

Abstract
A catalytic asymmetric double (1,3)-dipolar cycloaddition reaction has been developed. Using a chiral silver catalyst, enantioenriched pyrrolizidines can be prepared in one flask from inexpensive, commercially available starting materials. The pyrrolizidine products contain a variety of substitution patterns and as many as six stereogenic centers.
Introduction
Pyrrolizidines constitute a large family of biologically active natural products and synthetic pharmaceutical agents.1 They include plant-derived polyhydroxylated alkaloids such as alexine (1),2 hyacinthacine B2(2),3 and casuarine (3)4 – compounds that have garnered significant interest as glycosidase and glycosyl transfer inhibitors5 – as well as the lypophilic frog toxins exemplified by 223H (4).6 In addition, several synthetic pyrrolizidines have been reported as drug candidates. For example, pyrrolizidines 5 and 6 are selective antagonists of 5-hydroxytryptamine receptor 4 (5-HT4),7,8 while 7 is a potent antagonist of human neurokinin receptor 1 (hNK1).9 Although several strategies have been developed to prepare pyrrolizidines,10,11, they often require multi-step syntheses and do not readily provide access to a diverse array of substituent patterns. Moreover, the enantioselective synthesis of these frameworks can be challenging. Herein, we report the catalytic asymmetric preparation of pyrrolizidines from simple, inexpensive starting materials.12 This methodology enables the programmable incorporation of a variety of functional groups, and provides direct access to an array of highly substituted pyrrolizidines.
In the course of our synthetic studies toward the natural product acetylaranotin, we sought to prepare pyrrolidine 10 by a catalytic asymmetric (1,3)-dipolar cycloaddition (DCA) (Scheme 1).13 Although there are several reports of catalytic asymmetric (1,3)-DCAs between α-imino esters and acrylates,14,15,16 at the outset of our studies, there were no examples of enantioselective reactions between cinnamaldehyde-derived imines and simple, unsubstituted acrylates.17 This might be related to the instability of compounds such as 8, which are prone to polymerization upon standing. We were therefore pleased to find that adaptation of conditions originally developed by Oh and coworkers,18 which utilize brucin-OL (13, Table 1) as a chiral ligand, provided the desired pyrrolidine 10 in excellent ee, albeit in modest yield.
Scheme 1.

Isolation of pyrrolizidine 12.
Table 1.
Optimization of the catalytic asymmetric (1,3)-DCA reaction between glycinate imine 8 and tert-butyl acrylate (9).
 | |||||
|---|---|---|---|---|---|
| entry | catalyst/ligand/additivea | solvent | temp | yieldb | eec |
| (°C) | (%) | (%) | |||
| 1 | AgOAc, 14 | Et2O | 0 °C | 53 | −63 |
| 2 | AgClO4, 15, DABCO | PhMe | 0 °C | 59 | 45 |
| 3 | AgOAc, 16, DIPEAd | THF | −45 °C | 62 | 90 |
| 4 | AgOAc, 16, DIPEA | DCM | −45 °C | 36 | 78 |
| 5 | AgOAc, 16, DIPEA | CHCl3 | −45 °C | 5 | -- |
| 6 | AgOAc, 16, DIPEA | PhMe | −45 °C | 60 | 90 |
| 7 | AgOAc, 16, DIPEA | Et2O | −45 °C | 53 | 90 |
See Supporting Information for reaction details.
Isolated yield.
Determined by SFC using chiral stationary phase.
3 mol % each AgOAc and QUINAP, 10 mol % DIPEA.
A major side product formed in the reaction was isolated and characterized, and discovered to be pyrrolizidine 12.19,20 Pyrrolizidine 12 is presumably produced by condensation of pyrrolidine 10 with imine 8 or cinnamaldehyde (resulting from imine hydrolysis) to generate azomethine ylide 11, which undergoes a second highly diastereoselective (1,3)-DCA with 8. Pyrrolizidine 12 contains four stereogenic centers and is formed as a single diastereomer; the diastereoselectivity is consistent with an endo -selective (1,3)-DCA in which the dipolarophile approaches the face of azomethine ylide 11 opposite to the styrenyl and ester substituents. Given the importance of the pyrrolizidine framework in biologically active alkaloids and synthetic pharmaceutical agents, we sought to improve the yield and explore the substrate scope of this transformation.
Results and Discussion
Although our initial discovery of pyrrolizidine formation was in the context of the CuI/brucin-OL catalyzed (1,3)-DCA, the need for a 24-h catalyst generation period, coupled with variability in the yield of pyrrolizidine formation, led us to pursue other catalyst systems for the purposes of this methodological study. Given that the enantiomeric excess of pyrrolizidine 12 is established during the first (1,3)-DCA, we initially conducted a survey of several chiral catalyst systems16c,d,f for their ability to provide enantioenriched pyrrolidine 10; a selection of results are shown in Table 1. These studies revealed that good enantioinduction could be obtained using AgOAc (3 mol %) and (S )-QUINAP (16, 3 mol %) at −45 °C (Table 1, entry 3), conditions originally reported by Schreiber to catalyze (1,3)-DCA of aryl aldehyde-derived α-imino esters.16c,21 Whereas halogenated solvents resulted in low yields and modest enantioselectivity, ethereal solvents were more promising, with THF providing the highest combination of yield and ee.
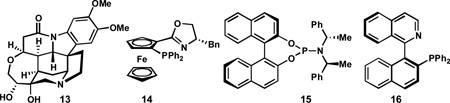
Having identified an operationally simple catalyst system to prepare pyrrolidine 10, we began to investigate pyrrolizidine formation. We were pleased to find that treatment of a mixture of cinnamaldehyde-derived α-imino ester 8, AgOAc (3 mol %), QUINAP (3 mol %) and DIPEA (10 mol %) with tert-butyl acrylate (9, 1.5 equiv) in THF at −45 °C for 24 h, followed by addition of cinnamaldehyde (1.0 equiv) and additional 9 (5.0 equiv) with warming to 23 °C provided pyrrolizidine 12 in 74% yield and 90% ee (Scheme 2).22,23 Notably, warmer temperatures are required for the second (1,3)-DCA, which proceeds very slowly at –45 °C. It is important that imine 8 is consumed before the reagents are added for the second cycloaddition; if 8 remains, it reacts rapidly and less selectively with tert-butyl acrylate upon warming to give 10,24 which can lead to the isolation of pyrrolizidine 12 in reduced ee.
Scheme 2.

Optimized conditions for preparation of pyrrolizidine 12.
It is frequently observed that reactions involving two, sequential catalytic asymmetric steps can benefit from a Horeau-type amplification of the ee.25,26 In the present case, no change in ee is observed for pyrrolizidine 12 relative to pyrrolidine 10. Indeed, exposure of racemic pyrrolidine 10 to 0.25 equivalents of cinnamaldehyde under otherwise standard conditions provided pyrrolizidine 12 in 25% yield and 0% ee, indicating that no kinetic resolution of pyrrolidine 10 occurs (Scheme 3). These data suggest that the diastereoselectivity of the second (1,3)-DCA is substrate-controlled. The enantioinduction for Ag-catalyzed asymmetric (1,3)-DCAs is hypothesized to result from two-point binding of the deprotonated α-imino ester to the chiral silver complex. However, azomethine ylide 11 (see Scheme 1) cannot achieve this two-point binding mode. Although the silver complex accelerates the rate of the second (1,3)-DCA – perhaps by Lewis acid activation of the acrylate – it does not exert any significant “matching” or “mismatching” effect with the chiral azomethine ylide.
Scheme 3.

Influence of chiral catalyst on second (1,3)-DCA.
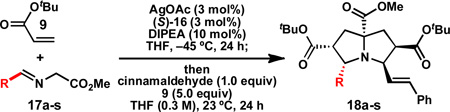
With conditions in hand to prepare pyrrolizidine 12 in good yield and high ee, attention turned to evaluating the substrate scope of the reaction (Table 2). A variety of substituted aryl aldehyde-derived α-imino esters 17 furnished the corresponding pyrrolizidines 18 in good yields with high enantioselectivity. Substitution at the o-, m-, and p-positions of the arene with both electron-donating and electron-withdrawing substituents is well-tolerated. In particular, α-imino esters bearing electron-withdrawing aryl substituents provide the pyrrolizidine products with uniformly high levels of enantioselectivity (Table 2, 18f–18j, 18m–18o). Notably, the α-imino ester derived from pyridine 3-carboxaldehyde is a suitable substrate, providing pyridyl-substituted pyrrolizidine 18r in good yield and good ee (entry 18). Alternatively, the 2-pyridyl α-imino ester provided pyrrolizidine 18s in low yield and modest ee (entry 19). It is possible that the proximal nitrogen results in an alternative binding mode between the azomethine ylide and the catalyst, decreasing the enantioselectivity during the first (1,3)-DCA.27 In addition, the pyrrolidine intermediate or the pyrrolizidine product (18s) might bind to and inhibit the silver catalyst.
Table 2.
Substrate scope: α-imino ester.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | yielda (%) |
eeb (%) |
entry | yielda (%) |
eeb (%) |
||
| 1 | 90 | 91 | 11d | 86 | 90 | ||
| 2 |  |
91 | 91 | 12 | 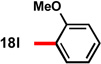 |
72 | 90 |
| 3c | 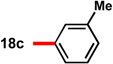 |
83 | 88 | 13 | 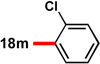 |
87 | 94 |
| 4c | 82 | 92 | 14 | 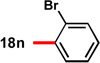 |
89 | 93 | |
| 5c |  |
78 | 88 | 15 |  |
82 | 92 |
| 6 | 87 | 93 | 15d |  |
76 | 92 | |
| 7 | 91 | 95 | 17c |  |
84 | 93 | |
| 8 | 89 | 92 | 18 | 90 | 90 | ||
| 9 | 70 | 96 | |||||
| 10 | 80 | 96 | 19 | 33 | 44 | ||
Isolated yield.
Determined by SFC using chiral stationary phase.
6 mol % each AgOAc and (S)-16 were employed.
6 mol % each AgOAc and (S)-16 were employed at 0.1 M reaction concentration.
In some cases, the pyrrolizidine products were obtained in lower ee using our standard reaction conditions (e.g. compounds 18c–e, 18q).28 It was determined that for these α-iminoesters, the first (1,3)-DCA proceeds slowly relative to the more electron-poor substrates; we reasoned that the remaining imine reacts with lower enantioselectively upon warming, which ultimately results in the formation of the pyrrolizidine with reduced ee. We hypothesized that increasing the catalyst loading should ensure complete consumption of the α-imino ester prior to warming the reaction for the second (1,3)-DCA. This hypothesis proved to be true; using 6 mol% each of AgOAc and (S)-16, pyrrolizidines 18c–e and 18q were obtained in good yields and high ee (Table 2, entries 3–5, and 17). Alternatively, the solubility of p -methoxy and 2-naphthyl α-iminoesters 17k and 17p was poor under our standard conditions;25 for these substrates the best results were obtained by lowering the overall reaction concentration (to 0.1 M versus 0.3 M) and increasing the catalyst loading to 6 mol% (Table 2, entries 11 and 16).
A variety of dipolarophiles can be used for the second (1,3)-DCA (Table 3). For example, use of E-1-nitro-2-phenylethylene as the second dipolarophile provides pyrrolizidine 19e in 89% yield and 90% ee. This compound contains six stereogenic centers and is isolated as a single diastereomer. Use of methyl methacrylate provides pyrrolizidine 19d, which contains an all-carbon quaternary center, in 91% yield and 90% ee.
Table 3.
Substrate scope: second dipolarophile.
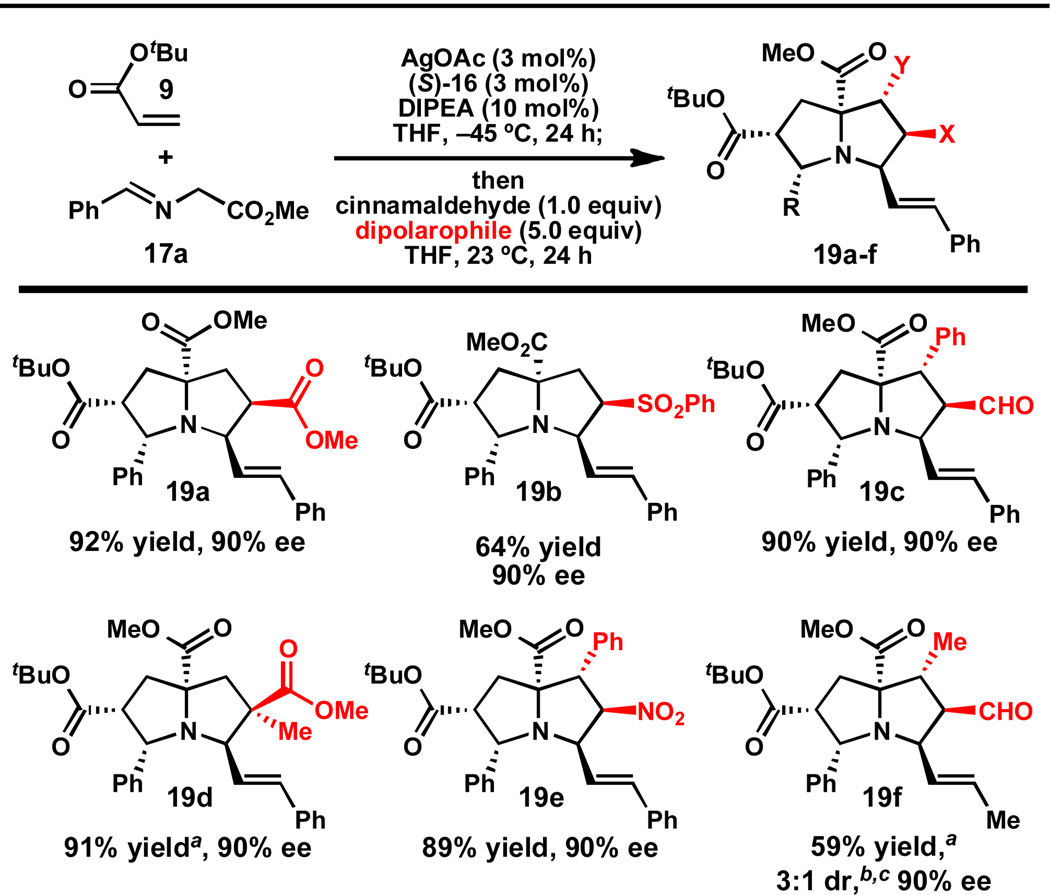 |
a10.0 equivs of dipolarophile were used.
cFor this reaction, crotonaldehyde was used instead of cinnamaldehyde.
Interestingly, α,β-unsaturated aldehydes appear to be uniquely well suited for generating the pyrrolidine-derived azomethine ylide for the second (1,3)-DCA. Attempts to employ aryl aldehydes (e.g. benzaldehyde) or alkyl aldehydes (e.g. 2-ethylbuytraldehyde) for the second (1,3)-DCA failed to provide the pyrrolizidine products in synthetically useful yields (see Supporting Information). We believe this unique reactivity of α,β-unsaturated aldehydes explains why standard catalytic, asymmetric (1,3)-DCAs are not plagued by unwanted pyrrolizidine formation, since α-imino esters derived from enals are rarely employed in methods development.
Using benzaldehyde-derived α-imino ester 17a, the catalytic asymmetric double (1,3)-DCA reaction has been conducted on gram-scale, providing pyrrolizidine ent-18a29 in 89% yield and slightly diminished ee (87%) (Scheme 3).30,31 Importantly, this compound can be selectively modified to give several intermediates capable of further derivitazation. For example, the more reactive and accessible methyl ester of ent-18a can be selectively saponified using LiOH or reduced using LiEt3BH to give carboxylic acid 20 or alcohol 21, respectively. Alternatively, the tert -butyl esters can be cleaved upon treatment with trifluoroacetic acid (TFA) to give dicarboxylic acid 22. Finally, the styrene of ent-18a can be oxidatively cleaved by ozonolysis and reduced in situ to provide amino alcohol 23 in 64% yield. These studies demonstrate that the individual functional groups of ent-18a can be chemoselectively modified.
Conclusions
In conclusion, a catalytic asymmetric double (1,3)-dipolar cycloaddition reaction has been developed. This reaction provides access to highly substituted, enantioenriched pyrrolizidines from inexpensive, commercially available starting materials. Depending on the second dipolarophile that is employed, pyrrolizidines containing as many as six stereogenic centers have been prepared with high levels of enantio- and diastereoselectivity. This methodology provides a versatile, programmable platform for the single-step synthesis of pyrrolizidines of unprecedented complexity. We expect that this reaction could be of use for the preparation of natural product analogues or new lead compounds for pharmaceutical studies.
Supplementary Material
Figure 1.

Pyrrolizidine-containing natural products and pharmaceutical agents.
Scheme 4.

Gram-scale synthesis of ent-18a and selective derivatization.
Acknowledgments
We thank the late Dr. Michael Day and Mr. Larry Henling for X-ray crystallographic structure determination, Dr. David vander Velde for assistance with NMR structure determination, as well as Prof. Brian Stoltz, Dr. Scott Virgil, and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment. Dr. Jacob Cha and Dr. Scott Virgil are acknowledged for assistance with QUINAP preparation. We also thank Sigma-Aldrich for a kind donation of chemicals. The Bruker KAPPA APEXII X-ray diffractometer was purchased through an award to the California Institute of Technology by the National Science Foundation (NSF) CRIF program (CHE-0639094). Fellowship support was provided by the Department of Defense (DoD) through the National Defense Science & Engineering Graduate Fellowship Program (J. A. C.), and by the NSF Graduate Research Fellowship Program (J. A. C. and A. D. L., Grant No. DGE-0703267). S. E. R. is a fellow of the Alfred P. Sloan Foundation and a Camille Dreyfus Teacher-Scholar. Financial support from the California Institute of Technology and the NIH (NIGMS RGM097582A) is gratefully acknowledged.
Footnotes
† Electronic Supplementary Information (ESI) available: experimental details, characterization data, X-ray data, and NMR spectral charts. See DOI: 10.1039/b000000x/
Notes and References
- 1.Hartmann T, Witte L. In: Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. Pergamon, Oxford: 1995. pp. 155–233. [Google Scholar]
- 2.Nash RJ, Fellows LE, Dring JV, Fleet GWJ, Derome AE, Hamor TA, Scofield AM, Watkin DJ. Tetrahedron Lett. 1988;29:2487. [Google Scholar]
- 3.Asano N, Kuroi H, Ikeda K, Kizu H, Kameda Y, Kato A, Adachi I, Watson AA, Nash RJ, Fleet GWJ. Tetrahedron: Asymm. 2000;11:1. [Google Scholar]
- 4.Nash RJ, Thomas PI, Waigh RD, Fleet GWJ, Wormald MR, Lilley PMD, Watkin DJ. Tetrahedron Lett. 1994;35:7849. [Google Scholar]
- 5.Asano N, Nash RJ, Molyneux RJ, Fleet GWJ. Tetrahedron: Asymm. 2000;11:1645. [Google Scholar]
- 6.(a) Daly JW. J. Nat. Prod. 1998;61:162. doi: 10.1021/np970460e. [DOI] [PubMed] [Google Scholar]; (b) Garraffo HM, Spande TF, Daly JW, Baldessari A, Gros EG. J. Nat. Prod. 1993;56:357. doi: 10.1021/np50093a008. [DOI] [PubMed] [Google Scholar]
- 7.Takeda M, Tsukamoto K, Mizutani Y, Suzuki T, Taniyama K K, K Jpn. J. Pharmacol. 1999:203. doi: 10.1254/jjp.79.203. [DOI] [PubMed] [Google Scholar]
- 8.Becker DP, Flynn DL, Moormann AE, Nosal R, Villamil CI, Loeffler R, Gullikson GW, Moummi C, Yang D-C. J. Med. Chem. 2006;49:1125. doi: 10.1021/jm0509501. [DOI] [PubMed] [Google Scholar]
- 9.Morriello J, DeVita RJ, Mills SG, Young JR, Lin P, Doss G, Chicchi GG, DeMartino J, Kurtz MM, Tsao K-LC, Carlson E, Townson K, Wheeldon A, Boyce S, Collinson N, Rupniak N, Moore S. Bioorg. Med. Chem. 2008;16:2156. doi: 10.1016/j.bmc.2007.11.081. [DOI] [PubMed] [Google Scholar]
- 10.Broggini G, Zecchi G. Synthesis. 1999:905. [Google Scholar]
- 11.Brandi A, Cardona F, Cicchi S, Cordero FM, Goti A. Chem. Eur. J. 2009;15:7808. doi: 10.1002/chem.200900707. [DOI] [PubMed] [Google Scholar]
- 12.The decarboxylative (1,3)-dipolar cycloaddition between proline and cinnamaldehyde to give the racemic pyrrolizidine has been reported: Kang T-R, Cheng Y, He L, Ye J, Liu Q-Z. Tetrahedron Lett. 2012;53:2552. Hong B-C, Liu K-L, Tsai C-W, Liao J-H. Tetrahedron Lett. 2008;49:5480.
- 13.Codelli JA, Puchlopek ALA, Reisman SE. J. Am. Chem. Soc. 2012;134:1930. doi: 10.1021/ja209354e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pandey G, Banerjee P, Gadre SR. Chem. Rev. 2006;106:4484. doi: 10.1021/cr050011g. [DOI] [PubMed] [Google Scholar]
- 15.The first highly enantioselective catalytic (1,3)-dipolar cycloadditions of azomethine ylides: Gothelf AS, Gothelf KV, Hazell RG, Jørgensen KA. Angew. Chem. Int. Ed. 2002;41:4236. doi: 10.1002/1521-3773(20021115)41:22<4236::AID-ANIE4236>3.0.CO;2-W. Longmire JM, Wang B, Zhang XM. J. Am. Chem. Soc. 2002;124:13400. doi: 10.1021/ja025969x.
- 16.Selected examples of metal-catalyzed enantioselective 1,3-DCAs between unsubstituted acrylates and azomethine ylides derived from α-(arylideneamino)esters: Yamashita Y, Imaizumi T, Kobayashi S. Angew. Chem. Int. Ed. 2011;50:4893. doi: 10.1002/anie.201008272. Shimizu K, Ogata K, Fukuzawa S-I. Tetrahedron Lett. 2010;51:5068. Wang C-J, Liang G, Xue Z-Y, Gao F. J. Am. Chem. Soc. 2008;130:17250. doi: 10.1021/ja807669q. Najera C, Retamosa MD, Sansano JM. Angew. Chem., Int. Ed. 2008;47:6055. doi: 10.1002/anie.200801690. Zeng W, Zhou Y-G. Org. Lett. 2005;7:5055. doi: 10.1021/ol0520370. Gao W, Zhang X, Raghunath M. Org. Lett. 2005;7:4241. doi: 10.1021/ol0516925. Chen C, Li XD, Schreiber SL. J. Am. Chem. Soc. 2003;125:10174. doi: 10.1021/ja036558z.
- 17.Kobayashi and coworkers reported the use of a cinnamaldehydederived α-iminoester in their recent manuscript describing exoselective (1,3)-dipolar cycloaddition. See ref. 16a.
- 18.Kim HY, Shih H-J, Knabe WE, Oh K. Angew. Chem., Int. Ed. 2009;48:7420. doi: 10.1002/anie.200903479. [DOI] [PubMed] [Google Scholar]
- 19.A single, non-enantioselective example of a related double (1,3)-DCA has been reported: Cui P, Xu L, Shi Z, Gan L. J. Org. Chem. 2011;76:4210. doi: 10.1021/jo200601y.
- 20.The structure of 12 was confirmed by single crystal X-ray diffraction of a derivative TFA salt. See Supporting Information.
- 21.Additional examples of Ag-catalyzed asymmetric (1,3)-DCA reactions of α-(arylideneamino)esters: Martin-Rodriguez M, Najera C, Sansano JM, de Cozar A, Cossio FP. Chem. Eur. Journal. 2011;17:14224. doi: 10.1002/chem.201101606. Chen Q-A, Wang D-S, Zhou Y-G. Chem. Commun. 2010;46:4043. doi: 10.1039/c001089h. Chen Q-A, Wang D-S, Zhou Y-G. Chem. Commun. 2010;46:4043. doi: 10.1039/c001089h. Wang C-J, Xue Z-Y, Liang G, Lu Z. Chem. Commun. 2009;45:2905. doi: 10.1039/b902556a. Liang G, Tong MC, Wang CJ. Adv. Synth. Cat. 2009;351:3101. Knopfel TF, Aschwanden P, Ichikawa T, Watanabe T, Carreira EM. Angew. Chem., Int. Ed., 2004;43:5971. doi: 10.1002/anie.200461286.
- 22.We have consistently found that pyrrolizidine 12 can be isolated in higher yields than the corresponding pyrrolidine 10. We attribute this to challenges in isolating and purifying the free N-H pyrrolidine.
- 23.Although the studies reported herein utilize co-catalytic quantities of DIPEA, at the request of a reviewer we have determined that pyrrolizidine 12 can be prepared with equal efficiency (75% yield, 90% ee) in the absence of DIPEA. Alternatively, the yield of pyrrolidine 10 is slightly reduced in the absence of DIPEA (56% yield, 91% ee).
- 24.The Ag(OAc)/QUINAP-catalyzed (1,3)-DCA between 8 and 9 at 23 °C furnishes pyrrolidine 10 in 77% ee.
- 25.Vigneron JP, Dhaenens M, Horeau A. Tetrahedron. 1973;29:1055. [Google Scholar]
- 26.(a) Masamune S, Choy W, Petersen J, Sita L. Angew. Chem. Int. Ed. 1985;24:1. [Google Scholar]; (b) Kolodiazhnyi OI. Tetrahedron. 2003;53:5953. [Google Scholar]
- 27.N-(2-Pyridylmethyl)imines have been shown to participate in Cu-catalyzed asymmetric (1,3)-DCA reactions: Padilla S, Tejero R, Adrio J, Carretero JC. Org. Lett. 2012;12:5608. doi: 10.1021/ol102605q.
- 28.Ee values obtained using standard conditions: 18c (80%), 18d (83%),18e (73%), 18q (84%), 18k (84%), 18p (82%).
- 29.(R)-QUINAP was used.
- 30.On 0.15 mmol scale, 18a could be prepared in 88% yield and 90% ee using 1 mol % catalyst in conjunction with a 48 h reaction period for the first (1,3)-DCA. Use of 1 mol% catalyst under standard conditions provided 18a in 64% yield and 86% ee.
- 31.Reducing the amount of t-butylacrylate used in the second (1,3)-DCA to 1.5 equiv provided 18a in 74% yield and 91% ee.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.