 4-nitrophenyl acetate, 4-methylumbelliferyl acetate and phenyl acetate. Based on the genetic context of the axe2 gene and its substrate specificity, Axe2 is an acetylxylan esterase. Based on bioinformatics analysis, Axe2 and its homologues do not belong to any known family in the CAZy database and thus represent a new family of carbohydrate esterases (Alalouf et al., 2011 ▶).
In this report we describe the crystallization of Axe2 and the preliminary crystallographic characterization of the resulting Axe2 crystals. Several crystal forms have been obtained from the recombinant WT (wild-type) enzyme, one of which (RB1 form) was found to be of sufficient quality and stability for high-resolution structural analysis. Similar crystals were obtained for the selenium derivative of Axe2 (Se-Axe2), where seven of the methionine residues have been replaced with selenium-methionine residues. A complete SAD (single-wavelength anomalous diffraction) data set at the selenium absorption edge and at a remote wavelength has been collected on the Se-Axe2 crystals to 1.85 and 1.70 Å resolution, respectively, together with a complete non-SAD data set which was collected on the WT-Axe2 crystals to 1.85 Å resolution. These data sets should be used for phase determination and a complete three-dimensional structure determination of the Axe2 protein.
2. Experimental
2.1. Purification of WT-Axe2
Wild-type Axe2 (WT-Axe2) was purified from an overexpressing strain of Escherichia coli. Briefly, the axe2 gene was cloned into the pET9d expression vector (Novagen) and overexpressed in E. coli BL21(DE3) cells grown in Terrific Broth medium. The extract was heat treated (333 K for 30 min), centrifuged and the soluble fraction was subject to gel filtration (Alalouf et al., 2011 ▶).
2.2. Production and purification of Se-Axe2
The selenium-methionine derivative of Axe2 (Se-Axe2) was produced using E. coli B834(DE3) (Met−)-competent cells transformed with the pET9d-axe2 vector and grown on defined medium consisting of the following in g l−1: KH2PO4, 6; Na2HPO4, 12; NH4Cl, 2; MgSO4
 7H2O, 0.5; FeSO4, 0.025; and 1 µg ml−1 of riboflavin, pyridoxine monohydrochloride and thiamin. The medium was supplemented with 4 g l−1 glucose, 50 µg ml−1 of amino acids (Asn, Asp, Cys, Glu, Ala, Arg, Val, Gln, Gly, His, Ile, Leu, Lys, Pro, Ser, Thr, Phe, Trp, Tyr), 50 µg ml−1 seleno-l-methionine and 30 µg ml−1 kanamycin. The culture was induced with IPTG (isopropyl β-d-1-thiogalactopyranoside) (0.4 mM final concentration) at an OD600 of 1.4–2.2 and further incubated to a final OD600 of 2.8–4.5. The protein was purified according to the procedure used for WT-Axe2 (Alalouf et al., 2011 ▶), but the heat treatment was done at 328–330 K for 30 min. Following gel filtration, the protein was >90% pure based on SDS–PAGE, with a total yield of about 80 mg protein per 1 l culture.
7H2O, 0.5; FeSO4, 0.025; and 1 µg ml−1 of riboflavin, pyridoxine monohydrochloride and thiamin. The medium was supplemented with 4 g l−1 glucose, 50 µg ml−1 of amino acids (Asn, Asp, Cys, Glu, Ala, Arg, Val, Gln, Gly, His, Ile, Leu, Lys, Pro, Ser, Thr, Phe, Trp, Tyr), 50 µg ml−1 seleno-l-methionine and 30 µg ml−1 kanamycin. The culture was induced with IPTG (isopropyl β-d-1-thiogalactopyranoside) (0.4 mM final concentration) at an OD600 of 1.4–2.2 and further incubated to a final OD600 of 2.8–4.5. The protein was purified according to the procedure used for WT-Axe2 (Alalouf et al., 2011 ▶), but the heat treatment was done at 328–330 K for 30 min. Following gel filtration, the protein was >90% pure based on SDS–PAGE, with a total yield of about 80 mg protein per 1 l culture.
2.3. Crystallization experiments
Crystallization experiments were set up immediately after the last purification step of the recombinant Axe2 protein. The purified protein was concentrated, using Centricon centrifugal concentrators (Millipore, Massachusetts, USA), to approximately 6 mg ml−1 and this protein solution was directly used for the crystallization experiments. All initial crystallization experiments were performed by the hanging-drop vapour-diffusion method, using an extensive series of different factorial screens (Jancarik & Kim, 1991 ▶). In general, these initial conditions were based on commercially available sets of screens. Once positive results were obtained, further refinement of these crystallization conditions was performed with specially prepared solutions, optimizing parameters such as pH, ionic strength and protein concentration (Almog et al., 1993 ▶, 1994 ▶; Teplitsky et al., 1997 ▶, 1999 ▶, 2000 ▶).
The final Axe2 protein crystallization drops were prepared by mixing the concentrated or quantitatively diluted protein solution (in the range of 3–6 mg ml−1) with an equal amount of each of the specific screen conditions, to a final drop volume of 5.0 µl. Each of these protein drops was suspended over a 1.0 ml reservoir solution in 4 × 6 VDX crystallization plates (Hampton Research, California, USA), for a period of about 1–10 d at a constant temperature of 293 K. A number of different crystal forms were obtained from these initial experiments, yet only one of them (RB1) was found to be suitable for further crystallographic analysis (see below).
All the diffraction data measurements were performed at the European Synchrotron Research Facility (ESRF, Grenoble, France). Processing, reduction, indexing, integration and scaling of the diffraction data were conducted using the DENZO and SCALEPACK crystallographic programs (Otwinowski, 1993 ▶; Otwinowski & Minor, 1997 ▶).
3. Results and discussion
3.1. Crystal forms of WT-Axe2
The initial crystallization screens indicated four different conditions for which reasonable single crystals could be obtained by the hanging-drop method. All of these hits resulted from Wizard Screen I (WSI) (Emerald BioSystems, Bainbridge Island, Washington, USA). These conditions were then refined, resulting in the following four optimal reservoir solutions: condition 1 (starting from WSI #18) 1.0–1.4 M K/Na tartrate, 0.3 M NaCl, 0.1 M imidazole buffer pH 7.0–7.4; condition 2 (WSI #20) 0.25 M NaH2PO4/1.0 M K2HPO4, 0.1 M imidazole buffer pH 8.0; condition 3 (WSI #21) 20%(w/v) PEG 8000, 0.1 M HEPES buffer pH 7.5; condition 4 (WSI #12) 20%(w/v) PEG 1000, 0.2 M Ca(OAc)2, 0.1 M imidazole buffer pH 8.0. The crystals obtained with the condition 1 reservoir solution appeared usually as rectangular boxes (all dimensions in the range of 0.05–0.3 mm) and were hence categorized as the RB1 crystal habit (Fig. 1 ▶). Similar crystals were obtained with the condition 2 reservoir solution, which were hence categorized as the RB2 crystal habit. The crystals obtained with the condition 3 reservoir solution appeared usually as triangular plates (typical dimensions of 0.2 × 0.2 × 0.03 mm), categorized as the TP1 crystal habit. The crystals obtained with the condition 4 reservoir solution appeared usually as elongated rods (typical dimensions of 0.6 × 0.05 × 0.02 mm), categorized as the ER1 crystal habit.
Figure 1.

Typical crystals of the RB1 crystal habit (WT-Axe2). It is noted that the shape of these crystals varies between rectangular boxes (left), almost perfect cubes and various polyhedral prisms (right). Crystals of the rectangular box shape were used for the measurement of the full diffraction data sets described here at 1.85–1.70 Å resolution.
Of the four crystal habits observed, it was demonstrated that the RB1 and RB2 habits belong to the same crystal form, with an identical space group and very similar unit-cell dimensions (see below), yet the RB1 crystals appeared to give a significantly better diffraction pattern. Crystals of the TP1 habit were demonstrated to belong to a primitive orthorhombic crystal system with average unit-cell dimensions of a = 67.1, b = 156.2 and c = 307.6 Å, but they showed a relatively smeared diffraction pattern that did not exceed the 3 Å resolution limit. The ER1 crystals appeared to diffract even worse (in terms of quality and resolution) than the TP1 crystals. Since the RB1 crystals proved to be more reproducible and better diffracting compared to the other crystal habits, it was decided to continue further crystallographic studies only with this type of crystal.
3.2. Crystallographic characterization of the RB1 crystal habit
The best RB1 crystals were obtained at a protein concentration of 6 µg µl−1 and a reservoir solution of 1.2 M K tartrate, 0.3 M NaCl, 0.1 M imidazole buffer pH 7.2. These crystals could be initially observed after about 1 d and grew to their full size after about an additional 2–3 d. Their final shape varied between almost perfect cubes (typical dimensions of about 0.2 × 0.2 × 0.2 mm) and elongated rectangular boxes (typical dimensions of about 0.4 × 0.3 × 0.2 mm, Fig. 1 ▶), with no apparent differences in their diffraction qualities.
Several RB1 crystals were used for a detailed crystallographic characterization and measurement of X-ray diffraction data at cryogenic conditions. These experiments were done using X-ray synchrotron radiation (λ = 0.95–0.98 Å) and a CCD area detector (MAR-225, MAR-Research Inc., USA) on the BM14 beamline of the ESRF. The crystal freezing procedure used for these experiments included a short soaking of the target crystal (about 20–40 s) in a cryoprotecting solution containing the original crystallization reservoir solution (1.2 M K tartrate, 0.3 M NaCl, 0.1 M imidazole buffer pH 7.2) and 20%(v/v) glycerol. Such pre-soaked crystals were then submitted to immediate flash-cooling, directly within a cold nitrogen-gas stream (100 K, Oxford Cryosystem).
The observed diffraction pattern of some of these crystals exceeded the 1.9 Å resolution limit and indicated that the crystals belong to a body-centred tetragonal crystal system (space group I422), with average crystallographic unit-cell dimensions of a = b = 110.2 and c = 213.1 Å. Different crystals of this crystallization batch gave similar unit-cell dimensions with an overall deviation from these average values of less than 0.2%.
3.3. X-ray diffraction data for WT-Axe2
One of the WT-Axe2 crystals (of the RB1 crystal habit) was used for a full high-resolution X-ray diffraction data measurement. An oscillation data set (Δϕ = 1.0°, 15 s exposure, 120 frames) was measured on a single crystal on the ESRF/BM14 beamline (λ = 0.954 Å, 100 K). The raw CCD diffraction images were processed with the DENZO and SCALEPACK software packages (Otwinowski, 1993 ▶). A total of 547 192 accepted reflections [F > 0σ(F)] were measured in the 30.0–1.85 Å resolution range, resulting in 55 865 independent reflections with 100.0% completeness to 1.85 Å resolution (100.0% completeness also for the highest-resolution shell of 1.88–1.85 Å). The overall redundancy in the combined data set was 9.8, the overall mosaicity was 0.31, the average 〈I/σ(I)〉 was 10.8 and the final squared R sym (or R merge) for the whole data was 5.8% (47.5% for the highest-resolution shell), indicating a full diffraction data set of relatively high quality (Table 1 ▶).
Table 1. Representative parameters from the crystallographic data measurement of Axe2.
Values in parentheses are for the outer diffraction shell.
| Crystal | Se-Axe2 (edge) | Se-Axe2 (remote HR) | WT-Axe2 |
|---|---|---|---|
| Crystal habit | RB1 | RB1 | RB1 |
| Beamline | BM14, ESRF | BM14, ESRF | BM14, ESRF |
| Wavelength (Å) | 0.978 | 0.954 | 0.954 |
| Space group | I422 | I422 | I422 |
| Unit-cell parameters (Å) | a = b = 110.42, c = 212.87 | a = b = 110.30, c = 213.09, | a = b = 109.83, c = 213.31 |
| Resolution range (Å) | 30.0–1.85 (1.88–1.85) | 30.0–1.70 (1.73–1.70) | 30.00–1.85 (1.88–1.85) |
| No. of reflections | |||
| Total | 1664726 | 590353 | 547192 |
| Unique | 56384 | 71600 | 55865 |
| Multiplicity | 29.5 (29.3) | 8.2 (8.1) | 9.8 (8.3) |
| 〈I〉/〈σ(I)〉 | 6.4 (5.2) | 8.3 (3.5) | 10.8 (3.3) |
| Mosaicity | 0.243 | 0.318 | 0.310 |
| Completenes (%) | 100 (100) | 98.9 (99.9) | 100 (100) |
| Linear R sym † (%) | 10.6 (67.5) | 7.7 (64.4) | 7.0 (61.1) |
| Square R sym ‡ (%) | 7.6 (51.2) | 5.6 (46.1) | 5.8 (47.5) |
Linear R
sym = 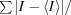
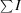 .
.
Square R
sym = 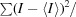
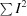 .
.
At this point it is possible to estimate the protein content of the crystallographic unit cell by a rough calculation of the water content in the crystal and the specific ratio of volume/protein (V M; Matthews, 1968 ▶). The range of V M values observed originally for soluble protein crystals was 1.68–3.5 Å3 Da−1 with a mean of 2.17 Å3 Da−1 (based on 116 proteins). A wider range and a mean of 2.69 Å3 Da−1 was calculated later, based on 10 471 proteins (Kantardjieff & Rupp, 2003 ▶). These mean V M values correspond to mean water contents of 43 and 47%, respectively. The volume of the WT-Axe2/RB1 crystallographic unit cell, as determined from the mean value of the cell dimensions at 100 K, is 2.57 × 106 Å3. Assuming that the V M value here is within the normal range, there should be two or three molecules of Axe2 monomers (219 amino acids; molecular weight 24 770 Da; Alalouf et al., 2011 ▶) in the crystallographic asymmetric unit. With two molecules in the RB1 asymmetric unit (32 in the unit cell), the calculated V M is 3.26 Å3 Da−1 and the corresponding solvent content in the crystals is 62.34%. With three molecules in the asymmetric unit (48 in the unit cell), the calculated V M is 2.18 Å3 Da−1 and the corresponding solvent content in the crystals is 43.51%. Both possibilities seem reasonable considering their calculated V M values and water content. Although the values of the second possibility (three monomers per asymmetric unit) are significantly closer to the mean values observed, the tetragonal crystal symmetry (I422) makes the first possibility (two monomers per asymmetric unit) more likely, as was also indicated by self-rotation calculations. The actual content of the asymmetric unit (and the unit cell) will be unequivocally resolved only when the crystallographic protein structure is fully determined.
3.4. X-ray diffraction data for Se-Axe2
The fully replaced selenium-methionine protein (Se-Axe2) was crystallized in the same conditions described for the WT enzyme and the resulting RB1 crystals were confirmed to be completely isomorphous with the corresponding WT-Axe2 crystals. One of these Se-Axe2 crystals was used for the measurement of two complete diffraction data sets, one at the selenium absorption edge (λ = 0.978 Å, 1.85 Å resolution, Table 1 ▶) and one at a remote wavelength (λ = 0.954 Å, 1.80 Å resolution, data not shown). Both data sets were collected on the same crystal on the ESRF/BM14 beamline (at 100 K). The data set at the selenium edge (Se-Axe2 edge) included 360 oscillation frames (Δϕ = 1.0°, 5 s exposure), resulting in a total of 1 664 726 accepted reflections [F > 0σ(F)] and 56 384 independent reflections in the 30.0–1.85 Å resolution range [100.0% completeness, overall redundancy 29.5, overall mosaicity 0.243, average 〈I/σ(I)〉 6.4, final square R sym 7.6%] (Table 1 ▶). Another Se-Axe2 crystal from the same crystallization batch was used for a fresh high-resolution data measurement at the same remote wavelength (λ = 0.954 Å, 1.70 Å resolution) (Fig. 2 ▶). These data (Se-Axe2 remote HR) included 198 oscillation frames (Δϕ = 0.5°, 5 s exposure), resulting in a total of 590 353 accepted reflections [F > 0σ(F)] and 71 600 independent reflections in the 30.0–1.70 Å resolution range [98.9% completeness, overall redundancy 8.2, overall mosaicity 0.32, average 〈I/σ(I)〉 8.3, final square R sym 5.6%] (Table 1 ▶). The parameters for both data sets indicate again a full diffraction data set of relatively high quality at the corresponding resolutions.
Figure 2.

X-ray diffraction pattern of Se-Axe2 (remote HR) obtained using a synchrotron source (BM14, ESRF). The outer circle corresponds to a 1.70 Å resolution limit. The insets represent magnified views of the sections indicated by the corresponding squares (left – medium resolution; right – high resolution).
Since the anomalous signal expected from seven selenium atoms per monomer of 219 amino-acid residues is relatively high, one should be able to use the three data sets collected on the two Se-Axe2 crystals for crystallographic phasing via the SAD methodology. Such phasing should lead to the structure determination of the Se-Axe2 protein at 1.85 Å resolution and its extension and refinement at 1.70 Å resolution. The structure of the Se-protein can then be used for the structural analysis of the WT protein (at 1.85 Å resolution), using the WT-Axe2 crystallographic data described above. Such analyses are currently in progress.
Acknowledgments
This work was supported by the Israel Science Foundation grant Nos. 500/10 and 152/11, the I-CORE Program of the Planning and Budgeting Committee, the Ministry of Environmental Protection and the Grand Technion Energy Program (GTEP), and comprises part of The Leona M. and Harry B. Helmsley Charitable Trust reports on Alternative Energy series of the Technion, Israel Institute of Technology and the Weizmann Institute of Science. NEC thanks the UK Engineering and Physical Sciences Research Council (EPSRC) grant Nos. EP/G027005 for financial support. We thank the staff at the European Synchrotron Research Facility (ESRF, BM14 beamline) for their helpful support in the X-ray synchrotron data measurement and analysis. The synchrotron experiments at the ESRF were supported also by the ESRF internal funding program. YS holds the Erwin and Rosl Pollak Chair in Biotechnology at the Technion.
References
- Akoh, C. C., Lee, G. C., Liaw, Y. C., Huang, T. H. & Shaw, J. F. (2004). Prog. Lipid Res. 43, 534–552. [DOI] [PubMed]
- Alalouf, O., Balazs, Y., Volkinshtein, M., Grimple, Y., Shoham, G. & Shoham, Y. (2011). J. Biol. Chem. 286, 41933–42001. [DOI] [PMC free article] [PubMed]
- Almog, O., Greenblatt, H. M., Spungin, A., Ben-Meir, D., Blumberg, S. & Shoham, G. (1993). J. Mol. Biol. 230, 342–344. [DOI] [PubMed]
- Almog, O., Klein, D., Braun, S. & Shoham, G. (1994). J. Mol. Biol. 235, 760–762. [DOI] [PubMed]
- Bar, M., Golan, G., Nechama, M., Zolotnitsky, G., Shoham, Y. & Shoham, G. (2004). Acta Cryst. D60, 545–549. [DOI] [PubMed]
- Ben-David, A., Bravman, T., Balazs, Y. S., Czjzek, M., Schomburg, D., Shoham, G. & Shoham, Y. (2007). ChemBioChem, 8, 2145–2151. [DOI] [PubMed]
- Biely, P., Mastihubová, M., Tenkanen, M., Eyzaguirre, J., Li, X. L. & Vršanská, M. (2011). J. Biotechnol. 151, 137–142. [DOI] [PubMed]
- Bravman, T., Zolotnitsky, G., Belakhov, V., Shoham, G., Henrissat, B., Baasov, T. & Shoham, Y. (2003). Biochemistry, 42, 10528–10536. [DOI] [PubMed]
- Brüx, C., Ben-David, A., Shallom-Shezifi, D., Leon, M., Niefind, K., Shoham, G., Shoham, Y. & Schomburg, D. (2006). J. Mol. Biol. 359, 97–109. [DOI] [PubMed]
- Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V. & Henrissat, B. (2009). Nucleic Acids Res. 37, D233–D238. [DOI] [PMC free article] [PubMed]
- Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., Gavin, O. L., Gunasekaran, P., Ceric, G., Forslund, K., Holm, L., Sonnhammer, E. L., Eddy, S. R. & Bateman, A. (2010). Nucleic Acids Res. 38, D211–D222. [DOI] [PMC free article] [PubMed]
- Gat, O., Lapidot, A., Alchanati, I., Regueros, C. & Shoham, Y. (1994). Appl. Environ. Microbiol. 60, 1889–1896. [DOI] [PMC free article] [PubMed]
- Golan, G., Shallom, D., Teplitsky, A., Zaide, G., Shulami, S., Baasov, T., Stojanoff, V., Thompson, A., Shoham, Y. & Shoham, G. (2004). J. Biol. Chem. 279, 3014–3024. [DOI] [PubMed]
- Hövel, K., Shallom, D., Niefind, K., Belakhov, V., Shoham, G., Baasov, T., Shoham, Y. & Schomburg, D. (2003). EMBO J. 22, 4922–4932. [DOI] [PMC free article] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409–411.
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Khasin, A., Alchanati, I. & Shoham, Y. (1993). Appl. Environ. Microbiol. 59, 1725–1730. [DOI] [PMC free article] [PubMed]
- Lapidot, A., Mechaly, A. & Shoham, Y. (1996). J. Biotechnol. 51, 259–264. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 56–62. Warrington: Daresbury Laboratory.
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Shallom, D., Belakhov, V., Solomon, D., Gilead-Gropper, S., Baasov, T., Shoham, G. & Shoham, Y. (2002). FEBS Lett. 514, 163–167. [DOI] [PubMed]
- Shallom, D., Belakhov, V., Solomon, D., Shoham, G., Baasov, T. & Shoham, Y. (2002). J. Biol. Chem. 277, 43667–43673. [DOI] [PubMed]
- Shallom, D., Golan, G., Shoham, G. & Shoham, Y. (2004). J. Bacteriol. 186, 6928–6937. [DOI] [PMC free article] [PubMed]
- Shallom, D., Leon, M., Bravman, T., Ben-David, A., Zaide, G., Belakhov, V., Shoham, G., Schomburg, D., Baasov, T. & Shoham, Y. (2005). Biochemistry, 44, 387–397. [DOI] [PubMed]
- Shulami, S., Gat, O., Sonenshein, A. L. & Shoham, Y. (1999). J. Bacteriol. 181, 3695–3704. [DOI] [PMC free article] [PubMed]
- Shulami, S., Zaide, G., Zolotnitsky, G., Langut, Y., Feld, G., Sonenshein, A. L. & Shoham, Y. (2007). Appl. Environ. Microbiol. 73, 874–884. [DOI] [PMC free article] [PubMed]
- Sjöström, E. (1993). Wood Chemistry, Fundamentals and Applications, 2nd ed. London: Academic Press.
- Solomon, V., Teplitsky, A., Shulami, S., Zolotnitsky, G., Shoham, Y. & Shoham, G. (2007). Acta Cryst. D63, 845–859. [DOI] [PubMed]
- Tabachnikov, O. & Shoham, Y. (2013). FEBS J. 280, 950–964. [DOI] [PubMed]
- Teplitsky, A., Feinberg, H., Gilboa, R., Lapidot, A., Mechaly, A., Stojanoff, V., Capel, M., Shoham, Y. & Shoham, G. (1997). Acta Cryst. D53, 608–611. [DOI] [PubMed]
- Teplitsky, A., Shulami, S., Moryles, S., Shoham, Y. & Shoham, G. (2000). Acta Cryst. D56, 181–184. [DOI] [PubMed]
- Teplitsky, A., Shulami, S., Moryles, S., Zaide, G., Shoham, Y. & Shoham, G. (1999). Acta Cryst. D55, 869–872. [DOI] [PubMed]
- Zaide, G., Shallom, D., Shulami, S., Zolotnitsky, G., Golan, G., Baasov, T., Shoham, G. & Shoham, Y. (2001). Eur. J. Biochem. 268, 3006–3016. [DOI] [PubMed]