

Abstract
Essential metals are crucial for the maintenance of cell homeostasis. Among the 23 elements that have known physiological functions in humans, 12 are metals, including iron (Fe) and manganese (Mn). Nevertheless, excessive exposure to these metals may lead to pathological conditions, including neurodegeneration. Similarly, exposure to metals that do not have known biological functions, such as mercury (Hg), also present great health concerns. This reviews focuses on the neurodegenerative mechanisms and effects of Fe, Mn and Hg. Oxidative stress (OS), particularly in mitochondria, is a common feature of Fe, Mn and Hg toxicity. However, the primary molecular targets triggering OS are distinct. Free cationic iron is a potent pro-oxidant and can initiate a set of reactions that form extremely reactive products, such as OH•. Mn can oxidize dopamine (DA), generating reactive species and also affect mitochondrial function, leading to accumulation of metabolites and culminating with OS. Cationic Hg forms have strong affinity for nucleophiles, such as –SH and –SeH. Therefore, they target critical thiol- and selenol-molecules with antioxidant properties. Finally, we address the main sources of exposure to these metals, their transport mechanisms into the brain, and therapeutic modalities to mitigate their neurotoxic effects.
1. Introduction
Analogous to carbon-based molecules, metals are crucial for the maintenance of cell homeostasis and preservation of life. They display important structural, regulatory and catalytic functions in different types of proteins, such as enzymes, receptors and transporters (Phipps, 2002). Among the 23 elements with known physiological functions, 12 are metals (sodium, magnesium, potassium, calcium, vanadium, chromium, manganese (Mn), iron (Fe), cobalt, copper, zinc, and molybdenum) (for a review, see (Fraga, 2005)). Nutritional deficiencies in specific trace-element metals [Fe (Cook et al., 1994; Goodnough, 2012), zinc (Chasapis et al., 2012), Mn (Takeda, 2003)], as well as genetic disorders leading to altered metal homeostasis (Kodama et al., 2012; Nandar and Connor, 2011), culminate in human diseases. At the other spectrum, exposures to toxic levels of essential metals, such as Mn (Racette et al., 2001), Fe (Schumann, 2001) and zinc (El Safty et al., 2008), may lead to pathological conditions. Of particular importance, oxidative stress and neurodegeneration have been reported as consequences of toxic exposures to essential metals, along with dyshomeostasis in essential metal metabolism (Bowman et al., 2011; Brewer, 2012; Jaiser and Winston, 2010).
Xenobiotic metals with no physiological functions, such as aluminum, cadmium, lead and mercury, are present in measurable concentrations in living organisms (Fraga, 2005). Such metals often enter organisms by molecular mimicry, utilizing inherent transporters for essential metals (Martinez-Finley et al., 2012). Environmental, occupational or intentional exposures to xenobiotic metals are frequently related to the development of toxicity and pathological conditions (Goyer, 1995; Valko et al., 2005). Notably, exposures to toxic metals, such as mercury, (Clarkson et al., 2003), lead (Fox et al., 2012) and aluminum (Bondy, 2010), have been related to the development of neuropathological conditions.
Among the aforementioned essential and non-essential metals, Fe, Mn and Hg have received considerable attention due to their ability to induce oxidative damage and neurodegeneration. Notably, the etiologies of neurodegenerative disease such as Parkinson’s disease (PD) and Alzheimer’s disease (AD) seem to be greatly dependent on environmental factors or on environmental/genetic interactions (Marras and Goldman, 2011). Of particular importance, specific metals have pro-oxidative properties and can perturb neurodegenerative genes by epigenetic events, leading to altered gene expression and late-onset neurodegenerative diseases (Kwok, 2010). Due to its ability to assume two oxidation states in biological systems [ferric (3+) and ferrous (2+)], Fe is an intrinsic producer of reactive oxygen species (ROS), leading to neuronal oxidative stress and neurodegeneration (Nunez et al., 2012). Fe dyshomeostasis has been reported as an important event mediating the physiopathogeny of PD and AD (Bartzokis et al., 2000; Jellinger, 1999). Analogous to Fe, Mn is also of concern due to its ability to cause manganism, an extrapyramidal syndrome resembling idiopathic PD (Benedetto et al., 2009). In contrast to Fe and Mn, Hg is a non-essential metal, whose neurotoxicological properties have been reported several decades ago secondary to environmental epidemic outbreaks (Bakir et al., 1973; Harada, 1978). Humans are continuously exposed to environmental and occupational mercury. Early-life exposures to this metal have been associated with long-lasting and enduring neurobehavioral and neurochemical deficits (Yorifuji et al., 2011). Moreover, in vitro experimental studies with neural cells have shown that mercury induces glial cell reactivity (a hallmark of brain inflammation), increases the expression of the amyloid precursor protein and stimulates the formation of insoluble beta-amyloid, which plays a crucial role in the pathogenesis of AD (Monnet-Tschudi et al., 2006). This review provides a synopsis on the chemical properties of Fe, manganese and mercury, as well as on their biological and toxicological aspects, highlighting oxidative stress as a pivotal event in mediating their toxicity. Particular emphasis is directed to their effects on the central nervous system (CNS).
2. Iron
2.1. Properties, Chemical Forms and Human Exposure
Iron (Fe) belongs to group VIII of periodic table and is one of the most abundant elements in the earth’s crust (Weber et al., 2006) and the most abundant of the transition metals in the periodic table (Wachtershauser, 2007). Therefore, Fe availability to living organism is high, which, added to its redox chemical properties (Bleackley and Macgillivray, 2011), likely contributes to its selection as a central element in mediating energy-related processes in living organisms (Turrens, 2003; Wachtershauser, 2007; Weber et al., 2006). Fe can exist in different oxidation states, varying from −2 to +6; however, within biological systems, it is bound to specific metalloproteins and is found in the +2 or +3 oxidation states; such change in its redox state is crucial to oxidative metabolism (Levi and Rovida, 2009). However, subtle changes in the folding of Fe-containing proteins can modify its coordination bond properties, which changes the physiological and/or pathological role played by the protein in cell biology (Patriarca et al., 2012). In the catalytic cycle of cytochrome P450, which is an important class of enzymes involved in the oxidative transformation and degradation of different xenobiotics and endogenous substrates, Fe is postulated to assume an Fe(IV)oxo (or ferryl) oxidation state (Rittle and Green, 2010). In contrast, the transport and storage of oxygen by hemoglobin and myoglobin in vertebrates does not involve change in the oxidation state of Fe2+ (Shikama, 2006).
In view of its widespread distribution in the earth’s crust, we are constantly exposed to Fe mainly via food intake. Normally, Fe absorption is physiologically regulated to avoid Fe toxicity (see bellow in section 2.2.). Sporadic accidental, intentional suicidal or occupational exposure to Fe may occur, but rarely has it been linked to neurotoxicity (Andersen, 2004; Anderson, 1994; Carlsson et al., 2008; Howland, 1996; Jang and Hoffman, 2011; Magdalan et al., 2011; Siew et al., 2008; Sipahi et al., 2002; Tseng et al., 2011). Within the context of neurodegeneration, there is no longitudinal study supporting that a single episode of exposure to toxic Fe levels results in delayed neurodegeneration. With respect to neurodegeneration, limited epidemiological evidence indicates that co-exposure to Fe and other toxic metals (Pb and Cu) presents a risk factor for PD (Gorell et al., 1997; 1999).
Biochemically, Fe2+ can be easily oxidized to Fe3+ and reduced back to Fe2+ after interaction with different oxidizing or reducing agents (Levi and Rovida, 2009). These changes in the oxidation state of Fe are crucial for energy production by many living organisms. In aerobic cells, Fe plays a vital role in the transport of electrons derived from food oxidation to molecular oxygen (O2) located at the end of respiratory chain (Levi and Rovida, 2009). Paradoxically, the redox properties of Fe determine its participation in potentially cytotoxic reactions. In fact, Fe2+can catalyze the decomposition of H2O2 with the formation of hydroxyl radical (OH•) (Figure 1), which is normally considered the most reactive and damaging intermediate formed during cellular metabolism (Gutteridge, 1984; Halliwell, 1984, 1992; Halliwell and Turrens, 2003 - Figure 1). Fe3+ can also be reduced back to Fe2+ after reacting with superoxide anion (O2•−) (Haber and Weiss, 1932). Consequently, in a pro-oxidant intracellular environment (particularly in mitochondria), the formation of O2•− can stimulate Fe2+-mediated H2O2 decomposition even in the presence of small catalytic amounts of free Fe (the coupling of these two reactions are depicted in Figure 1) (Halliwell, 1984; 1992; Halliwell and Gutteridge, 1984). Fe2+/Fe3+ are also involved in the propagation of lipid peroxidation, by a complex mechanism which has yet to be fully understood; however, it likely involves the direct interaction of Fe with molecular oxygen and ROS, such as organic peroxides (ROOH) formed in biological membranes (Minotti and Aust, 1989, 1992; Tadolini and Hakim, 1996).
Figure 1. Fe and Mitochondria Oxidative Sress.
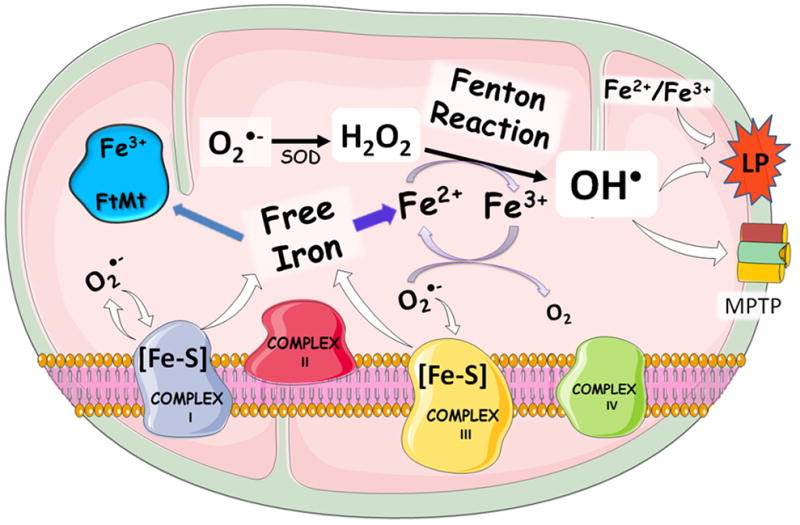
Fenton Reaction and hydroxyl radical formation are critical factors in Fe-induced mitochondrial toxicity; this type of reaction is thought to be central in neurodegeneration. Fe can start mitochondrial oxidative stress via interaction with different reactive oxygen species (ROS). Free Fe can be released from mitonchondrial Fe-sulfur clusters in complexes I and III upon interaction with ROS (in the figure it is shown the release of superoxide anion by these complexes and the potential oxidation of Fe-S cluster by O2•−; the oxidation of the Fe-sulfur clusters can increase the free Fe in the mitochondrial matrix. This can facilitate the operation of the toxic Haber-Weiss and Fenton reactions, feeding a general pro-oxidant cycle). The redox pair Fe2+-Fe3+ can also directly stimulate lipid peroxidation, which can intensify the oxidative stress and contribute to mitochondrial and cellular demise via mPTP formation. Free cationic Fe (regardless of the redox state) is the critical element for neurotoxicity and it can be buffered by intramitochondrial ferritin (FtMt), which acts as an antioxidant protein in the mitochondrial matrix.
Importantly, mitochondrial dysfunction elicited by different environmental or endogenous toxic agents (including Fe itself) can either initiate or propagate Fe release from non-toxic sites (i.e. Fe binding proteins), which may trigger and/or accelerate the progression of degenerative diseases (Beal, 1998; Horowitz and Greenamyre, 2010; Kumar et al., 2012; Mesquita et al., 2012; Sebastiani and Pantopoulos, 2011; Zecca et al., 2004). In mitochondria, the iron-sulfur clusters ([Fe-S]) found in complexes I and III of the electron transport chain (ETC) can be attacked by ROS, releasing free Fe to participate in the Fenton Reaction and other oxidative processes (Figure 1). Thus, Fe is an important player in cell toxicity and it can either initiate by itself a set of extremely oxidative toxic reactions, or nourish oxidative stress provoked by xenobiotics or endogenous metabolites. Of particular importance, Fe-mediated oxidative stress has been classically linked to apoptotic cell death (Ott et al., 2007; Wallace, 1999) and more recently to ferropoptosis, which represents a Fe-dependent form of non-apoptotic cell death (Dixon et al., 2012).
2.2. Transport, metabolism and excretion
As detailed above, Fe is highly abundant in the environment and its requirement for the proper human body functioning is normally exceeded after ingestion of western diets. In order to avoid Fe overload, the absorption of dietary Fe is tightly regulated by a complex and not yet fully understood interplay between Fe body burden and gastrointestinal absorptive mechanisms (De Domenico et al., 2008; Nunez, 2010). Fe transport into the enterocyte is adjusted to fulfill the body requirements of this element. The fine regulation of Fe absorption is extremely important because there are no cellular regulated processes for Fe excretion (De Domenico et al., 2008; Finberg, 2011; Fleming and Ponka, 2012; Mesquita et al., 2012).
In the human intestine, Fe is absorbed by different (at least 3) molecular mechanisms into the enterocyte, depending upon its chemical form and dietary source (Theil, 2011; West and Oates, 2008). There is a system that absorbs heme-Fe (normally derived from myoglobin from red meat or blood hemoglobin), which was formerly called heme carrier protein 1 (HCP1) due to its role in heme-Fe transport and absorption (Shayeghi et al., 2005). Experimental details on the modulation of heme-Fe absorption by these heme-transporters are poorly understood (Theil, 2011; West and Oates, 2008), but it is thought that the primary physiological role of the heme-transporters involves folate transport (Le Blanc et al., 2012). For this reason, the transporter involved in intestinal heme-Fe absorption is now named proton-coupled folate transport or PCFT/HCP1.
The literature also corroborates the existence of a clathrin-dependent, receptor-mediated endocytosis mechanism for mineralized Fe3+ in ferritin found in legume seeds, such as soybean (San Martin et al., 2008; Theil, 2011). There is a third system involved in non-heme Fe2+ derived from salts or chelators from supplements that is mediated by the divalent metal transporter 1 (DMT1), which works jointly with an Fe oxireductase (Dcytb, duodenal cytochorme b; (McKie et al., 2001). The Dcytb protein reduces Fe3+ to Fe2+ in the apical part of enterocytes (Figure 2, left), which allows absorption via DMT1. DMT1 mRNA transcripts have been found in a variety of tissues, indicating a universal role for this transport in Fe distribution in mammals (Mims and Prchal, 2005).
Figure 2. Mechanisms of Intestinal Fe Uptake.
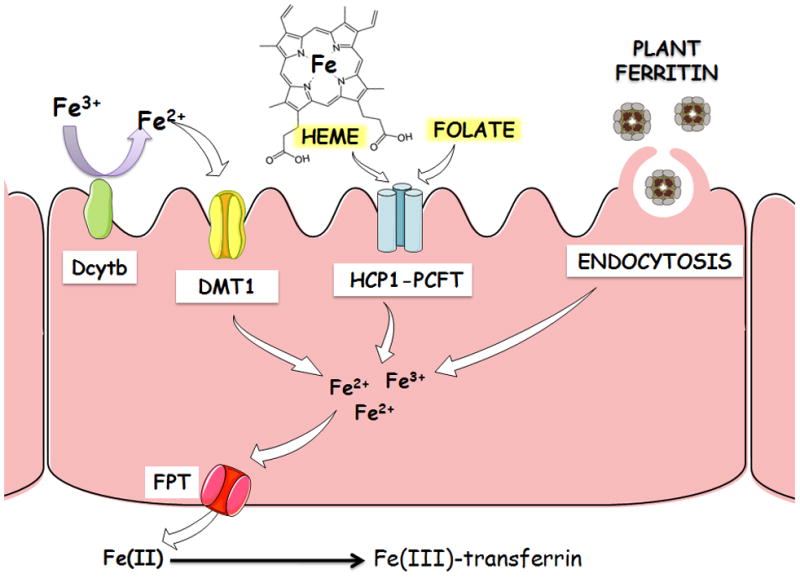
Fe can be absorbed in the enterocyte via distinc mechanisms: 1) Divalent Metal Transporter 1 (DMT1) and duodenal cytochrome b (Dcytb) Fe oxireductase system, which is involved in the absorption of free divalent Fe (Fe2+); 2) HCP1/PCFT or heme carrier protein 1 (HCP1)/proton-coupled folate transporter, which is involved in the absorption of heme-Fe and folate, and 3) a clathrin-dependent, receptor-mediated system that is involved in the absorption of vegetable-ferritin-bound Fe via endocytosis. After absorption, all forms of Fe are transformed to cationic Fe that can be exported from enterocytes by ferroportin (FTN). In the plasma, Fe2+ is oxidized by ceruloplasmin or hephaestin and binds to transferrin. Tranferrin can distribute Fe to all tissues of the body, including brain where Fe overloading contributes to neurodegeneration.
The export of absorbed Fe from enterocyte to the plasma is mediated by ferroportin (FPT), which is regulated by hepcidin and plays a crucial role in regulating plasma Fe levels (Nemeth and Ganz, 2006). In plasma, Fe2+ is oxidized to Fe3+ by ceruloplasmin or hephaestin and then binds to transferrin, which can distribute Fe to cells throughout the body. Fe3+-transferrin complex can interact with transferrin receptor 1, resulting in endocytosis and uptake of the transferrin-bound metal. Fe can then be transported to mitochondria and incorporated in heme prosthetic groups or into Fe-sulfur clusters (Finberg, 2011; Fleming and Ponka, 2012; Wang and Pantopoulos, 2011). Intramitochondrial free Fe can also be buffered by a specific mitochondrial ferritin (FtMt; Figure 1), which has an important physiological role as an antioxidant (Campanella et al., 2009; Santambrogio et al., 2007) (Figure 1).
The central role of mitochondria in heme biosynthesis highlights the importance of this organelle in Fe fate and metabolism. Physiologically, mitochondria have adapted to cope with Fe and to circumvent the potential toxicity of free cationic Fe forms (Levi and Rovida, 2009; Ott et al., 2007; Richardson et al., 2010). Since mitochondria are also important intracellular sites for ROS production (i.e. O2•− and H2O2) (Halliwell, 1992; Ott et al., 2007), the continued presence of Fe inside the mitochondrial matrix renders these organelles susceptible to damage by extremely reactive intermediates that can be formed after interaction of ROS with transitory free Fe2+ and Fe3+ (see Figure 1). In effect, mitochondrial Fe seems to play a fundamental role in neurodegeneration associated with several brain pathologies (Beal, 1998; Galaris and Pantopoulos, 2008; Horowitz and Greenamyre, 2010).
If the body burden of Fe is adequate and there is no requirement for this micronutrient, its absorption is negatively modulated by different mechanisms. As previously mentioned, the peptide hepcidin, which is synthesized as pro-hormone in the hepatocytes, is released into the blood in response to Fe intake. Hepcidin inhibits the intestinal absorption of Fe and its export from enterocytes (and also that derived from heme from red blood cells phagocytized by macrophages in the reticuloendothelial system). Hepcidin binds to ferroportin and stimulates its phosphorylation and degradation, modulating in this way the body burden of Fe and its availability for heme synthesis and erythropoiesis (Finberg, 2011; Nemeth and Ganz, 2006; Sebastiani and Pantopoulos, 2011; Wang and Pantopoulos, 2011). The absorption, distribution and storage of Fe are also regulated by the concerted interaction of Fe regulatory proteins (IRPs) and Fe responsive elements (IREs). IREs are located in the untranslated regions of mRNAs encoding protein involved in Fe handling and can interact with IRPs (Wang and Pantopoulos, 2011). For instance, the synthesis of Fe trafficking and storage proteins (DMT1, transferrin receptor and ferritin, etc.) is finely coordinated by IRPs and IREs in order to increase or decrease Fe absorption, depending upon the physiological requirements for Fe (Theil, 2011; Wang and Pantopoulos, 2011).
One important (but not fully explored) aspect on Fe homeostasis is how dietary or genetic Fe loading can modify the metabolism of proteins involved in Fe absorption, trafficking and storage in brain tissues. Clarifying such aspects would contribute on understanding how Fe participates in neurodegenerative processes; such knowledge may improve treatment options in a range of neurodegenerative disorders (Johnstone and Milward, 2010a; 2010b).
2.4. Fe and Neurodegeneration
As discussed above, free cationic Fe can be extremely toxic via disruption of mitochondrial function, and theoretically, Fe2+←→Fe3+ redox changes can be coupled with formation of extremely reactive species, such as hydroxyl radical (OH•). This molecule is highly reactive and its free existence is limited to its diffusion coefficient. In fact, OH• is expected to be found only close to its site of formation and in close proximity to Fe ions (Gutteridge, 1984). The formation of OH• can damage different biomolecules and start a vicious cycle of cellular damage (Figure 1). Furthermore, the redox pair Fe2+/Fe3+serves as an in vivo initiator of cytotoxic reactions, particularly, lipid peroxidation (Ryan and Aust, 1992; Welch et al., 2002).
With respect to neurodegeneration, a vast amount of literature data indicates that Fe is an important etiologic factor associated with oxidative stress induction and cell demise in pathological situations (Johnstone and Milward, 2010b; Jomova and Valko, 2011; Mesquita et al., 2012; Wu et al., 2012). Recently, it has been proposed that Fe could be a primary and unifying factor involved in the progression of different chronic neurodegenerative diseases, such as PD, Alzheimer’s and Huntington’s disease (Kell, 2010). In fact, there are numerous observation to support an early role for brain Fe overloading in the progression of neurodegenerative diseases (Rosas et al., 2012). However, temporal aspects on Fe-mediated initiation or progression of neuropathological conditions, as well as the exact role played by activation of Fe-triggered toxicological pathway(s), remain unknown (Andersen, 2004; Johnstone and Milward, 2010b; Kumar et al., 2012).
It is noteworthy that Fe deposition has been observed only in specific brain regions in patients with chronic degenerative diseases (Kell, 2010; Kumar et al., 2012; Rosas et al., 2012; Sian-Hulsmann et al., 2011). The basal ganglia represent a preferential site of Fe deposition in neurodegenerative diseases (Akatsu et al., 2012; Berg et al., 2001; Gregory and Hayflick, 2011). A similar phenomenon is also observed in a wide range of genetic diseases collectively named neurodegeneration with brain Fe accumulation (NBIA, such as Friedreich ataxia, pantothenate kinase 2-associated neurodegeneration, PLA2G6-associated neurodegeneration, FA2H-associated neurodegeneration, Kufor-Rakeb disease, aceruloplasminemia, and neuroferritinopathy (Gregory et al., 2009; McNeill et al., 2008; Schipper, 2012). These genetic diseases are characterized by Fe accumulation in basal ganglia and associated with mutations in proteins involved in Fe traffic or metabolism (Prohaska et al., 2012). However, as stated for the case of chronic Fe-associated degenerative brain diseases, little is known about the mechanisms that lead to brain Fe accumulation (Prohaska et al., 2012). Nevertheless, the study and understanding of the neuropathological modifications associated with the wide spectrum of NBIA diseases have indicated the existence of clinical, morphological and molecular features similar to those seen in chronic neurodegenerative diseases such as PD, Huntington’s and Alzheimer’s disease (Berg et al., 2001; Schneider et al., 2012).
As briefly noted above, the temporal relationship between Fe deposition and neurodegeneration has yet to be clearly established. Thus, in some diseases, Fe deposition can be the consequence and not the cause of neurodegeneration. Here we have a gap in knowledge, which indicates the need of mechanistic studies to determine the primary, secondary and tertiary factors involved in the initiation and progression of neurodegeneration in different Fe-associated brain pathologies. Most importantly, from a therapeutic point of view, the identification of a potential non-returning point of Fe neurotoxicity would be of great value in developing therapeutic and other interventional procedures that could delay the attainment of this point of cell demise. In short, although Fe (as Fe2+) is a central factor in Fenton reaction and, consequently, in OH• production, which is expected to damage biomolecules and contribute to neurodegeneration, there is no a direct or even an indirect method to accurately follow the chronology of Fenton’s reaction in a representative living model system of neurodegeneration. The assertion for the central role of Fe2+-Fe3+ (either as participants in Fenton reaction or as direct inductors of lipid peroxidation) in neurodegeneration is based largely on reactivity parameters derived from classical indirect procedures that are used to determine their occurrence in chemical pure systems. Thus, experimental in vitro and in vivo models designed to determine with precision the temporal role of Fenton reaction in neurodegeneration are highly needed. Furthermore, the role played by Fenton chemistry in the activation or inhibition of specific molecular and subcellular pathways that participate in Fe neurotoxicity is not fully understood. The ability of Fe (Fe2+:Fe3+) to initiate and propagate membrane lipid peroxidation adds an additional factor to these complex issues. In fact, we have no experimental indication on the proportional contribution of these specific reactions (Haber and Weiss, 1932; Halliwell and Gutteridge, 1984) either in simple or complex chemico-biological system(s).
Acute Brain Fe Overload
High amount of Fe can be acutely released in specific brain regions after local hemorrhage caused by brain trauma or after stroke episodes resulting from different etiologies (Carbonell and Rama, 2007; Halliwell, 1992; Raz et al., 2011; Wagner et al., 2003). After the hemorrhagic episode, erythrocytes are released inside the brain parenchyma, followed by hemolysis. Hemoglobin, heme and Fe are then released in the extracellular space, causing local Fe overloading (Halliwell, 1992). Although little is known about the fate of heme released from hemoglobin after brain hemorrhage, a recent study has indicated that hemoglobin and heme uptake was higher in neurons than in glial cells (Lara et al., 2009). Consequently, heme uptake by neurons after brain trauma or stroke contributes to Fe-associated neurodegeneration (Aronowski and Zhao, 2011).
Fe and Cell Death
At the molecular level, the primary toxicity of free Fe is associated with its redox properties, which can culminate in the production of ROS that will initiate a cascade of cytotoxic events. For instance, OH• can oxidize a variety of biomolecules, including thiol-containing proteins, and in the case of mitochondria this can lead to the formation of mitochondrial permeability transition pore (mPTP). mPTP formation will collapse membrane mitochondrial potential, increase intramitochondrial Ca2+, decrease ATP synthesis and in extreme cases result in cell death (Halestrap, 2009). The formation of mPTP can also trigger less dramatic changes in mitochondrial metabolism that can be associated with delayed apoptosis and/or necrosis (Kinnally et al., 2011). However, our knowledge on the role of Fe-induced oxidative stress on the activation of (a) particular cascade(s) of cellular or mitochondrial events that result in cell death is superficial. Recently, it was demonstrated that Fe is a key element involved in mitochondrial-induced oxidative stress and cell death(Dixon et al., 2012). This form of cell death, which was named ferroptosis, is morphologically, biochemically and genetically distinct from apoptosis, necrosis or autophagy, and can be activated by glutamate (Dixon et al., 2012). Accordingly, Fe can contribute to neurodegeneration by activating different cell death pathways.
2.5. Antidotal Strategies
Therapeutic approaches to treat neurodegeneration associated with Fe overload is limited and involve the use of chelating agents (Heli et al., 2011; Jomova and Valko, 2011; Miyajima et al., 1997; Molina-Holgado et al., 2007; Selim et al., 2011). However, treatment with these agents (including desferoxamine) may cause toxicity (Gassen and Youdim, 1997; Heli et al., 2011). Natural products, such as catechin and other polyphenols have been indicated as potential therapeutic agents against Fe toxicity, because of their simultaneous antioxidant and Fe-chelating properties (Mandel and Youdim, 2004; Reznichenko et al., 2006). The therapeutic efficacy of polyphenol compounds found in natural preparations used in folk medicine can be linked to these two general properties (Fibach and Rachmilewitz, 2010; Perron and Brumaghim, 2009).
3. Manganese
3.1. Properties and Chemical Forms and Human Exposure
Manganese (Mn) is one of the most abundant naturally occurring elements in the earth’s crust; it does not occur naturally in a pure state. Oxides, carbonates and silicates are the most important Mn-containing minerals. Mn exists in various chemical forms, oxidation states (Mn2+, Mn3+, Mn4+, Mn6+, Mn7+), salts (sulfate, chloride and gluconate) and chelates (aspartate, fumarate, succinate). More than 25 million tons are mined yearly, representing 5 million tons of the metal (Emsley, 2001). The versatile chemical properties of Mn have enabled its industrial usage in glass and ceramics, adhesives, welding, paint, gasoline anti-knock additives (methylcyclopentadienyl manganese tricarbonyl, MMT), just to name a few. Manganese dioxide is also used as a catalyst (Su et al., 2012). Mn is used to decolorize glass and make violet colored glass. Potassium permanganate is a potent oxidizer and used as a disinfectant. Other compounds with commercial applications are Mn oxide (MnO) and Mn carbonate (MnCO3), which have been present in fertilizers and ceramics, as well as in materials for making other Mn compounds. Mn is a paramagnetic metal, meaning that it has an unpaired electron in the outer shell and that it can be detected with MRI, Positron emission tomography (PET) and single-photon emission computed tomography (SPECT) (Aschner et al., 2007a; Inoue et al., 2011). These techniques allow for the tracking of Mn dynamics repeatedly in the same subject in vivo (Aschner et al., 2007a; Newland, 1999). Mn can also chemically interact with fluorophore fura-2, by quenching it and increasing its fluorescence, representing a new methodological approach for in vitro kinetic studies (Kwakye et al., 2011).
There are several sources of exposure to Mn, as follows:
Dietary exposure
The primary source of Mn for the general human population is diet. Adult dietary intake of Mn has been estimated to range from 0.9 to 10 mg Mn/day (ATSDR, 2000; Finley and Davis, 1999). Foods with Mn levels in excess of 30 mg/Kg include grains, rice and nuts. A cup of tea contains as much as 0.4 to 1.3 mg Mn (ATSDR, 2000). Another important source of Mn intake is the consumption of Mn-containing dietary supplements; tablets may contain 5–20 mg of Mn (NAS, 2001). Water concentrations of Mn typically range from 1–100 μg/L, with most values below 10 μg/L. Nevertheless, in some countries, such as Sweden, Mn concentrations in drinking water reach an average of 150 μg/L (Ljung and Vahter, 2007). Such elevated values pose the greatest potential risk to infants, in particular, as they have a higher retention of Mn and a more sensitive CNS than adults (Wasserman et al., 2006). Mn intake in milk is low; however, in formula-fed infants is much higher than that observed in their breast milk-fed counterparts, since levels of Mn in infant formulas may be substantially higher than those found in human milk (Krachler et al., 2000).
Airborne exposure
Inorganic Mn compounds are not volatile; however, they can exists as fumes, aerosols or suspended particulate matter (ATSDR, 2000). Atmospheric Mn derives from both anthropogenic and natural sources. Industries associated to Mn emissions include ferroalloy production, iron and steel foundries, metal fumes from welding, battery production and power plant and coke oven combustion (Aschner et al., 2005). Mn is also found in methylcyclopentadienyl manganese tricarbonyl (MMT), a fuel additive used in some unleaded gasoline (Davis, 1998). The use of this additive has been subject of much debate by regulatory agencies (Davis et al., 1998; Kaiser, 2003).
Parenteral Nutrition
Due to Mn essentiality, parenteral nutrition (PN) generally contains significant amounts of this trace element. However, many products contain Mn as ubiquitous contaminant (Hardy, 2009). There are several case reports of PN users that developed Mn neurotoxicity and showed high MRI intensity in the brain (Hardy, 2009). In PN patients, the normal intestinal regulatory mechanism is bypassed and the amount of Mn delivered via the intravenous route is 100% bioavailable. In addition, the normal pathway of elimination via the hepatobiliary system frequently is impaired because of PN-associated biliary stasis and obstructive jaundice. This may be especially important for parenterally fed infants who pass little or no stool and often show evidence of hepatic dysfunction and cholestasis (Aschner and Aschner, 2005). It also predisposes long-term PN patients to tissue accumulation and/or brain deposition of Mn, resulting in neurologic symptoms. However, a clear cause–effect relationship between PN-associated cholestasis and neurotoxicity has not been established and data about the temporal relationship between the dose and duration of Mn supplementation and increased Mn levels have been contradictory (Siepler et al., 2003).
Mn-containing drugs
A relatively new form of presumed Mn poisoning has been reported in drug-addicted subjects from Eastern Europe and the Baltic states who have intravenously injected self-prepared methcathinone hydrochloride(ephedrone), which is synthesized from pseudoephedrine hydrochloride using potassium permanganate as the oxidant (Zhingel et al., 1991). Ephedrone is relatively easily accessible for abuse. Its users develop an extrapyramidal syndrome and it is not known if this is caused by methcathinone itself, by side-ingredients (Mn), or both (Sikk et al., 2011). Neuroimaging studies with MRI have demonstrated Mn accumulation in the basal ganglia of these addicts (Sikk et al., 2010).
3.3. Transport, metabolism and excretion
As previously mentioned, the major source of Mn in humans is via dietary ingestion (Au et al., 2009). Approximately 3–5% of ingested Mn is absorbed, and the rest is excreted in the feces. Its uptake is tightly regulated and any excess of ingested Mn is readily excreted via the bile. In contrast, both pulmonary uptake and particulate transport via the olfactory bulb can lead to Mn deposition in the striatum and cerebellum and inflammation of the nasal epithelium (Roth, 2009).
Mn ions (Mn3+) bind to the same location as ferric ions (Fe3+) on the large glycoprotein molecule mucin, which is known to stabilize the ions preventing precipitation in the lumen of the gastro intestinal tract (Powell et al., 1999). Both metals are known to have an affinity for the intercellular metal binding molecule mobilferrin (Conrad et al., 1992). Absorption of metal ions into enterocytes is known to take place via transmembrane transporters. Gunshin et al. (1997) cloned the Divalent Metal Transporter1 (DMT1) from proximal small bowel, which avidly binds Fe2+ ions, but also has an affinity for Mn2+ and other cations. In this regard, it is important to mention that dietary Fe3+ is firstly reduced to Fe2+ by ascorbate or surface ferrireductases before being transported via DMT1 into the enterocytes (Mackenzie and Garrick, 2005). During Fe deficiency the number of transporters in enterocyte membranes is increased in order to maximize Fe absorption (Gunshin et al., 1997). This will inevitably result in increased Mn absorption, particularly in the absence of Fe. Fe has a strong influence on Mn homeostasis as both metals share the transporter, transferrin (Tf), binding and uptake via the Tf transporter and the divalent metal transporter, DMT1/NRAMP2. In rodents, Fe deficiency is associated with increased Mn absorption across the gastrointestinal tract, as well increased Mn brain deposition (Fitsanakis et al., 2008; Freeland-Graves and Lin, 1991; Garcia et al., 2007).
The exact identity of the carrier(s) involved in Mn transport into the brain is still controversial. In general, it is believed that at normal plasma concentrations, Mn enters into the CNS primarily across the capillary endothelium, whereas at high plasma concentrations, transport across the choroid plexus predominates (Murphy et al., 1991). How, and in what chemical form Mn is transported across the blood-brain barrier (BBB) has been addressed in a series of studies. Mn is absorbed in the GI tract as Mn2+, is oxidized to Mn3+ by liver and plasma ceruloplasmin and transported through the blood by transferrin (Tf) (Aschner and Gannon, 1994; Takeda et al., 1995). Although Tf-dependent Mn transport across the BBB has been documented (Aschner and Gannon, 1994), the majority of BBB transport occurs via the DMT1.
A critical regulator of brain Mn levels is the divalent metal transporter, DMT- 1/NRAMP-2. DMT-1 (also referred to as the DCT, or divalent cation transporter) is known to shuttle both Mn and Fe ions in the (+2) valence, as well as other divalent metals. Disruption of the orthologous DMT-1 gene in the rat or mouse results in significantly lower tissue levels and uptake of Mn and Fe in the brain (Chua and Morgan, 1997; Fleming et al., 1998). Notably, a recent study (Salazar et al., 2008) has shown that DMT1 contributes to neurodegeneration in an experimental model of PD. These authors observed an increased expression of a specific DMT1 isoform (DMT1/Nramp2/Slc11a2) in the substantia nigra of Parkinson’s disease patients. Moreover, the authors also showed that the administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP, a dopaminergic toxin used in experimental models of Parkinson’s disease) increased DMT1 expression in the ventral mesencephalon of mice, which was concomitant with iron accumulation, oxidative stress, and dopaminergic cell loss (Salazar et al., 2008).
Additional brain Mn transporters include the Mn-citrate transporters (MCT) and the Mn-bicarbonate symporters (Crossgrove et al., 2003). However, the relevance of these proteins to Mn transport in vivo is not completed understood. The Mn-bicarbonate symporters, ZIP-8 and ZIP-14, have been identified as members of the solute carrier-39, and are expressed on brain capillaries (He et al., 2006). These symporters utilize a HCO3− gradient as the driving force for Mn uptake across the plasma membrane.
Other possible mechanisms for Mn transport include the dopamine transporter (DAT). It is believed that DAT facilitates Mn transport into dopaminergic (DAergic) striatal neurons and that Mn accumulates in the globus pallidus via axonal transport (Anderson et al., 2007). As a result, blockage of the DAT in the striatum should attenuate Mn accumulation in striatal neurons and cause decreased Mn concentrations in the globus pallidus (Anderson et al., 2007). Finally, Mn transport via voltage regulated channels (Lucaciu et al., 1997), store-operated channels (Riccio et al., 2002), ionotropic glutamate receptor channels (Kannurpatti et al., 2000) (all Ca2+ channels) and choline transporters (Lockman et al., 2001) has also been described.
3.4. Mn and neurodegeneration
It has been known for more than 150 years that Mn can be a neurotoxic agent; its toxicity has been predominantly observed in occupational settings, following the accidental ingestion of large quantities or after chronic inhalation of high levels (Mergler et al., 1994). The brain is particularly susceptible to excess of this metal, but the mechanisms of toxicity are poorly understood. In humans, it has been postulated that there is a spectrum of neurobehavioral and neurophysiological effects associated with Mn toxicity, including subclinical and clinical symptoms (Mergler et al., 1994).
Mn neurotoxicity, or locura manganica, also referred to as manganism, is a neurologic disorder characterized by psychological and neurological abnormalities, which resemble Parkinson’s disease (Barbeau, 1984; Huang et al., 1989; Mena et al., 1967). Mn also damages brain areas distinct from those that are affected in PD (Calne et al., 1994; Olanow, 2004). The similarities between the clinical manifestations of PD and manganism include the presence of generalized bradykinesia and widespread rigidity and a characteristic “cock-walk” (Calne et al., 1994). There are also differences with respect to treatment response – although there may be an initial response to levodopa, the primary treatment option for PD, there is typically a failure to achieve a sustained therapeutic response in patients with manganism (Aschner et al., 2009; Calne et al., 1994). The similarities between the two disorders can be partially explained by the fact that the basal ganglia accumulate most of the excess Mn compared with other brain regions, and dysfunction in the basal ganglia is also involved in PD (Dobson et al., 2004).
Mn has also been linked to the etiology of other neurodegenerative diseases, such as Huntington’s disease, Alzheimer’s Disease, amyotrophic lateral sclerosis, as well reviewed by other authors (Aschner et al., 2009; Benedetto et al., 2009; Bowman et al., 2011; Zatta et al., 2003). Mechanisms mediating Mn-induced neurotoxicity, as well as their relationship with neurodegenerative diseases, are detailed as follows.
Dopamine oxidation
DA is one of the most abundant catecholamine within the brain. Chronic exposure to Mn has been shown to cause the degeneration of nigrostriatal DAergic neurons (Barbeau, 1984). Postnatal Mn exposure causes a decline in pre-synaptic DAergic functioning, reduced DA transporter expression and DA uptake in the striatum, and a long-lasting decrease in DA efflux (Huang et al., 2003; McDougall et al., 2008). In adult animal models, exposure to Mn inhibits DA neurotransmission and depletes striatal DA (Barceloux, 1999; Calne et al., 1994; Chen et al., 2006; Pal et al., 1999), thereby resulting in motor deficits (Guilarte, 2010).
Although it is generally accepted that free radicals play a key role in mediating Mn-induced DAergic neurodegeneration (Erikson et al., 2007), the precise mechanism of Mn-induced neurotoxicity remains unknown. One hypothesis invokes the ability of Mn to enhance ROS generation via quinone formation (Figure 3) (Graham, 1978). Indeed, the Mn-catalyzed autoxidation of DA involves redox cycling of Mn2+ and Mn3+ in a reaction that generates ROS and DA-o-quinone, thereby leading to oxidative damage (Donaldson et al., 1982; Reaney and Smith, 2005). Thus, elevated rate of autoxidation of cytoplasmic DA induced by Mn may contribute to DAergic cell death secondary to the formation of cytotoxic quinones and ROS (Graham, 1978).
Figure 3. Mn-induced dopamine (DA) oxidation: primary reactions involved in reactive oxygen species (ROS) and o-quinones radical generation.
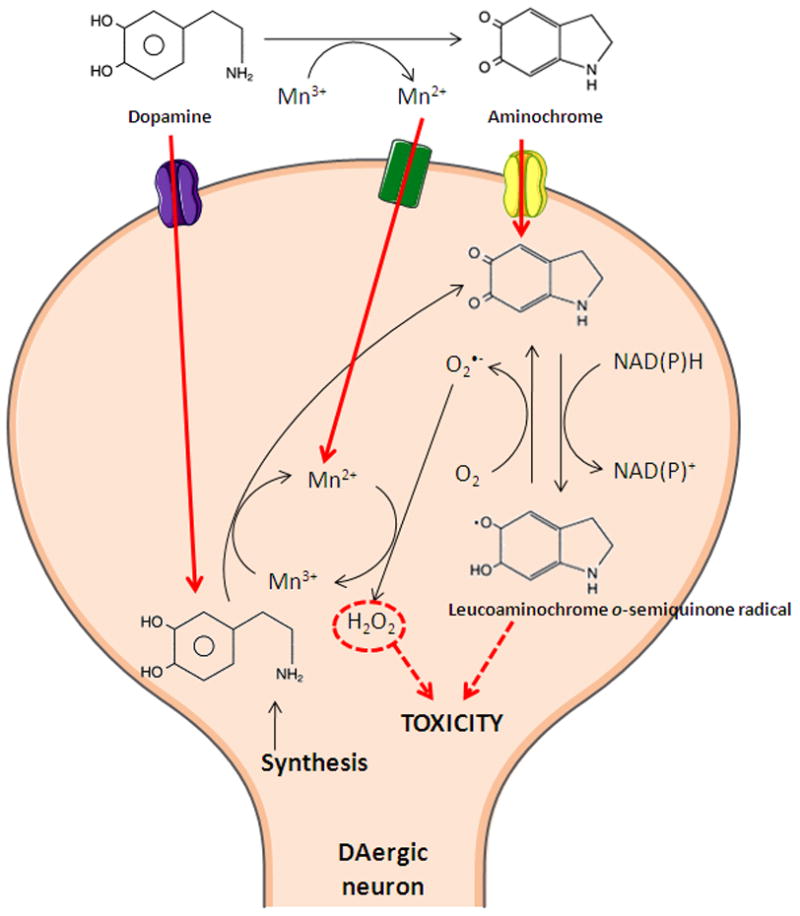
Mn-catalyzes the autoxidation of DA, involving the redox cycling of Mn2+ and Mn3+ in a reaction that generates ROS and DA-o-quinone or catalyzes the production of H2O2 inside the neurons, thereby leading to oxidative damage in DAergic neurons.
Mn-induced DA oxidation is a complex process involving several steps in which semi-quinone, aminochrome intermediates, L-cysteine or copper (Cu) and NADH are implicated (Segura-Aguilar, 1996; Segura-Aguilar and Lind, 1989). Mechanisms underlying semi-quinone and aminochrome-induced damage in the Mn-induced neurodegenerative process likely include: (i) NADH or NADPH depletion; (ii) inactivation of enzymes by oxidizing thiol groups or essential amino acids; (iii) formation of ROS. and (iv) lipid peroxidation. It is noteworthy that neither Mn2+ nor Mn3+ can generate hydroxyl radicals from hydrogen peroxide and/or superoxide via Fenton-type or Haber-Weiss-type reactions, while Mn2+ can scavenge and detoxify superoxide radicals (Archibald and Tyree, 1987; Donaldson et al., 1982).
Mitochondrial dysfunction
Intracellular Mn preferentially accumulates in the mitochondria, mainly as Mn2+ via the Ca2+ uniporter (Gavin et al., 1992; Gunter and Pfeiffer, 1990). Elevated intramitochondrial Mn interferes with oxidative respiration, leading to excessive production of ROS and consequently mitochondrial dysfunction (Gavin et al., 1992; Gunter and Pfeiffer, 1990). The ability of Mn to enhance oxidative stress is due to the transition of its oxidative state +2 to +3, which increases its pro-oxidant capacity (HaMai et al., 2001; Reaney and Smith, 2005). Superoxide produced in the mitochondrial electron transport chain (ETC) may catalyze this transition through a set of reactions similar to those mediated by SOD and thus lead to the increased oxidant capacity of the metal (Archibald and Tyree, 1987; Gunter and Pfeiffer, 1990). Superoxide radical can also form hydrogen peroxide (H2O2) by superoxide dismutase. This reaction is catalyzed by manganese (Mn)-superoxide dismutase (Mn-SOD) in the mitochondrial matrix. It also needs to be considered that Mn3+ has greater pro-oxidant potential than Mn2+, and its production in the mitochondria may also accentuate oxidative damage (Ali et al., 1995).
Mn can directly impair mitochondrial function by inhibiting the ETC (Gavin et al., 1992), resulting in reduced ATP production, increased leakage of electrons and increased O2•− production (Scholte, 1988). Although Mn3+ is more potent at inhibiting complex I (Archibald and Tyree, 1987), Mn2+ is the predominant species within cells and is largely bound to ATP (Gunter and Pfeiffer, 1990).
Mn interferes with calcium (Ca2+) homeostasis in mitochondria by inhibiting its efflux (Gavin et al., 1990; Spadoni et al., 2000). Oxidative stress generated by high Mn concentrations leads to the induction and opening of the mitochondrial permeability pore (MPT) pore, a Ca2+-dependent process, resulting in increased solubility to protons, ions and solutes, loss of the mitochondrial inner membrane potential (Δψm), impairment of oxidative phosphorylation and ATP synthesis and mitochondrial swelling (Gavin et al., 1990; Yin et al., 2008a; Zoratti and Szabo, 1995).
Astrocytosis
Astrocytes make up approximately 50% of the human brain volume (Chen et al., 2006) and assume many critical pathophysiological roles essential for normal neuronal activity, including glutamate uptake, glutamine release, K+ and H+ buffering, volume regulation and membrane–membrane mediated trophic cell signaling (Aschner and Gannon, 1994; Aschner et al., 2007a; Chen et al., 2006). Unlike neurons, astrocytes concentrate Mn to levels at least 50-fold higher than the culture media, thus functioning as the major homeostatic regulators and storage site for Mn (Aschner et al., 2009; Aschner and Gannon, 1994; Aschner et al., 2007a). Primate models of Mn toxicity have shown astrocytic pathological alterations (Alzheimer type II) (Olanow et al., 1996; Pentschew et al., 1963; Yamada et al., 1986), and exposure of cultured astrocytes to pathophysiologically relevant concentrations of Mn leads to a concentration- and time-dependent cell swelling, which appears to be a consequence of oxidative stress and changes in MPT (Rao and Norenberg, 2004). Increased accumulation of Mn in astrocytes has also been shown to alter glutamate homeostasis and elicit excitatory neurotoxicity (Erikson and Aschner, 2003). Thus, Mn decreases astrocytic glutamate uptake (Hazell and Butterworth, 1999; Hazell and Norenberg, 1998) and reduces the expression of the astrocytic glutamate:aspartate transporter (GLAST) (Erikson and Aschner, 2002), leading to increased extracellular glutamate levels, and neuronal excitability.
Mn has been implicated in the impairment of the glutamate-glutamine cycling, by deregulation of their turnover in astrocytes (Sidoryk-Wegrzynowicz et al., 2009). The functioning of this cycle is critical for normal brain function, once glutamine is the precursor of glutamate and GABA as well (Sidoryk-Wegrzynowicz et al., 2012). Expression of glutamine transporters was downregulated in Mn-exposed cultured astrocytes (Sidoryk-Wegrzynowicz et al., 2009), thus reducing glutamine uptake. As a consequence of this deregulation in glutamine transport, there is impairment in glutamine shuttling between neurons and astrocytes, altering the synthesis of glutamate and GABA (Sidoryk-Wegrzynowicz et al., 2009). Furthermore, Mn induces protein kinase C δ (PKC- isoform δ) activation, causing a decrease in glutamine uptake through two particular systems: SNAT3 and ASCT2 (Sidoryk-Wegrzynowicz et al., 2010). This process putatively promotes the initiation of the down-regulation of these transporters in astrocytes by the ubiquitin-mediated proteolytic system (Sidoryk-Wegrzynowicz et al., 2010). PKC activation by Mn exposure leads to reduced glutamate uptake, and inhibition of PKC reverses Mn-dependent down-regulation of glutamate influx, as well as increases GLT-1 and GLAST protein level in astrocytes (Sidoryk-Wegrzynowicz et al., 2011). Transfection of astrocytes with shRNA against PKCδ showed decreased sensitivity to Mn, corroborating the involvement of the PKCδ signaling (Sidoryk-Wegrzynowicz et al., 2011).
Interaction with Fe-containing enzymes
It is known that certain proteins have a degree of “promiscuity” in metal binding. However, most of these enzymes are active with only one metal as cofactor, although both metals can bind in vitro and in vivo. Fe(II) and Mn(II) bind weakly to most proteins and possess similar coordination preferences (Cotruvo and Stubbe, 2012). There are cases where enzymes, such as epimerases, are thought to use Fe2+ as a Lewis acid under normal growth conditions but switch to Mn2+ under oxidative stress. Estradiol dioxygenases have been found to use both Fe2+ and Mn2+ (Farquhar et al., 2011). Notably, a specific class of I ribonucleotide reductases (RNRs), which convert nucleotides in deoxynucleotides, have evolved unique biosynthetic pathways to control metallation (Stubbe and Cotruvo, 2011). For instance, Fe- and Mn-dependent superoxide dismutases (SODs) catalyze the disproportionation of superoxide using highly similar protein scaffolds and nearly identical active sites (Cotruvo and Stubbe, 2012). Despite the extensive homology between the isoforms, Mn- and Fe-SODs are only active with their cognate metal (Vance and Miller, 2001). Misincorporation of Fe into Mn-SOD or vice versa alters the redox potential of the enzyme’s active site and inhibits superoxide disproportionation (Beyer and Fridovich, 1991). Nevertheless, misincorporation of Fe into Mn-SOD does occur in vivo, as observed in Escherichia coli (Yang et al., 2006). Using mitochondria from Saccharomyces cerevisae, Naranuntarat and co-workers verified that Fe binds to SOD-2 when cells are starved for Mn, inactivating the enzyme (Naranuntarat et al., 2009).
Furthermore, in vivo chronic Mn exposure in rats receiving intraperitoneal injection of 6 mg/kg Mn as MnCl2 daily for 30 consecutive days led to a region-specific alteration in total aconitase in frontal cortex, striatum and substantia nigra (Zheng et al., 1998). Aconitase is an enzyme from the tricarboxylic acid cycle that possesses an iron-sulfur cluster. When the cellular Fe level is insufficient, cytoplasmic aconitase loses the fourth labile Fe and assumes a [3Fe-4S] configuration. In this state, the coordination chemistry of Mn closely resembles that of Fe, possibly allowing Mn to interact with Fe in both mitochondrial and cytoplasmic aconitases, thus altering cellular energy metabolism and Fe regulation (Zheng et al., 1998). Unzai et al. prepared a series of hybrid hemoglobins in which Fe from heme was replaced by different metals, Mn included, in the α or β subunits. None of the substituted hemoglobins reacted with dioxygen or carbon monoxide, suggesting that the putative substitution of Fe by Mn during ferropenic anemia would impair hemoglobin function (Unzai et al., 1998).
3.5. Antidotal Strategies
It remains controversial as to whether manganism, a parkinsonian-like syndrome, can be treated with levodopa (Lucchini et al., 2009; Racette et al., 2001). Accordingly, other therapeutic approaches using drugs and genomic evaluations have been investigated.
Because oxidative stress plays a crucial role in Mn-induced neurotoxicity, antioxidant compounds have been of great interest. It has been demonstrated that synthetic compounds such as organochalcogens 2-phenyl-1,2-benzisoselenazol-3[2H]-one (ebselen) and diethyl-2-phenyl-2 tellurophenyl vinylphosphonate (DPTVP) (Avila et al., 2010; Santos et al., 2012) mitigate Mn-induced neurotoxicity. These compounds, which possess strong antioxidant properties, caused improvement in motor activity in rats and attenuated Mn-induced brain ROS generation (Avila et al., 2010; Santos et al., 2012). In the nematode Caenorhabditis elegans, these compounds protected against Mn-induced oxidative stress, decreasing ROS levels and increasing the life-span of Mn-exposed worms (Avila et al., 2012). Another important antioxidant, lycopene, strongly inhibited lipid peroxidation induced by Mn in brain and liver by acting as an efficient chain-breaking antioxidant, trapping lipid radicals (Lebda et al., 2012).
In rodents, anti-inflammatory agents, such as indomethacin and para-aminosalicilic acid, reduced Mn-induced increase in oxidative stress (isoprostanes) and neuroinflammation (prostaglandin E2) (Milatovic et al., 2011; Santos et al., 2012). Notably, indomethacin protected against progressive spine degeneration and dendritic damage in striatal medium spiny neurons of mice exposed to Mn (Milatovic et al., 2011). This protection is probably mediated by the transcription factor NF-κB (Moreno et al., 2011). Using transgenic mice expressing a transcription factor fused to a green fluorescent protein (GFP), Moreno and co-workers showed that Mn exposure increased NF-κB reporter activity and nitric oxide synthase 2 (NOS2) expression in both microglia and astrocytes, and that these effects were prevented by supplementation with steroid 17β-estradiol. This steroid is one of the most active estrogen hormones possessing neuroprotective effects in both in vivo and in vitro models, and it has been shown to enhance astrocytic glutamate transporter function (Liang et al., 2002). Estrogen also decreased neuronal protein nitration in treated mice and inhibited apoptosis in striatal neurons cocultured with Mn-treated astrocytes in vitro (Moreno et al., 2011). Furthermore, tamoxifen, a estrogen related compound, effectively reversed glutamate transport inhibition in a Mn-induced model of glutamatergic deregulation, suggesting a potential therapeutic modality in neurodegenerative disorders which are characterized by altered glutamate homeostasis (Lee et al., 2012). In agreement with this study, Xu et al. showed that the pretreatment of rats with the NMDA (N-methyl-D-aspartate) antagonist MK801 protected neurons from Mn-induced glutamate excitotoxicity (Xu et al., 2010). Several studies have addressed genetic factors that mediate of Mn toxicity. Streifel and co-workers used mice lacking NOS, postulating that they would be protected from the neurotoxic effects of Mn. They found that loss of NOS2 reduced NO-induced peroxynitrite formation, thus attenuating Mn-related peroxynitrite adduct formation in the striatal-pallidum and substantia nigra pars reticulate. These mice showed attenuated alterations in neurobehavioral function and neurochemistry in vivo and also loss of NOS2 also prevented astrocyte-mediated neuronal apoptosis in vitro (Streifel et al., 2012). In C. elegans, Benedetto et al. observed that Mn-induced DAergic neurotoxicity requires the NADPH dual-oxidase BLI-3, suggesting that in vivo BLI-3 activity promotes the conversion of extracellular DA into toxic reactive species, which, in turn, can be taken up by DAT-1 in DAergic neurons, thus leading to oxidative stress and cell degeneration (Benedetto et al., 2010). BLI-3 knockout or inhibition may represent a novel strategy for mitigating Mn neurotoxicity. Expression of parkin, an E3 ubiquitin ligase also linked to PD, protects against Mn toxicity, as observed in SH-SY5Y cells (Roth, 2009). Conversely, deletion of parkin leads to increase in DMT-1 levels, thus causing increase in Mn uptake (Roth, 2009). Furthermore, it was reported in yeast that expression of PARK9, a gene linked to PD, protected cells from Mn toxicity (Gitler et al., 2009).
4. Mercury
4.1. Properties, Chemical Forms and Human Exposure
Mercury is a transition metal commonly named quicksilver due to its liquid and silvery characteristics. It is recognized by the symbol Hg, which comes from the Latin term hydrargyrum, meaning “watery silver”. It is present in the environment due to both natural (earth’s surface evaporation and volcanic eruptions) and anthropogenic (emissions from coal-burning power stations and incinerators) sources. As a result of specific reactions (i.e. oxidation, methylation), different chemical forms of Hg are present, such as elemental mercury (Hg0), inorganic (divalent and monovalent cationic forms; Hg2+ and Hg+) and organic (i.e. methylmercury; MeHg) mercury compounds. While human exposures to all environmentally existing forms of Hg have been documented, exposure to MeHg represents a major concern. Exposures to MeHg, which is present at high concentrations in seafood diets, are common and ubiquitous; MeHg has a higher entry rate into the CNS compared with inorganic mercurials, rendering it an important neurotoxicant (Aschner et al., 2007b; Debes et al., 2006). Occupational exposures to Hg (mainly in the form of elemental mercury, Hg°), due to its use in industry (Neghab et al., 2012) and artisanal gold mining (Lubick, 2010), are also of toxicological relevance. In addition, iatrogenic exposures to Hg continue to represent a concern. For example, dental amalgams (important source of Hg°) are still used (for a review, see (Clarkson and Magos, 2006)).
The toxic properties and target organs of Hg are dependent upon its chemical speciation. This review focuses on forms of Hg with major neurotoxicological relevance: (i) Primary focus is directed at MeHg, which occurs mainly from contaminated seafood; (ii) because of its efficient transport through the BBB, the neurotoxicological significance of mercury vapor, secondary to exposures from occupational settings and dental amalgam, is also discussed.
4.2. Transport, Metabolism and Excretion
4.2.1 Methylmercury
Methylmercury (MeHg; CH3Hg+) is an organic mercury compound found in the aquatic environment (Ullrich et al., 2007). The majority of MeHg is derived from the methylation of inorganic mercury, carried out mostly by aquatic microorganisms (Compeau and Bartha, 1985). MeHg is biomagnified in the aquatic food chain, reaching concentrations as high as 1ppm in predatory fish (Hintelmann, 2010). Accordingly, populations that rely on fish diets can be exposed to high MeHg levels (Clarkson et al., 2003). MeHg is well absorbed by the gastrointestinal tract (around 95%) (Miettinen, 1973). After absorption, more than 90% of MeHg in the blood is intracellular (bound to erythrocyte hemoglobin); the fraction present in the blood is about 6%, upon complete equilibrium between blood and tissues is reached (Kershaw et al., 1980). In humans orally exposed to MeHg, the percentage (of total) of inorganic Hg in the blood, breast milk and urine is 7%, 39% and 73%, respectively (IPCS, 1990), suggesting that inorganic Hg is an important excretable metabolite of MeHg. Additionally, experimental evidence shows that MeHg can also be excreted via the biliary route, likely complexed to glutathione (GSH), as a GSH mercaptide (CH3Hg-SG) (Ballatori et al., 1995).
The CNS represents the main target organ of MeHg toxicity reflecting its efficient transport into the brain. MeHg transport across the BBB, as well as its uptake by neural cells, occurs via a MeHg-L-cysteine complex, which is transported by the L-type neutral amino acid transporter (Kerper et al., 1992; Yin et al., 2008b). Of note, a high percentage of inorganic Hg (above 80%) was found in the brain of a 30 year old individual who was exposed to MeHg at 8 years of age (22 years before) (Davis et al., 1994). Neurohistological outcomes were cortical atrophy, neuronal loss and gliosis, most pronounced in the paracentral and parietooccipital regions. Before death, the most evident neurological signs were cortical blindness, diminished hand proprioception, choreoathetosis, and attention deficits. In this patient, the total Hg level (more that 80% as inorganic Hg) in the left occipital cortex was more that 50-fold the levels found in control individuals (Davis et al., 1994), indicating a high persistence of Hg in the brain after MeHg exposure. Although MeHg is well recognized as a neurotoxicant by acting at specific biomolecular sites (for a review, see (Farina et al., 2011a; Farina et al., 2011b), the dealkylation of MeHg into inorganic Hg likely accounts for Hg’s persistence in the brain, and potentially long-lasting neurological outcomes (Grandjean et al., 1997a; Ninomiya et al., 2005).
MeHg is transferred from the pregnant mother to the fetus, reaching the fetal brain. In an experimental study where pregnant mice were directly exposed to MeHg, Watanabe and collaborators (1999) detected higher levels of the metal in the fetuses brain when compared to the dams, indicating a high transplacental transport of MeHg, as well as a great retention in the fetus brain (Watanabe et al., 1999). MeHg seems to be actively transported from the maternal to the fetal blood as its cysteine conjugate via the neutral amino acid carrier system (Kajiwara et al., 1996). Its high entry in the developing brain is related, at least in part, to the lack of functional BBB (Costa et al., 2004; Manfroi et al., 2004).
There are epidemiological studies showing that maternal exposure to MeHg during pregnancy causes neurological deficits in their offspring (Grandjean et al., 1997b; Murata et al., 2004). Interestingly, exposure to MeHg during early fetal development is linked to subtle brain injury at levels much lower than those affecting the mature brain (Grandjean and Landrigan, 2006), most likely because it affects cell differentiation, migration and synaptogenesis (Theunissen et al., 2011; Zimmer et al., 2011)
4.2.2 Mercury vapor
The major sources of elemental mercury vapor (Hg0) exposure are occupational and dental amalgams. Hg0 is still used in industry in the production of caustic soda and chlorine, and in the manufacture of thermometers, thermostats, fluorescent light bulbs, batteries and manometers (for a review, see (Clarkson and Magos, 2006)). Artisanal miners are also exposed to Hg0 by inhaling vapors when they burn off the Hg that is used to amalgamate gold (Lubick, 2010). Dental amalgams have also been reported as an important source of Hg0 (Hursh et al., 1976), although it may also be ingested in a particulate form.
Once absorbed (mainly through the respiratory tract), Hg0 is oxidized mainly by erythrocyte catalase to mercurous (Hg+) and mercuric (Hg2+) ions, which are toxic to several organs (particularly the kidneys), but have limited access to the CNS. Conversely, a certain amount of blood Hg0 (not oxidized by blood catalase) passes through the BBB, reaching the CNS. Data on the distribution of brain Hg after Hg0 exposure are scarce. In an experimental study with squirrel monkeys, the profile of distribution was not homogeneous within the different encephalic structures; Hg was found in both glial cells and neurons mainly in the cortical areas and in the fiber systems (Warfvinge et al., 1994). After Hg0 exposure in man, urine and feces are the main pathways of Hg excretion (Tejning and Ohman, 1966). Because of the fast oxidation of Hg0 into Hg2+, the mercury excreted in feces is probably in the form of mercuric mercury (for a detailed review on Hg0 toxicokinetics, see (Clarkson and Magos, 2006)).
Although Hg0 exposure can cause toxicity to several organs (Clarkson and Magos, 2006; Goldwater, 1972), neurotoxicological signs are prevalent. In humans, common symptoms observed after occupational exposure to Hg0 include decreased strength and coordination, and increased tremor (Albers et al., 1988). Corroborating these findings, experimental data have reported motor-related neurological impairments in monkeys (Newland et al., 1996) and mice exposed to Hg0 (Yoshida et al., 2005).
4.3. Mercury and neurodegeneration
4.3.1 Methylmercury
Although not completely understood, the molecular mechanisms mediating MeHg-induced neurotoxicity and neurodegeneration are better known when compared with those of elemental Hg. Because MeHg is a monoalkylmercurial, its Hg atom is a monocation (CH3-Hg+), which possess electrophilic properties. As an electrophilic compound, MeHg interacts with and oxidizes nucleophilic groups of several biomolecules; sulfhydryl (thiol/thiolate; -SH/-S−) groups are important and relevant targets of MeHg in the biological systems. Accordingly, the interactions of MeHg with sulfhydryl-containing proteins (i.e. neurotransmitter receptors, transporters, antioxidant enzymes, etc.), as well as with nonprotein thiols (i.e. glutathione, cysteine), are crucial events in mediating its neurotoxicity (Clarkson et al., 2003; Sumi, 2008). By direct interaction with thiols, as well as indirect mechanisms (discussed latter), MeHg can modify the oxidation state of the -SH groups on proteins, modulating their functions (Kim et al., 2002). Consequently, the activities of several -SH-containing proteins whose roles are decisive for proper homeostasis of neuronal and glial cells [i.e., creatine kinase (Glaser et al., 2010), GSH reductase (Stringari et al., 2008), Ca2+-ATPase (Freitas et al., 1996), thioredoxin reductase(Branco et al., 2012), choline acetyltransferase and enolase (Kung et al., 1987)] are perturbed after MeHg exposure. Altered protein function has been posited as a causative factor in MeHg-induced neurotoxicity and neurodegeneration (Farina et al., 2012; Farina et al., 2011b).
In addition to -SH-containing proteins, nonprotein thiols (represented mainly by GSH, the major low-molecular-weight thiol) are also important molecular targets involved in MeHg-induced neurotoxicity. Knowledge on the direct chemical interaction between MeHg and GSH, as well as its importance in mercurial toxicity, dates several decades (Neville and Drakenberg, 1974). Such an interaction affects the deposition of MeHg in tissues (Richardson and Murphy, 1975) and modifies Hg excretion in the bile of MeHg-exposed rats (Osawa and Magos, 1974), indicating that this low-molecular-weight thiol compound modulates its toxicity. Based on these observations (Neville and Drakenberg, 1974; Osawa and Magos, 1974; Richardson and Murphy, 1975), studies on the toxicological relevance of MeHg x GSH interaction have shown that strategies to increase GSH levels are protective against MeHg-induced neurotoxicity (Kaur et al., 2006, 2011; Shanker et al., 2005). Moreover, several in vitro studies with isolated organelles or cultured cells (Franco et al., 2007; Ni et al., 2011), as well as in vivo studies in mice (Franco et al., 2006; Stringari et al., 2008), have shown that MeHg exposure causes GSH depletion. Because of the crucial role of GSH in maintaining redox homeostasis (Dringen et al., 2005), several aspects of MeHg-induced neurotoxicity have been ascribed to GSH depletion (for a review, see (Farina et al., 2011a)).
Based on the direct chemical interaction between GSH and MeHg, GSH depletion upon MeHg exposure (Franco et al., 2006; Stringari et al., 2008) represents an expected phenomenon. However, intracellular GSH concentrations in the mammalian cerebrum and cerebellum are in the milimolar (mM) range. Because decreased GSH levels have been reported in the cortices (cerebral and cerebellar) of MeHg-exposed animals whose cortical mercury levels were in the low micromolar (μM) range (Franco et al., 2006; Stringari et al., 2008), it is reasonable to assume that the simple MeHg-GSH interaction is not the only cause of MeHg-induced GSH oxidation. MeHg seems to induce the formation of ROS by GSH-independent mechanisms as well, leading to subsequent GSH oxidation (Franco et al., 2007; Mori et al., 2007). This event seems to be also important in terms of protein oxidation, where ROS generated from MeHg can modulate the redox state of proteins, thus affecting their function. A classical example of such phenomenon was described by Allen et al. (2001), who showed that MeHg induces the generation of hydrogen peroxide (a common endogenous ROS), which down regulates the activity of astrocytic glutamate transporters, culminating in excitotoxicity (Lockman et al., 2001).
In addition to -SH groups (from both protein and low-molecular weight sources), selenohydryl (selenol/selenolate; -SeH/-Se-) groups have also been reported as important targets mediating MeHg-induced neurotoxicity/neurodegeneration. From a molecular point of view, it is important to note that selenols are more nucleophilic than thiols, which could render selenoproteins preferential molecular targets of MeHg compared with -SH-containing proteins (Farina et al., 2012). Accordingly, a recent and growing body of evidence points to selenoproteins, such as GSH peroxidase and thioredoxin reductase, as critical and primary targets in mediating MeHg-induced neurotoxicity (Branco et al., 2012; Carvalho et al., 2008; Farina et al., 2009; Franco et al., 2009; Usuki et al., 2011). This is based on the higher affinity of Hg for selenols compared with thiols (Sugiura et al., 1976). Such affinity allows for the transference of MeHg from a thiol to a selenol biomolecule (MeHg-SR + RSeH ⇒ MeHg-SeR + RSH). This higher affinity of Hg for selenols also renders the selenium-mercury linkage relatively stable, even in the presence of high (i.e. mM) thiol concentrations. In agreement, nM concentrations of MeHg significantly decreased the activity of the selenoprotein GSH peroxidase-1 in cultured neurons (Farina et al., 2009), whose cytosolic GSH concentrations are in the mM range.
Based on the aforementioned, it is reasonable to assume that any selenoprotein can represent a potential molecular target for MeHg. Interestingly, GSH peroxidase-1 (Farina et al., 2009; Franco et al., 2009), GSH peroxidase-4 (Zemolin et al., 2012), thioredoxin reductase (Branco et al., 2012; Wagner et al., 2010), selenoprotein W (Kim et al., 2005) and 5′-deiodinase (Watanabe et al., 2007) are examples of selenoproteins whose activities were down-regulated by MeHg. Because of the crucial role of such selenoproteins in the maintenance of the cellular homeostasis (Lu and Holmgren, 2009), one might posit that the selenium-mercury interaction plays a pivotal role in MeHg-induced neurodegeneration. Although the complete understanding on this scheme has yet to be resolved, this hypothesis is reinforced by the fact that inorganic and organic selenium compounds mitigate MeHg-induced neurotoxicity (Farina et al., 2003a; Glaser et al., 2010; Kaur et al., 2009; Yin et al., 2011).
As already mentioned, the neurotoxicity induced by MeHg is related, at least in part, to changes in the redox state of nucleophilic groups (mainly thiols and selenols) from protein sources. These changes are likely responsible for two important events that occur in the CNS of MeHg-exposed animals, namely, oxidative stress (reviewed by (Farina et al., 2011a)) and glutamate dyshomeostasis (see below). From a mechanistic point of view, the altered redox state may represent a consequence of the direct interaction of the nucleophilic groups with MeHg, as well as a resultant from the pro-oxidative effects of ROS generated during MeHg exposure. Table 1 depicts several enzymes, transporters and receptors (most of them are sulfhydryl- or selenohydryl-containing proteins) as potential molecular targets of MeHg-induced neurotoxicity/neurodegeneration.
Table 1.
Potential proteins mediating MeHg-induced neurotoxicity
| Protein | Effect | Functions | Reference |
|---|---|---|---|
| 3-ketoacid-coenzyme A transferase I | ⇓* | Ketone body metabolism | Vendrell et al., 2007 |
| 5′-deiodinase | ⇓# | Thyroid hormone metabolism | Watanabe et al., 2007 |
| ASC cysteine transporter | ⇓# | Cysteine uptake | Shanker et al., 2001 |
| Astrocytic glutamine transporter | ⇓# | Glutamine uptake from synaptic cleft | Yin et al., 2011 |
| Choline acetyl transferase | ⇓# | Acetylcholine synthesis | Kung et al., 1987 |
| Creatine kinase | ⇓# | Energetic metabolism | Glaser et al., 2010 |
| Cytosolic phospholipase A2 | ⇑#* | Hydrolysis of membrane phospholipids (arachidonic acid releasing) | Shanker et al., 2004 |
| Enolase | ⇓# | Glycolitic pathway | Kung et al., 1987 |
| Glutamate transporters | ⇓# | Glutamate uptake | Aschner et al., 1990; Manfroiet al., 2004; Farina et al., 2003a |
| Glutathione peroxidase 1 | ⇓# | Peroxide detoxification | Farina et al., 2009 |
| Glutathione peroxidase 4 | ⇓#* | Peroxide detoxification | Zamolin et al., 2012 |
| Glutathione reductase | ⇓⇑# | Reduction of GSSG to GSH | Farina et al., 2005; Stringari et al., 2008 |
| Monoamine oxidase | ⇓# | dopamine, serotonin, and noradrenaline metabolism | Beyrouty et al., 2006 |
| Nitric oxide synthase | ⇑# | Nitric oxide synthesis | Herculano et al., 2006 |
| Nrf2 transcription factor | ⇑* | Modulation of antioxidant and phase 2 enzyme expression | Ni et al., 2011 |
| Phosphorylated-cofilin | ⇓* | Reorganization of actin filaments | Vendrell et al., 2010 |
| Non-phosphorylated-cofilin | ⇑* | ||
| Selenoprotein W | ⇓* | Not well-identified (antioxidant, response to stress, immunity) | Kim et al., 2005 |
| Thioredoxin reductase | ⇓# | Reduction of thioredoxin (antioxidant effect) | Wagner et al., 2010; Branco et al., 2012 |
| X(AG(-)) cysteine transporter | ⇓# | Cysteine uptake | Shanker et al., 2001 |
In vitro and in vivo experimental evidences indicate that the activities of several proteins (from neuronal, astrocytic and/or microglial source) are modulated after MeHg exposure, suggesting their role in MeHg-neurotoxicity. The arrows ⇑ or ⇓ mean positive or negative modulator effects, respectively.
indicates that the variable was measured at functional level (i.e. enzyme activity, transporter activity).
indicates that the variable was measured at expression level (protein or mRNA).
An established event in MeHg-induced neurotoxicity, which seems to result from the primary interaction of the electrophilic toxicant with nucleophilic groups, is glutamate dyshomeostasis (reviewed by (Aschner et al., 2007b)). Glutamate is the most important excitatory neurotransmitter in the mammalian CNS, serving crucial roles on development, learning, memory and response to injury (Fonnum, 1984). Due to its direct and indirect pro-oxidative properties, MeHg increases extracellular glutamate levels, which result from both inhibition of glutamate uptake (Aschner et al., 2000; Brookes and Kristt, 1989) and stimulation of its release into the synaptic cleft (Reynolds and Racz, 1987), culminating in excitotoxic events (Aschner et al., 2007b). Over-activation of the NMDA subtype glutamate receptors leads to an increased Na+ and Ca2+ influx, which is associated with the generation of oxidative stress and neurotoxicity (Lafon-Cazal et al., 1993). Indeed, glutamate-mediated increased intracellular Ca2+ concentrations leads to increased nitric oxide production (due to activation of neuronal nitric oxide synthase), as well as to mitochondrial collapse (Farina et al., 2011a). Notably, MeHg-induced Ca2+ and glutamate dyshomeostasis, as well as MeHg-induced ROS generation (oxidative stress), are events that contribute independently to neurotoxicity, but also represent inter-connected phenomena affecting each other. Figure 4 depicts the relationship between glutamate and calcium dyshomeostasis and oxidative stress in MeHg-mediating neurotoxicity.
Figure 4. MeHg-induced glutamate and calcium dyshomeostasis and oxidative stress.
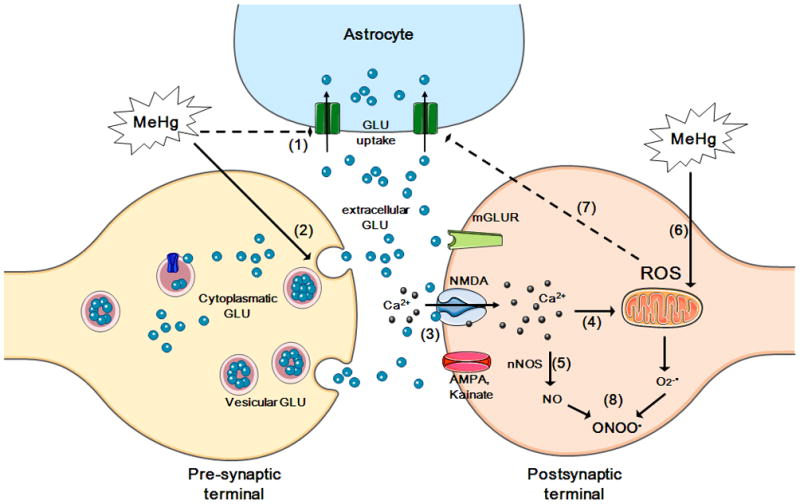
MeHg causes increased extracellular glutamate (GLU) levels via the inhibition of astrocytic glutamateuptake (event 1) and the stimulation of glutamate release from pre-synaptic terminals (event 2). Increased extracellular glutamate levels overactivate N-methyl D-aspartate (NMDA)-type glutamate receptors, increasing calcium influx into neurons (event 3). Increased levels of intracellular calcium, which can lead to mitochondrial collapse (event 4), activate neuronal nitric oxide synthase (nNOS) (event 5), thus increasing nitric oxide (NO) formation. MeHg affects the mitochondrial electron transfer chain (mainly at the level of complexes II–III) (event 6), leading to increased formation of reactive oxygen species [ROS; superoxide anion (O2•−) and hydrogen peroxide (H2O2)]. H2O2 can inhibit astrocyte glutamate transporters (event 7), contributing to the excitotoxic cycle. O2•− reacts with NO (event 8), generating peroxynitrite (ONOO•), a highly oxidative molecule. Adapted from Farina et al., 2011a.
An interesting aspect of MeHg neurotoxicology is its preferential affinity for specific regions/structures of the CNS, leading to particular histological and behavioral characteristics. Pathological analyses of MeHg-poisoned adult individuals from the Minamata Bay, Japan (where the major MeHg outbreak took place), showed that this mercurial does not uniformly affect the nervous system; commonly, the cerebral and cerebellar cortices are the regions more severely affected (Eto et al., 2010). Indeed, in adult Minamata patients, a significant neurodegeneration has been observed mainly in calcarine, temporal, pre- and postcentral cortices, as well as in the cerebellar hemispheres (Eto et al., 2010). These pathological observations are in agreement with the symptoms observed in Minamata disease patients, characterized by cerebellar ataxia, concentric constriction of their visual fields, and sensory disturbances (Uchino et al., 1995). Experimental studies with MeHg-exposed animals have also pointed to the cerebral and cerebellar cortices as preferential encephalic structures subjected to MeHg-neurodegeneration; moreover, similar symptoms (visual, motor and sensory disturbances) have been observed (Carvalho et al., 2007; Charleston et al., 1995; Dietrich et al., 2005).
The neurodegeneration detected in the cerebral and cerebellar cortices of Minamata patients and MeHg-exposed animals (Carvalho et al., 2007; Eto et al., 2010) is likely consequence of a relative short-term high dose exposure to this mercurial. However, it is noteworthy that fishing communities are commonly exposed to chronic low-dose exposures (Clarkson et al., 2003), which probably induce a more subtle (maybe “undetectable”) pattern of neurodegeneration/neurotoxicity. Human health concerns associated with these chronic exposures are of particular relevance taking into account (i) the absence of a factual non-observed adverse effect level (NOAEL) in terms of MeHg-induced neurotoxicity (mainly with respect to developmental toxicity) and (ii) the potential occurrence of a dangerous but silent pandemic of subclinical MeHg neurotoxicity (Grandjean and Landrigan, 2006).
4.3.1 Mercury vapor (elemental mercury)
Data on the molecular mechanisms mediating elemental mercury (Hg0)-induced neurotoxicity/neurodegeneration are scarce compared with those on MeHg. Hg0 (in contrast to MeHg) causes general toxicity in several tissues, such as lung, kidney and gastrointestinal tract, among others (Goldwater, 1972; Magos, 1967). Indeed, as previously mentioned, most of the absorbed Hg0 is oxidized in the blood to Hg2+, and subsequently targets several organs. However, a certain amount of blood Hg0 (not oxidized to Hg2+) passes through the BBB prior to this oxidation step, thus reaching the CNS. Of note, it is believed that the mercuric ion Hg2+ (generated within the CNS from Hg0 oxidation) is the proximate toxic chemical form because mercury vapor itself is unable to react with tissue ligands. Consequently, the oxidation of Hg0 to Hg2+ (in both blood and CNS) seems to be an important determinant on the degree and pattern of the toxic effects ofHg0 (Magos, 1967).
From a mechanistic point of view, it is important to note that Hg2+ (generated from Hg0 oxidation within the CNS) binds to -SH-containing ligands (Aschner and Aschner, 1990); this event likely dictates the neurotoxicity observed after Hg0exposure. In agreement, an experimental study in Hg0-exposed mice showed higher susceptibility to Hg0-induced behavioral changes in metallothionein (MT)-null compared with wild type animals (Yoshida et al., 2005). Based on the high affinity of Hg2+ for thiols, as well as on the fact that MTs are cysteine-rich intracellular proteins with great affinity for divalent metals, the results by Yoshida et al. (2005) indicate that the interaction of Hg2+(derived from Hg0) with-SH-containing ligands in the CNS likely represents an important event mediating toxicity.
In vitro studies aimed on Hg0-induced neurotoxicity have been carried out with Hg2+ (Albrecht and Matyja, 1996; Brookes and Kristt, 1989), as a surrogate of Hg0 since the latter is rapidly biotransformed to Hg2+. Cell culture-based studies (Brookes and Kristt, 1989) pointed to glutamate dyshomeostasis as a critical event mediating Hg2+-induced toxicity. In fact, sub- M concentrations of Hg2+ inhibited the clearance of extracellular glutamate both in astrocyte and spinal cord cultures, and reduced glutamine content and export in astrocyte cultures (Brookes and Kristt, 1989), indicating that Hg2+-induced neurotoxicity might be mediated by excitotoxic events. In agreement, Albrecht and Matyja (1996) not only observed decreased glutamate uptake, but also increased glutamate release in Hg2+-exposed cultured astrocytes, reinforcing the idea that Hg0/Hg2+-neurotoxicity may be mediated by excitotoxic activity of glutamate (Albrecht and Matyja, 1996). Interestingly, this study also reported that the inhibition of glutamate uptake was attenuated by addition to the cultures of a cell membrane-penetrating agent dithiothreitol (a dithiol agent), but not of GSH, which is not transported into the cells. These results reinforce that the intracellular thiol status is likely responsible for the effects of Hg2+ in mediating astrocyte glutamate dyshomeostasis. This hypothesis is reinforced by the fact that the activity of astrocyte glutamate transporters is sensitive to thiol agents (Volterra et al., 1994).
Although data on the mechanisms mediating Hg0-neurotoxicity are scarce, existing evidence suggests that changes in the redox state of -SH-containing proteins plays a critical role (Albrecht and Matyja, 1996; Aschner and Aschner, 1990; Brookes and Kristt, 1989; Yoshida et al., 2005). However, based on the high affinity of Hg2+ (herein, derived from Hg0) for selenols, it is reasonable to suggest that selenoproteins could also mediate the neurotoxic effects observed after Hg0 exposure. This idea is based on the higher affinity of Hg2+ for selenols compared with thiols (Sasakura and Suzuki, 1998). Carvalho and coworkers (2008) observed that the selenoprotein thioredoxin reductase (TrxR) is selectively inhibited by Hg2+ and concluded that the significant potency of the mercurial to bind to the selenol group in the active site of TrxR represents a major molecular mechanism of its toxicity (Carvalho et al., 2008). Because of the probable interaction between Hg2+ (derived from Hg0) and selenols in the CNS, the potential involvement of selenoproteins in the neurotoxicity elicited by Hg0 represents an important research field that deserves further attention. This is believed because (i) Hg2+ toxicity is antagonized by selenium compounds (Farina et al., 2003b; Yamamoto, 1985), (ii) Hg2+, which is generated in the SNC after Hg0 oxidation, inhibits the activity of selenoproteins by interacting with their selenol group (Carvalho et al., 2008), and (iii) miners occupationally exposed to Hg0hadlower levels of plasma selenium when compared with control individuals (Kobal et al., 2004).
4.4. Antidotal Strategies
Several compounds have been reported to protect against Hg toxicity in experimental in vitro and in vivo models. Vitamin E (Shichiri et al., 2007), thiol compounds (Falluel-Morel et al., 2012; Koh et al., 2002), natural products (Farina et al., 2005; Franco et al., 2010; Lapina et al., 2000; Lucena et al., 2007), vitamin K (Sakaue et al., 2011), chelating agents (Carvalho et al., 2007), Ca2+-channel blockers and glutamatergic antagonists (Ramanathan and Atchison, 2011), among others, have shown beneficial effects against mercurial toxicity. Although the aforementioned protective effects have been observed under experimental conditions, unfortunately, the clinical practice with Hg-exposed humans has shown the absence of an effective treatment that completely abolishes the toxic effects. In such cases, supportive care is given when necessary to maintain vital functions and the administration of chelator agents is performed in an attempt to assist the body’s ability to eliminate Hg from the tissues. However, these drugs have limited use because of incomplete efficacies in removing Hg from tissues and significant adverse side effects (Tchounwou et al., 2003).
A rapid antidotal intervention is required in high-dose acute exposures, which are commonly observed after occupational or intentional exposures to Hg0 (Bluhm et al., 1992; De Palma et al., 2008; Eyer et al., 2006). Different chelating agents, including penicillamine, dimercaprol, 2,3-dimercaptopropane-1-sulphonate (DMPS), and meso-2,3-dimercaptosuccinic acid (DMSA), have been administered in these cases (Eyer et al., 2006; Houeto et al., 1994); however, the desired beneficial results are generally not achieved. In fact, even though urinary Hg excretion could be significantly enhanced during chelation therapy, its efficacy on the disappearance of tissue Hg deposits seems to be negligible (Lin and Lim, 1993; Rodrigues et al., 1986).
Chelating therapy can also increase Hg excretion after MeHg exposure, which suggests its beneficial use as antidotal strategy for MeHg poisoning. Clarkson et al. (1981) studied the effects of three chelating agents (DMPS, D-penicillamine and N-acetyl-DL-penicillamine) and a thiolated resin in reducing the blood half-life (T 1/2) of MeHg during an outbreak of human poisoning. All four treatments significantly reduced the mean T 1/2 compared with placebo; DMPS was the most effective agent. Another study with healthy individuals showed that oral DMSA treatment produced a rise in urine Hg excretion of fish eaters; although a similar increase in renal Hg excretion was observed in non-fish eaters (Ruha et al., 2009). Existing evidence concerning the use of chelating therapy in MeHg poisoning indicates that chelators can remove MeHg from the body, but cannot reverse the damage to the CNS (Clarkson et al., 2003). This aspect is particularly important when considering the most common pattern of human MeHg exposure (low-dose/long-term exposures), which is observed in fish-eating populations. The relative short-term high-dose MeHg poisonings, such as those observed during the well known outbreaks in Minamata Bay (Harada, 1978) and Iraq (Bakir et al., 1973), do not represent the common profile of human MeHg poisoning. In fact, human exposures to MeHg in fishing communities generally occur over extended periods (months or years) due to long-term seafood intake. Thus, massive short-term MeHg exposures are not frequent and, consequently, antidotal clinical interventions (i.e., chelating therapy) are not usually necessary (and useful) in such cases. In fact, it is believed that the neurological impairments observed in humans chronically exposed to MeHg due to the ingestion of contaminated fish might not necessarily correlate with the Hg levels present in tissues. In line with this, an experimental study on the developmental exposure of mice to MeHg showed that cerebral biochemical parameters affected by MeHg exposure (i.e., lipid peroxidation, GSH levels, GPx and GR activities) remained changed in the MeHg-exposed animals even when the cerebral Hg concentration decreased to basal levels (Stringari et al., 2008). These results indicated the persistence of MeHg-induced cerebral biochemical changes even when the cerebral concentrations of the toxicant were undetectable, suggesting an enduring toxic mark. Such experimental observation (Stringari et al., 2008) appears to be closely related to permanent functional deficits observed at 14 years after prenatal MeHg exposure (Debes et al., 2006), where chelating therapy would probably have no beneficial effect.
There is a consensus that chelating therapy can significantly increase Hg excretion, at least in some specific cases (Bluhm et al., 1992; Clarkson et al., 1981; De Palma et al., 2008; Eyer et al., 2006). Of note, chelating therapy is greatly based on -SH-containing molecules, such as D-penicillamine, N-acetyl-DL-penicillamine, dimercaprol, DMPS, and DMSA. Based on the higher affinity of Hg for selenols when compared with thiols, one could ask: “why selenocompoundsare not used as potential chelating agents for human Hg poisoning”? To the best of our knowledge, there is no data on the potential antidotal effects of selenocompounds against Hg toxicity in humans. However, experimental evidence indicates that organic selenocompounds not only protect against mercurials’ toxicity (Farina et al., 2003a; Moretto et al., 2005; Yin et al., 2011), but also decrease Hg deposition in tissues (de Freitas et al., 2009). A comparative study on the effectiveness of thiol- and selenol-based compounds in reversing mercurial toxicity and in increasing Hg excretion is warranted.
5. Concluding Remarks
Metals are constantly present in our lives, as we ingest essential metals in food and as we are exposed to them in the air dust or in contaminated water or food. Interest in the toxicity of essential trace metals has evolved from the need for government regulatory agencies such as the United States Environmental Protection Agency (EPA) to set environmental standards for these metals, as well as classic toxic metals such as Hg. The metals discussed in this review can be readily absorbed from different sources, and reach the CNS thus affecting neurons and glial cells. The mechanisms of toxicity are still not clearly understood; however their clinical features are well described and remain of great concern. Understanding these mechanisms is essential in designing novel therapeutic approaches, including antioxidants with diverse modes of action. In fact, the efficacy of antioxidants as potential therapeutic agents against Fe, Mn and Hg highlights oxidative stress as a unifying feature in their neurotoxic effect. However, the primary events triggered by these metals are mediated via distinct molecular targets. A better understanding of these mechanisms will assist in the development of multifactorial approaches to blunt or delay the progression of disease.
Oxidative stress (OS), particularly in mitochondria, is a common feature of iron (Fe), Mn and Hg toxicity.
The primary molecular targets triggering of OS are distinct for each of the above metals.
These metals are transported by distinct transporters.1
Acknowledgments
The authors would like to thank their colleagues/co-authors who have contributed to several studies referenced in this review. These studies were funded in part by grants from (in alphabetical order): Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq, INCT for Excitotoxicity and Neuroprotection-MCT/CNPq and IBNnet/CNPq, Brazil (Daiana S. Ávila, João B. T. Rocha and Marcelo Farina); Fundação de Amparo à Pesquisa do Estado de Santa Catarina, Brazil (Marcelo Farina); Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul, Brazil (Daiana S. Ávila and João B. T. Rocha); US Public Health Service grants from the National Institute of Environemntal Health Sciences (NIEHS) R01 ES10563 and R01 ES07331 (Michael Aschner).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akatsu H, Hori A, Yamamoto T, Yoshida M, Mimuro M, Hashizume Y, Tooyama I, Yezdimer EM. Transition metal abnormalities in progressive dementias. Biometals. 2012;25:337–350. doi: 10.1007/s10534-011-9504-8. [DOI] [PubMed] [Google Scholar]
- Albers JW, Kallenbach LR, Fine LJ, Langolf GD, Wolfe RA, Donofrio PD, Alessi AG, Stolp-Smith KA, Bromberg MB. Neurological abnormalities associated with remote occupational elemental mercury exposure. Ann Neurol. 1988;24:651–659. doi: 10.1002/ana.410240510. [DOI] [PubMed] [Google Scholar]
- Albrecht J, Matyja E. Glutamate: a potential mediator of inorganic mercury neurotoxicity. Metab Brain Dis. 1996;11:175–184. doi: 10.1007/BF02069504. [DOI] [PubMed] [Google Scholar]
- Ali SF, Duhart HM, Newport GD, Lipe GW, Slikker W., Jr Manganese-induced reactive oxygen species: comparison between Mn+2 and Mn+3. Neurodegeneration. 1995;4:329–334. doi: 10.1016/1055-8330(95)90023-3. [DOI] [PubMed] [Google Scholar]
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10(Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- Anderson AC. Iron poisoning in children. Curr Opin Pediatr. 1994;6:289–294. doi: 10.1097/00008480-199406000-00010. [DOI] [PubMed] [Google Scholar]
- Anderson JG, Cooney PT, Erikson KM. Brain manganese accumulation is inversely related to gamma-amino butyric acid uptake in male and female rats. Toxicol Sci. 2007;95:188–195. doi: 10.1093/toxsci/kfl130. [DOI] [PubMed] [Google Scholar]
- Archibald FS, Tyree C. Manganese poisoning and the attack of trivalent manganese upon catecholamines. Arch Biochem Biophys. 1987;256:638–650. doi: 10.1016/0003-9861(87)90621-7. [DOI] [PubMed] [Google Scholar]
- Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42:1781–1786. doi: 10.1161/STROKEAHA.110.596718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner JL, Aschner M. Nutritional aspects of manganese homeostasis. Mol Aspects Med. 2005;26:353–362. doi: 10.1016/j.mam.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Aschner JL. Mercury neurotoxicity: mechanisms of blood-brain barrier transport. Neurosci Biobehav Rev. 1990;14:169–176. doi: 10.1016/s0149-7634(05)80217-9. [DOI] [PubMed] [Google Scholar]
- Aschner M, Erikson KM, Dorman DC. Manganese dosimetry: species differences and implications for neurotoxicity. Crit Rev Toxicol. 2005;35:1–32. doi: 10.1080/10408440590905920. [DOI] [PubMed] [Google Scholar]
- Aschner M, Erikson KM, Herrero Hernandez E, Tjalkens R. Manganese and its role in Parkinson’s disease: from transport to neuropathology. Neuromolecular Med. 2009;11:252–266. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Gannon M. Manganese (Mn) transport across the rat blood-brain barrier: saturable and transferrin-dependent transport mechanisms. Brain Res Bull. 1994;33:345–349. doi: 10.1016/0361-9230(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Aschner M, Guilarte TR, Schneider JS, Zheng W. Manganese: recent advances in understanding its transport and neurotoxicity. Toxicol Appl Pharmacol. 2007a;221:131–147. doi: 10.1016/j.taap.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Syversen T, Souza DO, Rocha JB, Farina M. Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Braz J Med Biol Res. 2007b;40:285–291. doi: 10.1590/s0100-879x2007000300001. [DOI] [PubMed] [Google Scholar]
- Aschner M, Yao CP, Allen JW, Tan KH. Methylmercury alters glutamate transport in astrocytes. Neurochem Int. 2000;37:199–206. doi: 10.1016/s0197-0186(00)00023-1. [DOI] [PubMed] [Google Scholar]
- ATSDR. (Agency of Toxic Substances and Disease Registry)- Toxicological profile for manganese. U.S. Department of Health and Human Services Public Health Service; 2000. [Google Scholar]
- Au C, Benedetto A, Anderson J, Labrousse A, Erikson K, Ewbank JJ, Aschner M. SMF-1, SMF-2 and SMF-3 DMT1 orthologues regulate and are regulated differentially by manganese levels in C. elegans. PLoS One. 2009;4:e7792. doi: 10.1371/journal.pone.0007792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila DS, Benedetto A, Au C, Manarin F, Erikson K, Soares FA, Rocha JB, Aschner M. Organotellurium and organoselenium compounds attenuate Mn-induced toxicity in Caenorhabditis elegans by preventing oxidative stress. Free Radic Biol Med. 2012;52:1903–1910. doi: 10.1016/j.freeradbiomed.2012.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila DS, Colle D, Gubert P, Palma AS, Puntel G, Manarin F, Noremberg S, Nascimento PC, Aschner M, Rocha JB, Soares FA. A possible neuroprotective action of a vinylic telluride against Mn-induced neurotoxicity. Toxicol Sci. 2010;115:194–201. doi: 10.1093/toxsci/kfq036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakir F, Damluji SF, Amin-Zaki L, Murtadha M, Khalidi A, al-Rawi NY, Tikriti S, Dahahir HI, Clarkson TW, Smith JC, Doherty RA. Methylmercury poisoning in Iraq. Science. 1973;181:230–241. doi: 10.1126/science.181.4096.230. [DOI] [PubMed] [Google Scholar]
- Ballatori N, Gatmaitan Z, Truong AT. Impaired biliary excretion and whole body elimination of methylmercury in rats with congenital defect in biliary glutathione excretion. Hepatology. 1995;22:1469–1473. [PubMed] [Google Scholar]
- Barbeau A. Manganese and extrapyramidal disorders (a critical review and tribute to Dr. George C. Cotzias) Neurotoxicology. 1984;5:13–35. [PubMed] [Google Scholar]
- Barceloux DG. Manganese. J Toxicol Clin Toxicol. 1999;37:293–307. doi: 10.1081/clt-100102427. [DOI] [PubMed] [Google Scholar]
- Bartzokis G, Sultzer D, Cummings J, Holt LE, Hance DB, Henderson VW, Mintz J. In vivo evaluation of brain iron in Alzheimer disease using magnetic resonance imaging. Arch Gen Psychiatry. 2000;57:47–53. doi: 10.1001/archpsyc.57.1.47. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta. 1998;1366:211–223. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- Benedetto A, Au C, Aschner M. Manganese-induced dopaminergic neurodegeneration: insights into mechanisms and genetics shared with Parkinson’s disease. Chem Rev. 2009;109:4862–4884. doi: 10.1021/cr800536y. [DOI] [PubMed] [Google Scholar]
- Benedetto A, Au C, Avila DS, Milatovic D, Aschner M. Extracellular dopamine potentiates mn-induced oxidative stress, lifespan reduction, and dopaminergic neurodegeneration in a BLI-3-dependent manner in Caenorhabditis elegans. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg D, Gerlach M, Youdim MB, Double KL, Zecca L, Riederer P, Becker G. Brain iron pathways and their relevance to Parkinson’s disease. J Neurochem. 2001;79:225–236. doi: 10.1046/j.1471-4159.2001.00608.x. [DOI] [PubMed] [Google Scholar]
- Beyer WF, Jr, Fridovich I. In vivo competition between iron and manganese for occupancy of the active site region of the manganese-superoxide dismutase of Escherichia coli. J Biol Chem. 1991;266:303–308. [PubMed] [Google Scholar]
- Blanusa M, Varnai VM, Piasek M, Kostial K. Chelators as antidotes of metal toxicity: therapeutic and experimental aspects. Curr Med Chem. 2005;12:2771–2794. doi: 10.2174/092986705774462987. [DOI] [PubMed] [Google Scholar]
- Bleackley MR, Macgillivray RT. Transition metal homeostasis: from yeast to human disease. Biometals. 2011;24:785–809. doi: 10.1007/s10534-011-9451-4. [DOI] [PubMed] [Google Scholar]
- Bluhm RE, Bobbitt RG, Welch LW, Wood AJ, Bonfiglio JF, Sarzen C, Heath AJ, Branch RA. Elemental mercury vapour toxicity, treatment, and prognosis after acute, intensive exposure in chloralkali plant workers. Part I: History, neuropsychological findings and chelator effects. Hum Exp Toxicol. 1992;11:201–210. doi: 10.1177/096032719201100308. [DOI] [PubMed] [Google Scholar]
- Bondy SC. The neurotoxicity of environmental aluminum is still an issue. Neurotoxicology. 2010;31:575–581. doi: 10.1016/j.neuro.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman AB, Kwakye GF, Hernandez EH, Aschner M. Role of manganese in neurodegenerative diseases. J Trace Elem Med Biol. 2011;25:191–203. doi: 10.1016/j.jtemb.2011.08.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco V, Canario J, Lu J, Holmgren A, Carvalho C. Mercury and selenium interaction in vivo: effects on thioredoxin reductase and glutathione peroxidase. Free Radic Biol Med. 2012;52:781–793. doi: 10.1016/j.freeradbiomed.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Copper excess, zinc deficiency, and cognition loss in Alzheimer’s disease. Biofactors. 2012;38:107–113. doi: 10.1002/biof.1005. [DOI] [PubMed] [Google Scholar]
- Brookes N, Kristt DA. Inhibition of amino acid transport and protein synthesis by HgCl2 and methylmercury in astrocytes: selectivity and reversibility. J Neurochem. 1989;53:1228–1237. doi: 10.1111/j.1471-4159.1989.tb07419.x. [DOI] [PubMed] [Google Scholar]
- Calne DB, Chu NS, Huang CC, Lu CS, Olanow W. Manganism and idiopathic parkinsonism: similarities and differences. Neurology. 1994;44:1583–1586. doi: 10.1212/wnl.44.9.1583. [DOI] [PubMed] [Google Scholar]
- Campanella A, Rovelli E, Santambrogio P, Cozzi A, Taroni F, Levi S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: hypothesis for a protective role in Friedreich ataxia. Hum Mol Genet. 2009;18:1–11. doi: 10.1093/hmg/ddn308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell T, Rama R. Iron, oxidative stress and early neurological deterioration in ischemic stroke. Curr Med Chem. 2007;14:857–874. doi: 10.2174/092986707780363014. [DOI] [PubMed] [Google Scholar]
- Carlsson M, Cortes D, Jepsen S, Kanstrup T. Severe iron intoxication treated with exchange transfusion. Arch Dis Child. 2008;93:321–322. doi: 10.1136/adc.2007.123240. [DOI] [PubMed] [Google Scholar]
- Carvalho CM, Chew EH, Hashemy SI, Lu J, Holmgren A. Inhibition of the human thioredoxin system. A molecular mechanism of mercury toxicity. J Biol Chem. 2008;283:11913–11923. doi: 10.1074/jbc.M710133200. [DOI] [PubMed] [Google Scholar]
- Carvalho MC, Franco JL, Ghizoni H, Kobus K, Nazari EM, Rocha JB, Nogueira CW, Dafre AL, Muller YM, Farina M. Effects of 2,3-dimercapto-1-propanesulfonic acid (DMPS) on methylmercury-induced locomotor deficits and cerebellar toxicity in mice. Toxicology. 2007;239:195–203. doi: 10.1016/j.tox.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Charleston JS, Body RL, Mottet NK, Vahter ME, Burbacher TM. Autometallographic determination of inorganic mercury distribution in the cortex of the calcarine sulcus of the monkey Macaca fascicularis following long-term subclinical exposure to methylmercury and mercuric chloride. Toxicol Appl Pharmacol. 1995;132:325–333. doi: 10.1006/taap.1995.1114. [DOI] [PubMed] [Google Scholar]
- Chasapis CT, Loutsidou AC, Spiliopoulou CA, Stefanidou ME. Zinc and human health: an update. Arch Toxicol. 2012;86:521–534. doi: 10.1007/s00204-011-0775-1. [DOI] [PubMed] [Google Scholar]
- Chen MK, Lee JS, McGlothan JL, Furukawa E, Adams RJ, Alexander M, Wong DF, Guilarte TR. Acute manganese administration alters dopamine transporter levels in the non-human primate striatum. Neurotoxicology. 2006;27:229–236. doi: 10.1016/j.neuro.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Chua AC, Morgan EH. Manganese metabolism is impaired in the Belgrade laboratory rat. J Comp Physiol [B] 1997;167:361–369. doi: 10.1007/s003600050085. [DOI] [PubMed] [Google Scholar]
- Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit Rev Toxicol. 2006;36:609–662. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- Clarkson TW, Magos L, Cox C, Greenwood MR, Amin-Zaki L, Majeed MA, Al-Damluji SF. Tests of efficacy of antidotes for removal of methylmercury in human poisoning during the Iraq outbreak. J Pharmacol Exp Ther. 1981;218:74–83. [PubMed] [Google Scholar]
- Clarkson TW, Magos L, Myers GJ. The toxicology of mercury--current exposures and clinical manifestations. N Engl J Med. 2003;349:1731–1737. doi: 10.1056/NEJMra022471. [DOI] [PubMed] [Google Scholar]
- Compeau GC, Bartha R. Sulfate-reducing bacteria: principal methylators of mercury in anoxic estuarine sediment. Appl Environ Microbiol. 1985;50:498–502. doi: 10.1128/aem.50.2.498-502.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad ME, Umbreit JN, Moore EG, Rodning CR. Newly identified iron-binding protein in human duodenal mucosa. Blood. 1992;79:244–247. [PubMed] [Google Scholar]
- Cook JD, Skikne BS, Baynes RD. Iron deficiency: the global perspective. Adv Exp Med Biol. 1994;356:219–228. doi: 10.1007/978-1-4615-2554-7_24. [DOI] [PubMed] [Google Scholar]
- Costa LG, Aschner M, Vitalone A, Syversen T, Soldin OP. Developmental neuropathology of environmental agents. Annu Rev Pharmacol Toxicol. 2004;44:87–110. doi: 10.1146/annurev.pharmtox.44.101802.121424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotruvo JA, Jr, Stubbe J. Metallation and mismetallation of iron and manganese proteins in vitro and in vivo: the class I ribonucleotide reductases as a case study. Metallomics. 2012;4:1020–1036. doi: 10.1039/c2mt20142a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford S, Davis K, Saddler C, Joseph J, Catapane EJ, Carroll MA. The Ability of PAS, Acetylsalicylic Acid and Calcium Disodium EDTA to Protect Against the Toxic Effects of Manganese on Mitochondrial Respiration in Gill of Crassostrea virginica. In Vivo. 2011;33:7–14. [PMC free article] [PubMed] [Google Scholar]
- Crossgrove JS, Allen DD, Bukaveckas BL, Rhineheimer SS, Yokel RA. Manganese distribution across the blood-brain barrier. I. Evidence for carrier-mediated influx of managanese citrate as well as manganese and manganese transferrin. Neurotoxicology. 2003;24:3–13. doi: 10.1016/s0161-813x(02)00089-x. [DOI] [PubMed] [Google Scholar]
- Davis JM. Methylcyclopentadienyl manganese tricarbonyl: health risk uncertainties and research directions. Environ Health Perspect. 1998;106(Suppl 1):191–201. doi: 10.1289/ehp.98106s1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JM, Jarabek AM, Mage DT, Graham JA. The EPA health risk assessment of methylcyclopentadienyl manganese tricarbonyl (MMT) Risk Anal. 1998;18:57–70. doi: 10.1111/j.1539-6924.1998.tb00916.x. [DOI] [PubMed] [Google Scholar]
- Davis LE, Kornfeld M, Mooney HS, Fiedler KJ, Haaland KY, Orrison WW, Cernichiari E, Clarkson TW. Methylmercury poisoning: long-term clinical, radiological, toxicological, and pathological studies of an affected family. Ann Neurol. 1994;35:680–688. doi: 10.1002/ana.410350608. [DOI] [PubMed] [Google Scholar]
- De Domenico I, McVey Ward D, Kaplan J. Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat Rev Mol Cell Biol. 2008;9:72–81. doi: 10.1038/nrm2295. [DOI] [PubMed] [Google Scholar]
- de Freitas AS, Funck VR, Rotta Mdos S, Bohrer D, Morschbacher V, Puntel RL, Nogueira CW, Farina M, Aschner M, Rocha JB. Diphenyl diselenide, a simple organoselenium compound, decreases methylmercury-induced cerebral, hepatic and renal oxidative stress and mercury deposition in adult mice. Brain Res Bull. 2009;79:77–84. doi: 10.1016/j.brainresbull.2008.11.001. [DOI] [PubMed] [Google Scholar]
- De Palma G, Mariotti O, Lonati D, Goldoni M, Catalani S, Mutti A, Locatelli C, Apostoli P. Toxicokinetics and toxicodynamics of elemental mercury following self-administration. Clin Toxicol (Phila) 2008;46:869–876. doi: 10.1080/15563650802136241. [DOI] [PubMed] [Google Scholar]
- Debes F, Budtz-Jorgensen E, Weihe P, White RF, Grandjean P. Impact of prenatal methylmercury exposure on neurobehavioral function at age 14 years. Neurotoxicol Teratol. 2006;28:536–547. doi: 10.1016/j.ntt.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Dietrich MO, Mantese CE, Anjos Gd, Souza DO, Farina M. Motor impairment induced by oral exposure to methylmercury in adult mice. Environmental Toxicology and Pharmacology. 2005;19:169–175. doi: 10.1016/j.etap.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson AW, Erikson KM, Aschner M. Manganese neurotoxicity. Ann N Y Acad Sci. 2004;1012:115–128. doi: 10.1196/annals.1306.009. [DOI] [PubMed] [Google Scholar]
- Donaldson J, McGregor D, LaBella F. Manganese neurotoxicity: a model for free radical mediated neurodegeneration? Can J Physiol Pharmacol. 1982;60:1398–1405. doi: 10.1139/y82-208. [DOI] [PubMed] [Google Scholar]
- Dringen R, Pawlowski PG, Hirrlinger J. Peroxide detoxification by brain cells. J Neurosci Res. 2005;79:157–165. doi: 10.1002/jnr.20280. [DOI] [PubMed] [Google Scholar]
- El Safty A, El Mahgoub K, Helal S, Abdel Maksoud N. Zinc toxicity among galvanization workers in the iron and steel industry. Ann N Y Acad Sci. 2008;1140:256–262. doi: 10.1196/annals.1454.007. [DOI] [PubMed] [Google Scholar]
- Emsley J. Nature’s Building Blocks—An A–Z Guide to the Elements. New York: Oxford University Press; 2001. [Google Scholar]
- Erikson K, Aschner M. Manganese causes differential regulation of glutamate transporter (GLAST) taurine transporter and metallothionein in cultured rat astrocytes. Neurotoxicology. 2002;23:595–602. doi: 10.1016/s0161-813x(02)00012-8. [DOI] [PubMed] [Google Scholar]
- Erikson KM, Aschner M. Manganese neurotoxicity and glutamate-GABA interaction. Neurochem Int. 2003;43:475–480. doi: 10.1016/s0197-0186(03)00037-8. [DOI] [PubMed] [Google Scholar]
- Erikson KM, Dorman DC, Lash LH, Aschner M. Manganese inhalation by rhesus monkeys is associated with brain regional changes in biomarkers of neurotoxicity. Toxicol Sci. 2007;97:459–466. doi: 10.1093/toxsci/kfm044. [DOI] [PubMed] [Google Scholar]
- Eto K, Marumoto M, Takeya M. The pathology of methylmercury poisoning (Minamata disease) Neuropathology. 2010 doi: 10.1111/j.1440-1789.2010.01119.x. [DOI] [PubMed] [Google Scholar]
- Eyer F, Felgenhauer N, Pfab R, Drasch G, Zilker T. Neither DMPS nor DMSA is effective in quantitative elimination of elemental mercury after intentional IV injection. Clin Toxicol (Phila) 2006;44:395–397. doi: 10.1080/15563650600671795. [DOI] [PubMed] [Google Scholar]
- Falluel-Morel A, Lin L, Sokolowski K, McCandlish E, Buckley B, DiCicco-Bloom E. N-acetyl cysteine treatment reduces mercury-induced neurotoxicity in the developing rat hippocampus. J Neurosci Res. 2012;90:743–750. doi: 10.1002/jnr.22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina M, Aschner M, Rocha JB. Oxidative stress in MeHg-induced neurotoxicity. Toxicol Appl Pharmacol. 2011a;256:405–417. doi: 10.1016/j.taap.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina M, Aschner M, Rocha JB. Redox State in Mediating Methylmercury Neurotoxicity. In: Aschner MA, Ceccatelli S, editors. Methylmercury and Neurotoxicity. Vol. 2. Springer; 2012. pp. 101–125. [Google Scholar]
- Farina M, Brandao R, de Lara FS, Pagliosa LB, Soares FA, Souza DO, Rocha JB. Profile of nonprotein thiols, lipid peroxidation and delta-aminolevulinate dehydratase activity in mouse kidney and liver in response to acute exposure to mercuric chloride and sodium selenite. Toxicology. 2003b;184:179–187. doi: 10.1016/s0300-483x(02)00576-0. [DOI] [PubMed] [Google Scholar]
- Farina M, Campos F, Vendrell I, Berenguer J, Barzi M, Pons S, Sunol C. Probucol increases glutathione peroxidase-1 activity and displays long-lasting protection against methylmercury toxicity in cerebellar granule cells. Toxicol Sci. 2009;112:416–426. doi: 10.1093/toxsci/kfp219. [DOI] [PubMed] [Google Scholar]
- Farina M, Franco JL, Ribas CM, Meotti FC, Missau FC, Pizzolatti MG, Dafre AL, Santos AR. Protective effects of Polygala paniculata extract against methylmercury-induced neurotoxicity in mice. J Pharm Pharmacol. 2005;57:1503–1508. doi: 10.1211/jpp.57.11.0017. [DOI] [PubMed] [Google Scholar]
- Farina M, Frizzo ME, Soares FA, Schwalm FD, Dietrich MO, Zeni G, Rocha JB, Souza DO. Ebselen protects against methylmercury-induced inhibition of glutamate uptake by cortical slices from adult mice. Toxicol Lett. 2003a;144:351–357. doi: 10.1016/s0378-4274(03)00242-x. [DOI] [PubMed] [Google Scholar]
- Farina M, Rocha JB, Aschner M. Mechanisms of methylmercury-induced neurotoxicity: evidence from experimental studies. Life Sci. 2011b;89:555–563. doi: 10.1016/j.lfs.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar ER, Emerson JP, Koehntop KD, Reynolds MF, Trmcic M, Que L., Jr In vivo self-hydroxylation of an iron-substituted manganese-dependent extradiol cleaving catechol dioxygenase. J Biol Inorg Chem. 2011;16:589–597. doi: 10.1007/s00775-011-0760-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fibach E, Rachmilewitz EA. The role of antioxidants and iron chelators in the treatment of oxidative stress in thalassemia. Ann N Y Acad Sci. 2010;1202:10–16. doi: 10.1111/j.1749-6632.2010.05577.x. [DOI] [PubMed] [Google Scholar]
- Finberg KE. Unraveling mechanisms regulating systemic iron homeostasis. Hematology Am Soc Hematol Educ Program. 2011;2011:532–537. doi: 10.1182/asheducation-2011.1.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley JW, Davis CD. Manganese deficiency and toxicity: are high or low dietary amounts of manganese cause for concern? Biofactors. 1999;10:15–24. doi: 10.1002/biof.5520100102. [DOI] [PubMed] [Google Scholar]
- Fitsanakis VA, Zhang N, Anderson JG, Erikson KM, Avison MJ, Gore JC, Aschner M. Measuring brain manganese and iron accumulation in rats following 14 weeks of low-dose manganese treatment using atomic absorption spectroscopy and magnetic resonance imaging. Toxicol Sci. 2008;103:116–124. doi: 10.1093/toxsci/kfn019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A. 1998;95:1148–1153. doi: 10.1073/pnas.95.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366:348–359. doi: 10.1056/NEJMra1004967. [DOI] [PubMed] [Google Scholar]
- Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984;42:1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- Fox DA, Grandjean P, de Groot D, Paule MG. Developmental origins of adult diseases and neurotoxicity: Epidemiological and experimental studies. Neurotoxicology. 2012;33:810–816. doi: 10.1016/j.neuro.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga CG. Relevance, essentiality and toxicity of trace elements in human health. Mol Aspects Med. 2005;26:235–244. doi: 10.1016/j.mam.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Franco JL, Braga HC, Stringari J, Missau FC, Posser T, Mendes BG, Leal RB, Santos AR, Dafre AL, Pizzolatti MG, Farina M. Mercurial-induced hydrogen peroxide generation in mouse brain mitochondria: protective effects of quercetin. Chem Res Toxicol. 2007;20:1919–1926. doi: 10.1021/tx7002323. [DOI] [PubMed] [Google Scholar]
- Franco JL, Posser T, Dunkley PR, Dickson PW, Mattos JJ, Martins R, Bainy AC, Marques MR, Dafre AL, Farina M. Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radic Biol Med. 2009;47:449–457. doi: 10.1016/j.freeradbiomed.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Franco JL, Posser T, Missau F, Pizzolatti MG, Santos ARS, Souza DO, Aschner M, Rocha JBT, Dafre AL, Farina M. Structure–activity relationship of flavonoids derived from medicinal plants in preventing methylmercury-induced mitochondrial dysfunction. Environmental Toxicology and Pharmacology. 2010;30:272–278. doi: 10.1016/j.etap.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco JL, Teixeira A, Meotti FC, Ribas CM, Stringari J, Garcia Pomblum SC, Moro AM, Bohrer D, Bairros AV, Dafre AL, Santos AR, Farina M. Cerebellar thiol status and motor deficit after lactational exposure to methylmercury. Environ Res. 2006;102:22–28. doi: 10.1016/j.envres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Freeland-Graves JH, Lin PH. Plasma uptake of manganese as affected by oral loads of manganese, calcium, milk, phosphorus, copper, and zinc. J Am Coll Nutr. 1991;10:38–43. doi: 10.1080/07315724.1991.10718124. [DOI] [PubMed] [Google Scholar]
- Freitas AJ, Rocha JB, Wolosker H, Souza DO. Effects of Hg2+ and CH3Hg+ on Ca2+ fluxes in rat brain microsomes. Brain Res. 1996;738:257–264. doi: 10.1016/s0006-8993(96)00781-0. [DOI] [PubMed] [Google Scholar]
- Galaris D, Pantopoulos K. Oxidative stress and iron homeostasis: mechanistic and health aspects. Crit Rev Clin Lab Sci. 2008;45:1–23. doi: 10.1080/10408360701713104. [DOI] [PubMed] [Google Scholar]
- Garcia SJ, Gellein K, Syversen T, Aschner M. Iron deficient and manganese supplemented diets alter metals and transporters in the developing rat brain. Toxicol Sci. 2007;95:205–214. doi: 10.1093/toxsci/kfl139. [DOI] [PubMed] [Google Scholar]
- Gassen M, Youdim MB. The potential role of iron chelators in the treatment of Parkinson’s disease and related neurological disorders. Pharmacol Toxicol. 1997;80:159–166. doi: 10.1111/j.1600-0773.1997.tb00390.x. [DOI] [PubMed] [Google Scholar]
- Gavin CE, Gunter KK, Gunter TE. Manganese and calcium efflux kinetics in brain mitochondria. Relevance to manganese toxicity. Biochem J. 1990;266:329–334. doi: 10.1042/bj2660329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin CE, Gunter KK, Gunter TE. Mn2+ sequestration by mitochondria and inhibition of oxidative phosphorylation. Toxicol Appl Pharmacol. 1992;115:1–5. doi: 10.1016/0041-008x(92)90360-5. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, Lindquist S. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser V, Leipnitz G, Straliotto MR, Oliveira J, dos Santos VV, Wannmacher CM, de Bem AF, Rocha JB, Farina M, Latini A. Oxidative stress-mediated inhibition of brain creatine kinase activity by methylmercury. Neurotoxicology. 2010;31:454–460. doi: 10.1016/j.neuro.2010.05.012. [DOI] [PubMed] [Google Scholar]
- Goldwater LJ. Mercury; a history of quicksilver. York Press; 1972. [Google Scholar]
- Goodnough LT. Iron deficiency syndromes and iron-restricted erythropoiesis (CME) Transfusion. 2012;52:1584–1592. doi: 10.1111/j.1537-2995.2011.03495.x. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposures to metals as risk factors for Parkinson’s disease. Neurology. 1997;48:650–658. doi: 10.1212/wnl.48.3.650. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology. 1999;20:239–247. [PubMed] [Google Scholar]
- Goyer RA. Nutrition and metal toxicity. Am J Clin Nutr. 1995;61:646S–650S. doi: 10.1093/ajcn/61.3.646S. [DOI] [PubMed] [Google Scholar]
- Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol. 1978;14:633–643. [PubMed] [Google Scholar]
- Grandjean P, Landrigan PJ. Developmental neurotoxicity of industrial chemicals. Lancet. 2006;368:2167–2178. doi: 10.1016/S0140-6736(06)69665-7. [DOI] [PubMed] [Google Scholar]
- Grandjean P, Weihe P, White RF, Debes F, Araki S, Yokoyama K, Murata K, Sorensen N, Dahl R, Jorgensen PJ. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol Teratol. 1997a;19:417–428. doi: 10.1016/s0892-0362(97)00097-4. [DOI] [PubMed] [Google Scholar]
- Grandjean P, Weihe P, White RF, Debes F, Araki S, Yokoyama K, Murata K, SØRensen N, Dahl R, JØRgensen PJ. Cognitive Deficit in 7-Year-Old Children with Prenatal Exposure to Methylmercury. Neurotoxicol Teratol. 1997b;19:417–428. doi: 10.1016/s0892-0362(97)00097-4. [DOI] [PubMed] [Google Scholar]
- Gregory A, Hayflick SJ. Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep. 2011;11:254–261. doi: 10.1007/s11910-011-0181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009;46:73–80. doi: 10.1136/jmg.2008.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilarte TR. Manganese and Parkinson’s disease: a critical review and new findings. Environ Health Perspect. 2010;118:1071–1080. doi: 10.1289/ehp.0901748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Gutteridge JM. Reactivity of hydroxyl and hydroxyl-like radicals discriminated by release of thiobarbituric acid-reactive material from deoxy sugars, nucleosides and benzoate. Biochem J. 1984;224:761–767. doi: 10.1042/bj2240761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber F, Weiss J. Über die Katalyse des Hydroperoxydes (On the catalysis of hydroperoxide) Naturwissenschaften. 1932;20:948–950. [Google Scholar]
- Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxygen radicals: a commonsense look at their nature and medical importance. Med Biol. 1984;62:71–77. [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. Role of iron in oxygen radical reactions. Methods Enzymol. 1984;105:47–56. doi: 10.1016/s0076-6879(84)05007-2. [DOI] [PubMed] [Google Scholar]
- HaMai D, Campbell A, Bondy SC. Modulation of oxidative events by multivalent manganese complexes in brain tissue. Free Radic Biol Med. 2001;31:763–768. doi: 10.1016/s0891-5849(01)00639-6. [DOI] [PubMed] [Google Scholar]
- Harada M. Congenital Minamata disease: intrauterine methylmercury poisoning. Teratology. 1978;18:285–288. doi: 10.1002/tera.1420180216. [DOI] [PubMed] [Google Scholar]
- Hardy G. Manganese in parenteral nutrition: who, when, and why should we supplement? Gastroenterology. 2009;137:S29–35. doi: 10.1053/j.gastro.2009.08.011. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Butterworth RF. Hepatic encephalopathy: An update of pathophysiologic mechanisms. Proc Soc Exp Biol Med. 1999;222:99–112. doi: 10.1046/j.1525-1373.1999.d01-120.x. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Norenberg MD. Ammonia and manganese increase arginine uptake in cultured astrocytes. Neurochem Res. 1998;23:869–873. doi: 10.1023/a:1022411012512. [DOI] [PubMed] [Google Scholar]
- He L, Girijashanker K, Dalton TP, Reed J, Li H, Soleimani M, Nebert DW. ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: characterization of transporter properties. Mol Pharmacol. 2006;70:171–180. doi: 10.1124/mol.106.024521. [DOI] [PubMed] [Google Scholar]
- Heli H, Mirtorabi S, Karimian K. Advances in iron chelation: an update. Expert Opin Ther Pat. 2011;21:819–856. doi: 10.1517/13543776.2011.569493. [DOI] [PubMed] [Google Scholar]
- Hintelmann H. Organomercurials. Their formation and pathways in the environment. Met Ions Life Sci. 2010;7:365–401. doi: 10.1039/BK9781847551771-00365. [DOI] [PubMed] [Google Scholar]
- Horowitz MP, Greenamyre JT. Mitochondrial iron metabolism and its role in neurodegeneration. J Alzheimers Dis. 2010;20(Suppl 2):S551–568. doi: 10.3233/JAD-2010-100354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houeto P, Sandouk P, Baud FJ, Levillain P. Elemental mercury vapour toxicity: treatment and levels in plasma and urine. Hum Exp Toxicol. 1994;13:848–852. doi: 10.1177/096032719401301205. [DOI] [PubMed] [Google Scholar]
- Howland MA. Risks of parenteral deferoxamine for acute iron poisoning. J Toxicol Clin Toxicol. 1996;34:491–497. doi: 10.3109/15563659609028006. [DOI] [PubMed] [Google Scholar]
- Huang CC, Chu NS, Lu CS, Wang JD, Tsai JL, Tzeng JL, Wolters EC, Calne DB. Chronic manganese intoxication. Arch Neurol. 1989;46:1104–1106. doi: 10.1001/archneur.1989.00520460090018. [DOI] [PubMed] [Google Scholar]
- Huang CC, Weng YH, Lu CS, Chu NS, Yen TC. Dopamine transporter binding in chronic manganese intoxication. J Neurol. 2003;250:1335–1339. doi: 10.1007/s00415-003-0214-1. [DOI] [PubMed] [Google Scholar]
- Hursh JB, Cherian MG, Clarkson TW, Vostal JJ, Mallie RV. Clearance of mercury (HG-197, HG-203) vapor inhaled by human subjects. Arch Environ Health. 1976;31:302–309. doi: 10.1080/00039896.1976.10667240. [DOI] [PubMed] [Google Scholar]
- Inoue T, Majid T, Pautler RG. Manganese enhanced MRI (MEMRI): neurophysiological applications. Rev Neurosci. 2011;22:675–694. doi: 10.1515/RNS.2011.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IPCS. INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY- methylmercury. 1990. [Google Scholar]
- Jaiser SR, Winston GP. Copper deficiency myelopathy. J Neurol. 2010;257:869–881. doi: 10.1007/s00415-010-5511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang DH, Hoffman RS. Heavy metal chelation in neurotoxic exposures. Neurol Clin. 2011;29:607–622. doi: 10.1016/j.ncl.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. The role of iron in neurodegeneration: prospects for pharmacotherapy of Parkinson’s disease. Drugs Aging. 1999;14:115–140. doi: 10.2165/00002512-199914020-00004. [DOI] [PubMed] [Google Scholar]
- Johnstone D, Milward EA. Genome-wide microarray analysis of brain gene expression in mice on a short-term high iron diet. Neurochem Int. 2010a;56:856–863. doi: 10.1016/j.neuint.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Johnstone D, Milward EA. Molecular genetic approaches to understanding the roles and regulation of iron in brain health and disease. J Neurochem. 2010b;113:1387–1402. doi: 10.1111/j.1471-4159.2010.06697.x. [DOI] [PubMed] [Google Scholar]
- Jomova K, Valko M. Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr Pharm Des. 2011;17:3460–3473. doi: 10.2174/138161211798072463. [DOI] [PubMed] [Google Scholar]
- Kaiser J. Manganese: a high-octane dispute. Science. 2003;300:926–928. doi: 10.1126/science.300.5621.926. [DOI] [PubMed] [Google Scholar]
- Kajiwara Y, Yasutake A, Adachi T, Hirayama K. Methylmercury transport across the placenta via neutral amino acid carrier. Arch Toxicol. 1996;70:310–314. doi: 10.1007/s002040050279. [DOI] [PubMed] [Google Scholar]
- Kannurpatti SS, Joshi PG, Joshi NB. Calcium sequestering ability of mitochondria modulates influx of calcium through glutamate receptor channel. Neurochem Res. 2000;25:1527–1536. doi: 10.1023/a:1026602100160. [DOI] [PubMed] [Google Scholar]
- Kaur P, Aschner M, Syversen T. Glutathione modulation influences methyl mercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. Neurotoxicology. 2006;27:492–500. doi: 10.1016/j.neuro.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Kaur P, Aschner M, Syversen T. Biochemical factors modulating cellular neurotoxicity of methylmercury. J Toxicol. 2011;2011:721987. doi: 10.1155/2011/721987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P, Evje L, Aschner M, Syversen T. The in vitro effects of selenomethionine on methylmercury-induced neurotoxicity. Toxicol In Vitro. 2009;23:378–385. doi: 10.1016/j.tiv.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Kell DB. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch Toxicol. 2010;84:825–889. doi: 10.1007/s00204-010-0577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerper LE, Ballatori N, Clarkson TW. Methylmercury transport across the blood-brain barrier by an amino acid carrier. Am J Physiol. 1992;262:R761–765. doi: 10.1152/ajpregu.1992.262.5.R761. [DOI] [PubMed] [Google Scholar]
- Kershaw TG, Clarkson TW, Dhahir PH. The relationship between blood levels and dose of methylmercury in man. Arch Environ Health. 1980;35:28–36. doi: 10.1080/00039896.1980.10667458. [DOI] [PubMed] [Google Scholar]
- Kim SO, Merchant K, Nudelman R, Beyer WF, Jr, Keng T, DeAngelo J, Hausladen A, Stamler JS. OxyR: a molecular code for redox-related signaling. Cell. 2002;109:383–396. doi: 10.1016/s0092-8674(02)00723-7. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Chai YG, Ryu JC. Selenoprotein W as molecular target of methylmercury in human neuronal cells is down-regulated by GSH depletion. Biochemical and Biophysical Research Communications. 2005;330:1095–1102. doi: 10.1016/j.bbrc.2005.03.080. [DOI] [PubMed] [Google Scholar]
- Kinnally KW, Peixoto PM, Ryu SY, Dejean LM. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta. 2011;1813:616–622. doi: 10.1016/j.bbamcr.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobal AB, Horvat M, Prezelj M, Briski AS, Krsnik M, Dizdarevic T, Mazej D, Falnoga I, Stibilj V, Arneric N, Kobal D, Osredkar J. The impact of long-term past exposure to elemental mercury on antioxidative capacity and lipid peroxidation in mercury miners. J Trace Elem Med Biol. 2004;17:261–274. doi: 10.1016/S0946-672X(04)80028-2. [DOI] [PubMed] [Google Scholar]
- Kodama H, Fujisawa C, Bhadhprasit W. Inherited copper transport disorders: biochemical mechanisms, diagnosis, and treatment. Curr Drug Metab. 2012;13:237–250. doi: 10.2174/138920012799320455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh AS, Simmons-Willis TA, Pritchard JB, Grassl SM, Ballatori N. Identification of a mechanism by which the methylmercury antidotes N-acetylcysteine and dimercaptopropanesulfonate enhance urinary metal excretion: transport by the renal organic anion transporter-1. Mol Pharmacol. 2002;62:921–926. doi: 10.1124/mol.62.4.921. [DOI] [PubMed] [Google Scholar]
- Krachler M, Prohaska T, Koellensperger G, Rossipal E, Stingeder G. Concentrations of selected trace elements in human milk and in infant formulas determined by magnetic sector field inductively coupled plasma-mass spectrometry. Biol Trace Elem Res. 2000;76:97–112. doi: 10.1385/BTER:76:2:97. [DOI] [PubMed] [Google Scholar]
- Kumar H, Lim HW, More SV, Kim BW, Koppula S, Kim IS, Choi DK. The role of free radicals in the aging brain and Parkinson’s disease: convergence and parallelism. Int J Mol Sci. 2012;13:10478–10504. doi: 10.3390/ijms130810478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung MP, Kostyniak P, Olson J, Malone M, Roth JA. Studies of the in vitro effect of methylmercury chloride on rat brain neurotransmitter enzymes. J Appl Toxicol. 1987;7:119–121. doi: 10.1002/jat.2550070208. [DOI] [PubMed] [Google Scholar]
- Kwakye GF, Li D, Kabobel OA, Bowman AB. Cellular fura-2 manganese extraction assay (CFMEA) Curr Protoc Toxicol. 2011;Chapter 12(Unit12):18. doi: 10.1002/0471140856.tx1218s48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JB. Role of epigenetics in Alzheimer’s and Parkinson’s disease. Epigenomics. 2010;2:671–682. doi: 10.2217/epi.10.43. [DOI] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Lapina VA, Sheshko PM, Pankovets EA, Dontsov AE. Phytosorbent prepared from sunflower seed husks prevents mercuric chloride accumulation in kidney and muscle of adult rabbits. Arch Environ Health. 2000;55:48–50. doi: 10.1080/00039890009603385. [DOI] [PubMed] [Google Scholar]
- Lara FA, Kahn SA, da Fonseca AC, Bahia CP, Pinho JP, Graca-Souza AV, Houzel JC, de Oliveira PL, Moura-Neto V, Oliveira MF. On the fate of extracellular hemoglobin and heme in brain. J Cereb Blood Flow Metab. 2009;29:1109–1120. doi: 10.1038/jcbfm.2009.34. [DOI] [PubMed] [Google Scholar]
- Le Blanc S, Garrick MD, Arredondo M. Heme carrier protein 1 transports heme and is involved in heme-Fe metabolism. Am J Physiol Cell Physiol. 2012;302:C1780–1785. doi: 10.1152/ajpcell.00080.2012. [DOI] [PubMed] [Google Scholar]
- Lebda MA, El-Neweshy MS, El-Sayed YS. Neurohepatic toxicity of subacute manganese chloride exposure and potential chemoprotective effects of lycopene. Neurotoxicology. 2012;33:98–104. doi: 10.1016/j.neuro.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Lee E, Sidoryk-Wegrzynowicz M, Farina M, Rocha JB, Aschner M. Estrogen Attenuates Manganese-Induced Glutamate Transporter Impairment in Rat Primary Astrocytes. Neurotox Res. 2012 doi: 10.1007/s12640-012-9347-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi S, Rovida E. The role of iron in mitochondrial function. Biochim Biophys Acta. 2009;1790:629–636. doi: 10.1016/j.bbagen.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Liang Z, Valla J, Sefidvash-Hockley S, Rogers J, Li R. Effects of estrogen treatment on glutamate uptake in cultured human astrocytes derived from cortex of Alzheimer’s disease patients. J Neurochem. 2002;80:807–814. doi: 10.1046/j.0022-3042.2002.00779.x. [DOI] [PubMed] [Google Scholar]
- Lin JL, Lim PS. Massive oral ingestion of elemental mercury. J Toxicol Clin Toxicol. 1993;31:487–492. doi: 10.3109/15563659309000417. [DOI] [PubMed] [Google Scholar]
- Ljung K, Vahter M. Time to re-evaluate the guideline value for manganese in drinking water? Environ Health Perspect. 2007;115:1533–1538. doi: 10.1289/ehp.10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockman PR, Roder KE, Allen DD. Inhibition of the rat blood-brain barrier choline transporter by manganese chloride. J Neurochem. 2001;79:588–594. doi: 10.1046/j.1471-4159.2001.00589.x. [DOI] [PubMed] [Google Scholar]
- Lu J, Holmgren A. Selenoproteins. J Biol Chem. 2009;284:723–727. doi: 10.1074/jbc.R800045200. [DOI] [PubMed] [Google Scholar]
- Lubick N. Mercury alters immune system response in artisanal gold miners. Environ Health Perspect. 2010;118:A243. doi: 10.1289/ehp.118-a243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucaciu CM, Dragu C, Copaescu L, Morariu VV. Manganese transport through human erythrocyte membranes. An EPR study. Biochim Biophys Acta. 1997;1328:90–98. doi: 10.1016/s0005-2736(97)00039-4. [DOI] [PubMed] [Google Scholar]
- Lucchini RG, Martin CJ, Doney BC. From manganism to manganese-induced parkinsonism: a conceptual model based on the evolution of exposure. Neuromolecular Med. 2009;11:311–321. doi: 10.1007/s12017-009-8108-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucena GM, Franco JL, Ribas CM, Azevedo MS, Meotti FC, Gadotti VM, Dafre AL, Santos AR, Farina M. Cipura paludosa extract prevents methyl mercury-induced neurotoxicity in mice. Basic Clin Pharmacol Toxicol. 2007;101:127–131. doi: 10.1111/j.1742-7843.2007.00091.x. [DOI] [PubMed] [Google Scholar]
- Mackenzie B, Garrick MD. Iron Imports. II. Iron uptake at the apical membrane in the intestine. Am J Physiol Gastrointest Liver Physiol. 2005;289:G981–G986. doi: 10.1152/ajpgi.00363.2005. [DOI] [PubMed] [Google Scholar]
- Magdalan J, Zawadzki M, Sozanski T. Fulminant hepatic failure in woman with iron and non-steroidal anti-inflammatory drug intoxication. Hum Exp Toxicol. 2011;30:1106–1111. doi: 10.1177/0960327110386392. [DOI] [PubMed] [Google Scholar]
- Magos L. Mercury--blood interaction and mercury uptake by the brain after vapor exposure. Environ Res. 1967;1:323–337. doi: 10.1016/0013-9351(67)90023-0. [DOI] [PubMed] [Google Scholar]
- Mandel S, Youdim MB. Catechin polyphenols: neurodegeneration and neuroprotection in neurodegenerative diseases. Free Radic Biol Med. 2004;37:304–317. doi: 10.1016/j.freeradbiomed.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Manfroi CB, Schwalm FD, Cereser V, Abreu F, Oliveira A, Bizarro L, Rocha JB, Frizzo ME, Souza DO, Farina M. Maternal milk as methylmercury source for suckling mice: neurotoxic effects involved with the cerebellar glutamatergic system. Toxicol Sci. 2004;81:172–178. doi: 10.1093/toxsci/kfh201. [DOI] [PubMed] [Google Scholar]
- Marras C, Goldman SM. Genetics meets environment: evaluating gene-environment interactions in neurologic diseases. Semin Neurol. 2011;31:553–561. doi: 10.1055/s-0031-1299793. [DOI] [PubMed] [Google Scholar]
- Martinez-Finley EJ, Chakraborty S, Fretham SJ, Aschner M. Cellular transport and homeostasis of essential and nonessential metals. Metallomics. 2012;4:593–605. doi: 10.1039/c2mt00185c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall SA, Reichel CM, Farley CM, Flesher MM, Der-Ghazarian T, Cortez AM, Wacan JJ, Martinez CE, Varela FA, Butt AE, Crawford CA. Postnatal manganese exposure alters dopamine transporter function in adult rats: Potential impact on nonassociative and associative processes. Neuroscience. 2008;154:848–860. doi: 10.1016/j.neuroscience.2008.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755–1759. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- McNeill A, Birchall D, Hayflick SJ, Gregory A, Schenk JF, Zimmerman EA, Shang H, Miyajima H, Chinnery PF. T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology. 2008;70:1614–1619. doi: 10.1212/01.wnl.0000310985.40011.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena I, Marin O, Fuenzalida S, Cotzias GC. Chronic manganese poisoning. Clinical picture and manganese turnover. Neurology. 1967;17:128–136. doi: 10.1212/wnl.17.2.128. [DOI] [PubMed] [Google Scholar]
- Mergler D, Huel G, Bowler R, Iregren A, Belanger S, Baldwin M, Tardif R, Smargiassi A, Martin L. Nervous system dysfunction among workers with long-term exposure to manganese. Environ Res. 1994;64:151–180. doi: 10.1006/enrs.1994.1013. [DOI] [PubMed] [Google Scholar]
- Mesquita SD, Ferreira AC, Sousa JC, Santos NC, Correia-Neves M, Sousa N, Palha JA, Marques F. Modulation of iron metabolism in aging and in Alzheimer’s disease: relevance of the choroid plexus. Front Cell Neurosci. 2012;6:25. doi: 10.3389/fncel.2012.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen JK. Absorption and elimination of dietary mercury (Hg2+) and methylmercury in man. In: Miller MW, Clarkson TW, editors. Mercury, Mercurials and Mercaptans. Springfield: 1973. [Google Scholar]
- Milatovic D, Gupta RC, Yu Y, Zaja-Milatovic S, Aschner M. Protective effects of antioxidants and anti-inflammatory agents against manganese-induced oxidative damage and neuronal injury. Toxicol Appl Pharmacol. 2011;256:219–226. doi: 10.1016/j.taap.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mims MP, Prchal JT. Divalent metal transporter 1. Hematology. 2005;10:339–345. doi: 10.1080/10245330500093419. [DOI] [PubMed] [Google Scholar]
- Minotti G, Aust SD. The role of iron in oxygen radical mediated lipid peroxidation. Chem Biol Interact. 1989;71:1–19. doi: 10.1016/0009-2797(89)90087-2. [DOI] [PubMed] [Google Scholar]
- Minotti G, Aust SD. Redox cycling of iron and lipid peroxidation. Lipids. 1992;27:219–226. doi: 10.1007/BF02536182. [DOI] [PubMed] [Google Scholar]
- Miyajima H, Takahashi Y, Kamata T, Shimizu H, Sakai N, Gitlin JD. Use of desferrioxamine in the treatment of aceruloplasminemia. Ann Neurol. 1997;41:404–407. doi: 10.1002/ana.410410318. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Hider RC, Gaeta A, Williams R, Francis P. Metals ions and neurodegeneration. Biometals. 2007;20:639–654. doi: 10.1007/s10534-006-9033-z. [DOI] [PubMed] [Google Scholar]
- Monnet-Tschudi F, Zurich MG, Boschat C, Corbaz A, Honegger P. Involvement of environmental mercury and lead in the etiology of neurodegenerative diseases. Rev Environ Health. 2006;21:105–117. doi: 10.1515/reveh.2006.21.2.105. [DOI] [PubMed] [Google Scholar]
- Moreno JA, Streifel KM, Sullivan KA, Hanneman WH, Tjalkens RB. Manganese-induced NF-kappaB activation and nitrosative stress is decreased by estrogen in juvenile mice. Toxicol Sci. 2011;122:121–133. doi: 10.1093/toxsci/kfr091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretto MB, Funchal C, Santos AQ, Gottfried C, Boff B, Zeni G, Pureur RP, Souza DO, Wofchuk S, Rocha JB. Ebselen protects glutamate uptake inhibition caused by methyl mercury but does not by Hg2+ Toxicology. 2005;214:57–66. doi: 10.1016/j.tox.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Mori N, Yasutake A, Hirayama K. Comparative study of activities in reactive oxygen species production/defense system in mitochondria of rat brain and liver, and their susceptibility to methylmercury toxicity. Arch Toxicol. 2007;81:769–776. doi: 10.1007/s00204-007-0209-2. [DOI] [PubMed] [Google Scholar]
- Murata K, Weihe P, Budtz-Jorgensen E, Jorgensen PJ, Grandjean P. Delayed brainstem auditory evoked potential latencies in 14-year-old children exposed to methylmercury. J Pediatr. 2004;144:177–183. doi: 10.1016/j.jpeds.2003.10.059. [DOI] [PubMed] [Google Scholar]
- Murphy VA, Wadhwani KC, Smith QR, Rapoport SI. Saturable transport of manganese(II) across the rat blood-brain barrier. J Neurochem. 1991;57:948–954. doi: 10.1111/j.1471-4159.1991.tb08242.x. [DOI] [PubMed] [Google Scholar]
- Nandar W, Connor JR. HFE gene variants affect iron in the brain. J Nutr. 2011;141:729S–739S. doi: 10.3945/jn.110.130351. [DOI] [PubMed] [Google Scholar]
- Naranuntarat A, Jensen LT, Pazicni S, Penner-Hahn JE, Culotta VC. The interaction of mitochondrial iron with manganese superoxide dismutase. J Biol Chem. 2009;284:22633–22640. doi: 10.1074/jbc.M109.026773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAS. Panel on Micronutrients. 2001 Available at www.nap.edu/books/0309072794/html/
- Neghab M, Norouzi MA, Choobineh A, Kardaniyan MR, Zadeh JH. Health effects associated with long-term occupational exposure of employees of a chlor-alkali plant to mercury. Int J Occup Saf Ergon. 2012;18:97–106. doi: 10.1080/10803548.2012.11076920. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323–342. doi: 10.1146/annurev.nutr.26.061505.111303. [DOI] [PubMed] [Google Scholar]
- Neville GA, Drakenberg T. Mercuric mercury and methylmercury complexes of glutathione. Acta Chem Scand B. 1974;28:473–477. doi: 10.3891/acta.chem.scand.28b-0473. [DOI] [PubMed] [Google Scholar]
- Newland MC. Animal models of manganese’s neurotoxicity. Neurotoxicology. 1999;20:415–432. [PubMed] [Google Scholar]
- Newland MC, Warfvinge K, Berlin M. Behavioral consequences of in utero exposure to mercury vapor: alterations in lever-press durations and learning in squirrel monkeys. Toxicol Appl Pharmacol. 1996;139:374–386. doi: 10.1006/taap.1996.0178. [DOI] [PubMed] [Google Scholar]
- Ni M, Li X, Yin Z, Sidoryk-Wegrzynowicz M, Jiang H, Farina M, Rocha JB, Syversen T, Aschner M. Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia. 2011;59:810–820. doi: 10.1002/glia.21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya T, Imamura K, Kuwahata M, Kindaichi M, Susa M, Ekino S. Reappraisal of somatosensory disorders in methylmercury poisoning. Neurotoxicol Teratol. 2005;27:643–653. doi: 10.1016/j.ntt.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Nunez MT. Regulatory mechanisms of intestinal iron absorption-uncovering of a fast-response mechanism based on DMT1 and ferroportin endocytosis. Biofactors. 2010;36:88–97. doi: 10.1002/biof.84. [DOI] [PubMed] [Google Scholar]
- Nunez MT, Urrutia P, Mena N, Aguirre P, Tapia V, Salazar J. Iron toxicity in neurodegeneration. Biometals. 2012;25:761–776. doi: 10.1007/s10534-012-9523-0. [DOI] [PubMed] [Google Scholar]
- Olanow CW. Manganese-induced parkinsonism and Parkinson’s disease. Ann N Y Acad Sci. 2004;1012:209–223. doi: 10.1196/annals.1306.018. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Good PF, Shinotoh H, Hewitt KA, Vingerhoets F, Snow BJ, Beal MF, Calne DB, Perl DP. Manganese intoxication in the rhesus monkey: a clinical, imaging, pathologic, and biochemical study. Neurology. 1996;46:492–498. doi: 10.1212/wnl.46.2.492. [DOI] [PubMed] [Google Scholar]
- Osawa M, Magos L. The chemical form of the methylmercury complex in the bile of the rat. Biochem Pharmacol. 1974;23:1903–1905. doi: 10.1016/0006-2952(74)90199-3. [DOI] [PubMed] [Google Scholar]
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- Pal PK, Samii A, Calne DB. Manganese neurotoxicity: a review of clinical features, imaging and pathology. Neurotoxicology. 1999;20:227–238. [PubMed] [Google Scholar]
- Patriarca A, Polticelli F, Piro MC, Sinibaldi F, Mei G, Bari M, Santucci R, Fiorucci L. Conversion of cytochrome c into a peroxidase: inhibitory mechanisms and implication for neurodegenerative diseases. Arch Biochem Biophys. 2012;522:62–69. doi: 10.1016/j.abb.2012.03.028. [DOI] [PubMed] [Google Scholar]
- Pentschew A, Ebner FF, Kovatch RM. Experimental Manganese Encephalopathy in Monkeys. A Preliminary Report. J Neuropathol Exp Neurol. 1963;22:488–499. doi: 10.1097/00005072-196307000-00010. [DOI] [PubMed] [Google Scholar]
- Perron NR, Brumaghim JL. A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem Biophys. 2009;53:75–100. doi: 10.1007/s12013-009-9043-x. [DOI] [PubMed] [Google Scholar]
- Phipps DA. The Biological Role of Metals. CAMBRIDGE UNIVERSITY PRESS; 2002. [Google Scholar]
- Powell JJ, Jugdaohsingh R, Thompson RP. The regulation of mineral absorption in the gastrointestinal tract. Proc Nutr Soc. 1999;58:147–153. doi: 10.1079/pns19990020. [DOI] [PubMed] [Google Scholar]
- Prohaska R, Sibon OC, Rudnicki DD, Danek A, Hayflick SJ, Verhaag EM, Vonk JJ, Margolis RL, Walker RH. Brain, blood, and iron: perspectives on the roles of erythrocytes and iron in neurodegeneration. Neurobiol Dis. 2012;46:607–624. doi: 10.1016/j.nbd.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racette BA, McGee-Minnich L, Moerlein SM, Mink JW, Videen TO, Perlmutter JS. Welding-related parkinsonism: clinical features, treatment, and pathophysiology. Neurology. 2001;56:8–13. doi: 10.1212/wnl.56.1.8. [DOI] [PubMed] [Google Scholar]
- Ramanathan G, Atchison WD. Ca2+ entry pathways in mouse spinal motor neurons in culture following in vitro exposure to methylmercury. Neurotoxicology. 2011;32:742–750. doi: 10.1016/j.neuro.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao KV, Norenberg MD. Manganese induces the mitochondrial permeability transition in cultured astrocytes. J Biol Chem. 2004;279:32333–32338. doi: 10.1074/jbc.M402096200. [DOI] [PubMed] [Google Scholar]
- Raz E, Jensen JH, Ge Y, Babb JS, Miles L, Reaume J, Grossman RI, Inglese M. Brain iron quantification in mild traumatic brain injury: a magnetic field correlation study. AJNR Am J Neuroradiol. 2011;32:1851–1856. doi: 10.3174/ajnr.A2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaney SH, Smith DR. Manganese oxidation state mediates toxicity in PC12 cells. Toxicol Appl Pharmacol. 2005;205:271–281. doi: 10.1016/j.taap.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Reynolds JN, Racz WJ. Effects of methylmercury on the spontaneous and potassium-evoked release of endogenous amino acids from mouse cerebellar slices. Can J Physiol Pharmacol. 1987;65:791–798. doi: 10.1139/y87-127. [DOI] [PubMed] [Google Scholar]
- Reznichenko L, Amit T, Zheng H, Avramovich-Tirosh Y, Youdim MB, Weinreb O, Mandel S. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (−)-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer’s disease. J Neurochem. 2006;97:527–536. doi: 10.1111/j.1471-4159.2006.03770.x. [DOI] [PubMed] [Google Scholar]
- Riccio A, Mattei C, Kelsell RE, Medhurst AD, Calver AR, Randall AD, Davis JB, Benham CD, Pangalos MN. Cloning and functional expression of human short TRP7, a candidate protein for store-operated Ca2+ influx. J Biol Chem. 2002;277:12302–12309. doi: 10.1074/jbc.M112313200. [DOI] [PubMed] [Google Scholar]
- Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, Sheftel AD, Ponka P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A. 2010;107:10775–10782. doi: 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Murphy SD. Effect of glutathione depletion on tissue deposition of methylmercury in rats. Toxicol Appl Pharmacol. 1975;31:505–519. doi: 10.1016/0041-008x(75)90274-4. [DOI] [PubMed] [Google Scholar]
- Rittle J, Green MT. Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- Rodrigues IM, Hopkinson ND, Harris RI. Pulmonary embolism associated with self-administration of mercury. Hum Toxicol. 1986;5:287–289. doi: 10.1177/096032718600500415. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Chen YI, Doros G, Salat DH, Chen NK, Kwong KK, Bush A, Fox J, Hersch SM. Alterations in Brain Transition Metals in Huntington Disease: An Evolving and Intricate Story. Arch Neurol. 2012 doi: 10.1001/archneurol.2011.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JA. Are there common biochemical and molecular mechanisms controlling manganism and parkisonism. Neuromolecular Med. 2009;11:281–296. doi: 10.1007/s12017-009-8088-8. [DOI] [PubMed] [Google Scholar]
- Ruha AM, Curry SC, Gerkin RD, Caldwell KL, Osterloh JD, Wax PM. Urine mercury excretion following meso-dimercaptosuccinic acid challenge in fish eaters. Arch Pathol Lab Med. 2009;133:87–92. doi: 10.5858/133.1.87. [DOI] [PubMed] [Google Scholar]
- Ryan TP, Aust SD. The role of iron in oxygen-mediated toxicities. Crit Rev Toxicol. 1992;22:119–141. doi: 10.3109/10408449209146308. [DOI] [PubMed] [Google Scholar]
- Sakaue M, Mori N, Okazaki M, Kadowaki E, Kaneko T, Hemmi N, Sekiguchi H, Maki T, Ozawa A, Hara S, Arishima K, Yamamoto M. Vitamin K has the potential to protect neurons from methylmercury-induced cell death in vitro. J Neurosci Res. 2011;89:1052–1058. doi: 10.1002/jnr.22630. [DOI] [PubMed] [Google Scholar]
- Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nunez MT, Garrick MD, Raisman-Vozari R, Hirsch EC. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc Natl Acad Sci U S A. 2008;105:18578–18583. doi: 10.1073/pnas.0804373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martin CD, Garri C, Pizarro F, Walter T, Theil EC, Nunez MT. Caco-2 intestinal epithelial cells absorb soybean ferritin by mu2 (AP2)-dependent endocytosis. J Nutr. 2008;138:659–666. doi: 10.1093/jn/138.4.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santambrogio P, Biasiotto G, Sanvito F, Olivieri S, Arosio P, Levi S. Mitochondrial ferritin expression in adult mouse tissues. J Histochem Cytochem. 2007;55:1129–1137. doi: 10.1369/jhc.7A7273.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AP, Lucas RL, Andrade V, Mateus ML, Milatovic D, Aschner M, Batoreu MC. Protective effects of ebselen (Ebs) and para-aminosalicylic acid (PAS) against manganese (Mn)-induced neurotoxicity. Toxicol Appl Pharmacol. 2012;258:394–402. doi: 10.1016/j.taap.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Sasakura C, Suzuki KT. Biological interaction between transition metals (Ag, Cd and Hg), selenide/sulfide and selenoprotein P. J Inorg Biochem. 1998;71:159–162. doi: 10.1016/s0162-0134(98)10048-x. [DOI] [PubMed] [Google Scholar]
- Schipper HM. Neurodegeneration with brain iron accumulation - clinical syndromes and neuroimaging. Biochim Biophys Acta. 2012;1822:350–360. doi: 10.1016/j.bbadis.2011.06.016. [DOI] [PubMed] [Google Scholar]
- Schneider SA, Hardy J, Bhatia KP. Syndromes of neurodegeneration with brain iron accumulation (NBIA): an update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov Disord. 2012;27:42–53. doi: 10.1002/mds.23971. [DOI] [PubMed] [Google Scholar]
- Scholte HR. The biochemical basis of mitochondrial diseases. J Bioenerg Biomembr. 1988;20:161–191. doi: 10.1007/BF00768393. [DOI] [PubMed] [Google Scholar]
- Schumann K. Safety aspects of iron in food. Ann Nutr Metab. 2001;45:91–101. doi: 10.1159/000046713. [DOI] [PubMed] [Google Scholar]
- Sebastiani G, Pantopoulos K. Disorders associated with systemic or local iron overload: from pathophysiology to clinical practice. Metallomics. 2011;3:971–986. doi: 10.1039/c1mt00082a. [DOI] [PubMed] [Google Scholar]
- Segura-Aguilar J. Peroxidase activity of liver microsomal vitamin D 25-hydroxylase and cytochrome P450 1A2 catalyzes 25-hydroxylation of vitamin D3 and oxidation of dopamine to aminochrome. Biochem Mol Med. 1996;58:122–129. doi: 10.1006/bmme.1996.0039. [DOI] [PubMed] [Google Scholar]
- Segura-Aguilar J, Lind C. On the mechanism of the Mn3(+)-induced neurotoxicity of dopamine:prevention of quinone-derived oxygen toxicity by DT diaphorase and superoxide dismutase. Chem Biol Interact. 1989;72:309–324. doi: 10.1016/0009-2797(89)90006-9. [DOI] [PubMed] [Google Scholar]
- Selim M, Yeatts S, Goldstein JN, Gomes J, Greenberg S, Morgenstern LB, Schlaug G, Torbey M, Waldman B, Xi G, Palesch Y. Safety and tolerability of deferoxamine mesylate in patients with acute intracerebral hemorrhage. Stroke. 2011;42:3067–3074. doi: 10.1161/STROKEAHA.111.617589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanker G, Syversen T, Aschner JL, Aschner M. Modulatory effect of glutathione status and antioxidants on methylmercury-induced free radical formation in primary cultures of cerebral astrocytes. Brain Res Mol Brain Res. 2005;137:11–22. doi: 10.1016/j.molbrainres.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, Frazer DM, Anderson GJ, Vulpe CD, Simpson RJ, McKie AT. Identification of an intestinal heme transporter. Cell. 2005;122:789–801. doi: 10.1016/j.cell.2005.06.025. [DOI] [PubMed] [Google Scholar]
- Shichiri M, Takanezawa Y, Uchida K, Tamai H, Arai H. Protection of cerebellar granule cells by tocopherols and tocotrienols against methylmercury toxicity. Brain Res. 2007;1182:106–115. doi: 10.1016/j.brainres.2007.08.084. [DOI] [PubMed] [Google Scholar]
- Shikama K. Nature of the FeO2 bonding in myoglobin and hemoglobin: A new molecular paradigm. Prog Biophys Mol Biol. 2006;91:83–162. doi: 10.1016/j.pbiomolbio.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Sian-Hulsmann J, Mandel S, Youdim MB, Riederer P. The relevance of iron in the pathogenesis of Parkinson’s disease. J Neurochem. 2011;118:939–957. doi: 10.1111/j.1471-4159.2010.07132.x. [DOI] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz M, Lee E, Albrecht J, Aschner M. Manganese disrupts astrocyte glutamine transporter expression and function. J Neurochem. 2009;110:822–830. doi: 10.1111/j.1471-4159.2009.06172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz M, Lee E, Aschner M. Mechanism of Mn(II)-mediated dysregulation of glutamine-glutamate cycle: focus on glutamate turnover. J Neurochem. 2012;122:856–867. doi: 10.1111/j.1471-4159.2012.07835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz M, Lee ES, Ni M, Aschner M. Manganese-induced downregulation of astroglial glutamine transporter SNAT3 involves ubiquitin-mediated proteolytic system. Glia. 2010;58:1905–1912. doi: 10.1002/glia.21060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz M, Wegrzynowicz M, Lee E, Bowman AB, Aschner M. Role of astrocytes in brain function and disease. Toxicol Pathol. 2011;39:115–123. doi: 10.1177/0192623310385254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepler JK, Nishikawa RA, Diamantidis T, Okamoto R. Asymptomatic hypermanganesemia in long-term home parenteral nutrition patients. Nutr Clin Pract. 2003;18:370–373. doi: 10.1177/0115426503018005370. [DOI] [PubMed] [Google Scholar]
- Siew SS, Kauppinen T, Kyyronen P, Heikkila P, Pukkala E. Exposure to iron and welding fumes and the risk of lung cancer. Scand J Work Environ Health. 2008;34:444–450. doi: 10.5271/sjweh.1296. [DOI] [PubMed] [Google Scholar]
- Sikk K, Haldre S, Aquilonius SM, Taba P. Manganese-Induced Parkinsonism due to Ephedrone Abuse. Parkinsons Dis. 2011;2011:865319. doi: 10.4061/2011/865319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikk K, Taba P, Haldre S, Bergquist J, Nyholm D, Askmark H, Danfors T, Sorensen J, Thurfjell L, Raininko R, Eriksson R, Flink R, Farnstrand C, Aquilonius SM. Clinical, neuroimaging and neurophysiological features in addicts with manganese-ephedrone exposure. Acta Neurol Scand. 2010;121:237–243. doi: 10.1111/j.1600-0404.2009.01189.x. [DOI] [PubMed] [Google Scholar]
- Sipahi T, Karakurt C, Bakirtas A, Tavil B. Acute iron ingestion. Indian J Pediatr. 2002;69:947–949. doi: 10.1007/BF02726009. [DOI] [PubMed] [Google Scholar]
- Spadoni F, Stefani A, Morello M, Lavaroni F, Giacomini P, Sancesario G. Selective vulnerability of pallidal neurons in the early phases of manganese intoxication. Exp Brain Res. 2000;135:544–551. doi: 10.1007/s002210000554. [DOI] [PubMed] [Google Scholar]
- Streifel KM, Moreno JA, Hanneman WH, Legare ME, Tjalkens RB. Gene deletion of nos2 protects against manganese-induced neurological dysfunction in juvenile mice. Toxicol Sci. 2012;126:183–192. doi: 10.1093/toxsci/kfr335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringari J, Nunes AK, Franco JL, Bohrer D, Garcia SC, Dafre AL, Milatovic D, Souza DO, Rocha JB, Aschner M, Farina M. Prenatal methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicol Appl Pharmacol. 2008;227:147–154. doi: 10.1016/j.taap.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe J, Cotruvo JA., Jr Control of metallation and active cofactor assembly in the class Ia and Ib ribonucleotide reductases: diiron or dimanganese? Curr Opin Chem Biol. 2011;15:284–290. doi: 10.1016/j.cbpa.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su HY, Gorlin Y, Man IC, Calle-Vallejo F, Norskov JK, Jaramillo TF, Rossmeisl J. Identifying active surface phases for metal oxide electrocatalysts: a study of manganese oxide bi-functional catalysts for oxygen reduction and water oxidation catalysis. Phys Chem Chem Phys. 2012;14:14010–14022. doi: 10.1039/c2cp40841d. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Hojo Y, Tamai Y, Tanaka H. Selenium protection against mercury toxicity. Binding of methylmercury by the selenohydryl-containing ligand. Journal of the American Chemical Society. 1976;98:2339–2341. doi: 10.1021/ja00424a059. [DOI] [PubMed] [Google Scholar]
- Sumi D. Biological Effects of and Responses to Exposure to Electrophilic Environmental Chemicals. Journal of Health Science. 2008;54:6. [Google Scholar]
- Tadolini B, Hakim G. The mechanism of iron (III) stimulation of lipid peroxidation. Free Radic Res. 1996;25:221–227. doi: 10.3109/10715769609149047. [DOI] [PubMed] [Google Scholar]
- Takeda A. Manganese action in brain function. Brain Res Brain Res Rev. 2003;41:79–87. doi: 10.1016/s0165-0173(02)00234-5. [DOI] [PubMed] [Google Scholar]
- Takeda A, Sawashita J, Okada S. Biological half-lives of zinc and manganese in rat brain. Brain Res. 1995;695:53–58. doi: 10.1016/0006-8993(95)00916-e. [DOI] [PubMed] [Google Scholar]
- Tchounwou PB, Ayensu WK, Ninashvili N, Sutton D. Environmental exposure to mercury and its toxicopathologic implications for public health. Environ Toxicol. 2003;18:149–175. doi: 10.1002/tox.10116. [DOI] [PubMed] [Google Scholar]
- Tejning S, Ohman H. Uptake, excretion and retention of metallic mercury in chloralkali workers Hygiene toxicology occupational diseases. proceedings of the] 15th international congress on occupational medicine; Viena. 1966. pp. 239–242. [Google Scholar]
- Theil EC. Iron homeostasis and nutritional iron deficiency. J Nutr. 2011;141:724S–728S. doi: 10.3945/jn.110.127639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen PT, Pennings JL, Robinson JF, Claessen SM, Kleinjans JC, Piersma AH. Time-response evaluation by transcriptomics of methylmercury effects on neural differentiation of murine embryonic stem cells. Toxicol Sci. 2011;122:437–447. doi: 10.1093/toxsci/kfr134. [DOI] [PubMed] [Google Scholar]
- Tseng YJ, Chen CH, Chen WK, Dong-Zong H. Hand warmer related corrosive injury. Clin Toxicol (Phila) 2011;49:870–871. doi: 10.3109/15563650.2011.607460. [DOI] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino M, Okajima T, Eto K, Kumamoto T, Mishima I, Ando M. Neurologic features of chronic Minamata disease (organic mercury poisoning) certified at autopsy. Intern Med. 1995;34:744–747. doi: 10.2169/internalmedicine.34.744. [DOI] [PubMed] [Google Scholar]
- Unzai S, Eich R, Shibayama N, Olson JS, Morimoto H. Rate constants for O2 and CO binding to the alpha and beta subunits within the R and T states of human hemoglobin. J Biol Chem. 1998;273:23150–23159. doi: 10.1074/jbc.273.36.23150. [DOI] [PubMed] [Google Scholar]
- Usuki F, Yamashita A, Fujimura M. Post-transcriptional defects of antioxidant selenoenzymes cause oxidative stress under methylmercury exposure. J Biol Chem. 2011;286:6641–6649. doi: 10.1074/jbc.M110.168872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- Vance CK, Miller AF. Novel insights into the basis for Escherichia coli superoxide dismutase’s metal ion specificity from Mn-substituted FeSOD and its very high E(m) Biochemistry. 2001;40:13079–13087. doi: 10.1021/bi0113317. [DOI] [PubMed] [Google Scholar]
- Volterra A, Trotti D, Tromba C, Floridi S, Racagni G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J Neurosci. 1994;14:2924–2932. doi: 10.1523/JNEUROSCI.14-05-02924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachtershauser G. On the chemistry and evolution of the pioneer organism. Chem Biodivers. 2007;4:584–602. doi: 10.1002/cbdv.200790052. [DOI] [PubMed] [Google Scholar]
- Wagner C, Sudati JH, Nogueira CW, Rocha JB. In vivo and in vitro inhibition of mice thioredoxin reductase by methylmercury. Biometals. 2010;23:1171–1177. doi: 10.1007/s10534-010-9367-4. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–652. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. 2011;434:365–381. doi: 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfvinge K, Hua J, Logdberg B. Mercury distribution in cortical areas and fiber systems of the neonatal and maternal adult cerebrum after exposure of pregnant squirrel monkeys to mercury vapor. Environ Res. 1994;67:196–208. doi: 10.1006/enrs.1994.1074. [DOI] [PubMed] [Google Scholar]
- Wasserman GA, Liu X, Parvez F, Ahsan H, Levy D, Factor-Litvak P, Kline J, van Geen A, Slavkovich V, LoIacono NJ, Cheng Z, Zheng Y, Graziano JH. Water manganese exposure and children’s intellectual function in Araihazar, Bangladesh. Environ Health Perspect. 2006;114:124–129. doi: 10.1289/ehp.8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe C, Yoshida K, Kasanuma Y, Kun Y, Satoh H. In utero methylmercury exposure differentially affects the activities of selenoenzymes in the fetal mouse brain. Environ Res. 1999;80:208–214. doi: 10.1006/enrs.1998.3889. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Yamamura T, Watanabe M, Yasuo S, Nakao N, Dawson A, Ebihara S, Yoshimura T. Hypothalamic expression of thyroid hormone-activating and -inactivating enzyme genes in relation to photorefractoriness in birds and mammals. Am J Physiol Regul Integr Comp Physiol. 2007;292:R568–572. doi: 10.1152/ajpregu.00521.2006. [DOI] [PubMed] [Google Scholar]
- Weber KA, Achenbach LA, Coates JD. Microorganisms pumping iron: anaerobic microbial iron oxidation and reduction. Nat Rev Microbiol. 2006;4:752–764. doi: 10.1038/nrmicro1490. [DOI] [PubMed] [Google Scholar]
- Welch KD, Davis TZ, Van Eden ME, Aust SD. Deleterious iron-mediated oxidation of biomolecules. Free Radic Biol Med. 2002;32:577–583. doi: 10.1016/s0891-5849(02)00760-8. [DOI] [PubMed] [Google Scholar]
- West AR, Oates PS. Mechanisms of heme iron absorption: current questions and controversies. World J Gastroenterol. 2008;14:4101–4110. doi: 10.3748/wjg.14.4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Ding T, Sun J. Neurotoxic potential of iron oxide nanoparticles in the rat brain striatum and hippocampus. Neurotoxicology. 2012 doi: 10.1016/j.neuro.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Xu Z, Jia K, Xu B, He A, Li J, Deng Y, Zhang F. Effects of MK-801, taurine and dextromethorphan on neurotoxicity caused by manganese in rats. Toxicol Ind Health. 2010;26:55–60. doi: 10.1177/0748233709359275. [DOI] [PubMed] [Google Scholar]
- Yamada M, Ohno S, Okayasu I, Okeda R, Hatakeyama S, Watanabe H, Ushio K, Tsukagoshi H. Chronic manganese poisoning: a neuropathological study with determination of manganese distribution in the brain. Acta Neuropathol. 1986;70:273–278. doi: 10.1007/BF00686083. [DOI] [PubMed] [Google Scholar]
- Yamamoto I. Effect of various amounts of selenium on the metabolism of mercuric chloride in mice. Biochem Pharmacol. 1985;34:2713–2720. doi: 10.1016/0006-2952(85)90572-6. [DOI] [PubMed] [Google Scholar]
- Yang M, Cobine PA, Molik S, Naranuntarat A, Lill R, Winge DR, Culotta VC. The effects of mitochondrial iron homeostasis on cofactor specificity of superoxide dismutase 2. EMBO J. 2006;25:1775–1783. doi: 10.1038/sj.emboj.7601064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Aschner JL, dos Santos AP, Aschner M. Mitochondrial-dependent manganese neurotoxicity in rat primary astrocyte cultures. Brain Res. 2008a;1203:1–11. doi: 10.1016/j.brainres.2008.01.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Jiang H, Syversen T, Rocha JB, Farina M, Aschner M. The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter. J Neurochem. 2008b;107:1083–1090. doi: 10.1111/j.1471-4159.2008.05683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Lee E, Ni M, Jiang H, Milatovic D, Rongzhu L, Farina M, Rocha JB, Aschner M. Methylmercury-induced alterations in astrocyte functions are attenuated by ebselen. Neurotoxicology. 2011;32:291–299. doi: 10.1016/j.neuro.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorifuji T, Debes F, Weihe P, Grandjean P. Prenatal exposure to lead and cognitive deficit in 7- and 14-year-old children in the presence of concomitant exposure to similar molar concentration of methylmercury. Neurotoxicol Teratol. 2011;33:205–211. doi: 10.1016/j.ntt.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Watanabe C, Horie K, Satoh M, Sawada M, Shimada A. Neurobehavioral changes in metallothionein-null mice prenatally exposed to mercury vapor. Toxicol Lett. 2005;155:361–368. doi: 10.1016/j.toxlet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Zatta P, Lucchini R, van Rensburg SJ, Taylor A. The role of metals in neurodegenerative processes: aluminum, manganese, and zinc. Brain Res Bull. 2003;62:15–28. doi: 10.1016/s0361-9230(03)00182-5. [DOI] [PubMed] [Google Scholar]
- Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- Zemolin AP, Meinerz DF, de Paula MT, Mariano DO, Rocha JB, Pereira AB, Posser T, Franco JL. Evidences for a role of glutathione peroxidase 4 (GPx4) in methylmercury induced neurotoxicity in vivo. Toxicology. 2012;302:60–67. doi: 10.1016/j.tox.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Zheng W, Ren S, Graziano JH. Manganese inhibits mitochondrial aconitase: a mechanism of manganese neurotoxicity. Brain Res. 1998;799:334–342. doi: 10.1016/s0006-8993(98)00481-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhingel K, Dovensky W, Crossman A, Allen A. Ephedrone: 2-methylamino-1-phenylpropan-1-one. Journal of Forensic Sciences. 1991;36:915–920. [Google Scholar]
- Zimmer B, Schildknecht S, Kuegler PB, Tanavde V, Kadereit S, Leist M. Sensitivity of dopaminergic neuron differentiation from stem cells to chronic low-dose methylmercury exposure. Toxicol Sci. 2011;121:357–367. doi: 10.1093/toxsci/kfr054. [DOI] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]