

Abstract
Over the past few decades, it has become increasingly clear that the lethality of cancers depends on more than the malignant cells themselves. The environment those malignant cells are exposed to is just as important a determinant of their behavior. Head and neck squamous cell carcinoma (HNSCC) is both common and deadly. It is the 6th most frequently occurring cancers, and prognosis is still generally poor. Recent evidence indicates that activated fibroblasts residing within the tumor stroma play a significant role in promoting the aggressive spread often seen in head and neck cancer. Tumor associated fibroblasts (TAFs) have also been implicated in facilitating angiogenesis and suppressing the normal anti-tumor function of immune cells. Studying the signaling molecules involved in these processes will facilitate the development of promising targets and inhibitors to prevent tumor-associated fibroblasts from exerting their reinforcing effects on the tumor. In this article, we review the recent literature on the signals used in tumor associated fibroblast communication, with a focus on potential therapeutic targets. Further, we highlight the lead candidates for TAF-targeted therapeutic interventions. Future anticancer strategies may achieve better results than current approaches by targeting the support cells in tumor stroma in addition to the cancerous cells.
Keywords: Fibroblasts, Head and Neck Cancer, tumor stroma, invasion, proliferation
Introduction
In recent years it has become apparent that tumors are complex structures formed from mutually supporting parts. The malignant cells do not grow in isolation; rather they communicate with and are supported by stromal cells. Therefore, a full understanding of tumor progression requires studying the tumor stroma in addition to the malignant parenchymal cells. One of the main elements of the stroma, and a major contributor to the extracellular environment of solid tumors, is the tumor associated fibroblast (TAF). In many different types of cancer, the secretion of factors by TAFs correlates with poor prognosis [1], leading to the idea that TAFs could be the targets of new drugs to interrupt the supportive relationship between stroma and tumor cells.
Soluble factors secreted by TAFs have been implicated in the progression of tumors in many different organs, including breast and pancreas [1]. While some of the factors identified in tumor-stroma communication are similar among cancer types, others are unique; suggesting that the best drug targets might be specific to particular cancers. For example, hepatocyte growth factor (HGF) and insulin-like growth factor 1 (IGF1) have been implicated in prostate cancer, while the cytokine CXCL12 has been implicated in breast cancer [1]. Transforming growth factor beta (TGFβ), however, has been identified as a key mediator in prostate, breast, and pancreatic cancer [1]. In order to design the optimal therapeutic agent for any given malignancy, it is necessary to study the communication between stroma and parenchyma in that cancer.
Head and neck cancer is a malignancy that is both common and deadly. Oral cancer is the 11th most common type of cancer worldwide, and laryngeal cancer is the 20th most common [2]. If all head and neck cancers are counted together, they are the 6th most common neoplasm in the world [3]. Survival is still less than 50% for head and neck cancer, and this has not improved over the last four decades [2, 4]. When detected early, survival is much better (75% at 5 years), but the majority of patients present with advanced metastatic disease at diagnosis [3]. The tendency of head and neck squamous cell carcinoma (HNSCC) to invade and metastasize aggressively plays a major role in its morbidity and mortality.
Several reports suggest that TAFs play a significant role HNSCC progression. Expression profiles of fibroblasts from cancer free oral mucosa, dysplastic epithelium, and malignant epithelium are all reliably different from each other [5]. TAFs are also frequently found in the lymph node metastases of HNSCC tumors [6]. It is unclear whether TAFs migrate to distant sites along with the cancer cells, or if the cancer cells recruit new TAFs upon arrival in the lymph node. In either case, TAFs are a critical part of HNSCC tumor progression [6]. Various studies have implicated TAFs in promoting HNSCC metastasis [7, 8], invasion [9, 10], angiogenesis [11, 12], immune escape [11, 12], and proliferation [13, 14]. Lack of TAFs or TAF-produced factors significantly impairs the ability of HNSCC cells to engage in all of these processes [9–14]. The purpose of this review is to provide an overview of the signaling mechanisms triggered by TAFs reported to influence HNSCC tumor progression (Figure 1). The molecular interactions between the tumor and stromal fibroblasts are complex. A comprehensive compilation of TAF-tumor signaling is important to fully grasp the mechanisms contributing to tumor progression and to facilitate the development of potent therapeutic strategies to reduce the morbidity and mortality caused by HNSCC. The promise of targeting fibroblasts is illustrated in a study where inhibition of the fibroblast growth factor receptor (FGFR) reduced growth of HNSCC tumors transplated into mice, possibly due to a reduction in the number of murine fibroblasts within the tumor mass [15].
Figure 1.
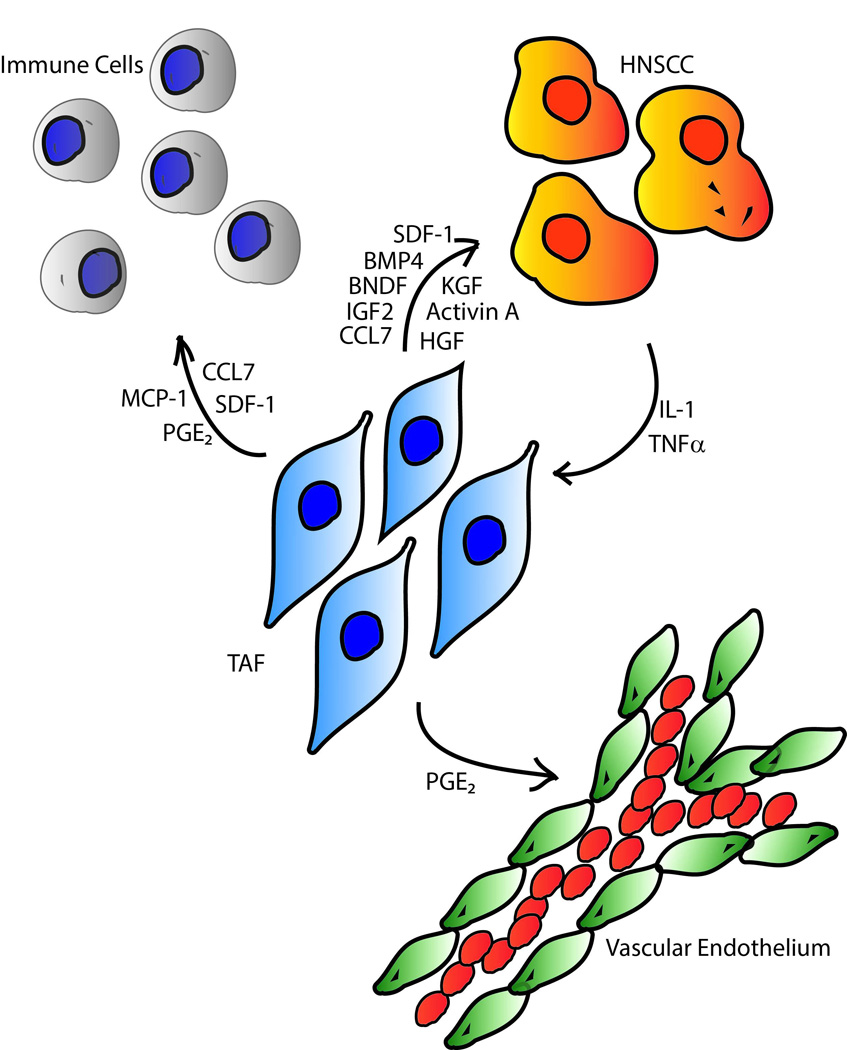
Schematic showing the paracrine factors involved in TAFs-mediated tumor progression. Arrows point from the cell that secretes the mediator to the cell it acts upon.
HNSCC tumors arise at multiple sites in the head and neck region including tongue, floor of the mouth, pharynx, and larynx. There may be significant heterogeneity in the signaling pathways used by different types of HNSCCs and their associated fibroblasts. Some of the molecules discussed in this paper have been implicated in the progression of HNSCC arising from various sites, while most have only been studied in HNSCC tumors from a specific site. When selecting molecules for further development as drug targets, it may be helpful to pick out molecules which are implicated in HNSCC across all sites. It is also important to note that some studies used patient derived TAFs, while others used transformed normal fibroblasts. Although exposure to HNSCC cells can induce a TAF-like phenotype in normal fibroblasts [7], there has not yet been a study rigorously comparing the behavior of transformed normal fibroblasts and TAFs. Although studies using primary TAFs might more closely reflect the situation in vivo, further studies comparing the phenotypes of primary TAFs vs. human fibroblast derived from other sources such as foreskin are required. Table 1 lists the source of the fibroblasts used in various reports examining molecules involved in promoting HNSCC tumor progression. In this review, we will we discuss the molecules implicated in transforming normal fibroblasts or mesenchymal stem cells into TAFs and the impact of these signaling molecules (listed in Table 2) on invasion/metastasis, proliferation, angiogenesis, and immune escape by HNSCC cells.
Table 1.
Origin of the fibroblasts used in studies reviewed in this article
| Secreted Factor | Source of fibroblasts | Reference |
|---|---|---|
| Gene expression study | Patient derived oral SCC | [5] |
| BDNF | Cancer-free human oral fibroblasts activated by exposure to oral SCC | [7] |
| HGF | Cancer-free human oral fibroblasts activated by exposure to oral SCC | [9] |
| CCL7 | Patient derived oral SCC | [10] |
| PGE2 | Cancer-free human dermal fibroblasts activated by exposure to pharyngeal or tongue SCC | [11] |
| IL-1β | Cancer-free human oral fibroblasts activated by exposure to oral SCC | [12] |
| KGF | Patient derived oral SCC | [13] |
| Activin A | Patient derived lingual SCC | [14] |
| TGFβ | Cancer-free human oral fibroblasts activated by exposure to oral SCC | [21] |
| TGFβ | Patient derived HNSCCs, various locations | [22] |
| BMP4 | Patient derived lingual, tonsilar and laryngeal SCC | [24] |
| HGF | Patient-derived lingual, tonsillar and laryngeal SCC | [25] |
| MMPs | Patient derived lingual and pharyngeal SCC | [26] |
| MMPs | Cancer-free human oral fibroblasts activated by exposure to oral SCC | [27] |
| MMPs | Patient derived oral and pharyngeal SCC | [28] |
| MMPs | Human gingival fibroblast from cancer free subjects | [29] |
Table 2.
Molecular signals, categorized by the mechanism they use to facilitate tumor progression
Induction of TAF phenotype
TAFs are distinct from normal fibroblasts (NFs) from cancer-free patients in a variety of ways. Normal fibroblasts are quiescent but maintain tissue architecture by secreting extracellular matrix (ECM) components [1]. If damage occurs to the surrounding tissue, paracrine signals trigger fibroblast activation facilitating wound healing. They begin to express α-smooth muscle actin, increase production of ECM components, and begin proliferating [1, 16]. After wounds heal, reactive fibroblasts undergo apoptosis (some may also revert to quiescence), but TAFs retain their activated phenotype [1]. This has led to the characterization of tumors as “wounds that do not heal” [16].
TAFs can originate from either normal fibroblasts in the immediate vicinity of the tumor or from circulating bone marrow-derived mesenchymal stem cells (MSCs) [17]. MSCs have been found to contribute a significant number of cells to the tumor stroma [17–19]. Once they arrive in the tumor periphery, the MSCs are exposed to factors secreted from the cancer cells, which induce differentiation into TAFs [20]. So far little work has been done to investigate what factors drive the MSC to TAF transformation, but exposure to exogenous transforming growth factor beta (TGFβ) has been found to partially mimic the effects of direct exposure to cancer cells [20].
Recent reports also suggest that cancer cells are capable of transforming normal fibroblasts into TAFs. Co-culture of normal fibroblasts with cancer cells has been found to produce specific changes in gene expression patterns of fibroblasts [12]. Treatment of normal fibroblasts with interleukin-1 beta (IL-1β) or TGFβ causes them to recapitulate these changes in gene expression [12, 21]. Furthermore, IL-1β or TGFβ inhibition reduced the gene expression changes induced by co-culturing fibroblasts with HNSCC cells [12, 21].
It appears that the TAFs themselves may play a role in the induction of TAF phenotype, helping to transform their normal fibroblast neighbors or newly arrived MSCs. TGFβ-1 has been identified in expression profile assays as a molecule whose production is substantially upregulated in TAFs as compared to normal fibroblasts [22]. TGFβ-1 is the most effective form of TGFβ in inducing the TAF phenotype in normal fibroblasts and MSCs [20, 21]. It appears that the transformation of fibroblasts to TAFs can start a positive feedback loop; each fibroblast that is converted into a TAF starts producing TGFβ-1 and influencing other normal fibroblasts and MSCs in turn transform into a TAF phenotype.
In summary, tumor cells secrete TGFβ and IL-1β, inducing TAF phenotype in local fibroblasts. The cancer cells could also co-opt MSCs by exposing them to TGFβ as they migrate into the damaged area. Blocking TGFβ or IL-1β might therefore be very useful in preventing the production of tumor-supporting TAFs.
The Role of TAFs in Tumor Invasion
HNSCC is known for its tendency to invade local structures aggressively and metastasize to distant sites. In fact, the majority of patients diagnosed with advanced non-metastatic disease will go on to develop metastases in spite of treatment [23]. Once the disease becomes metastatic, there are no good treatment options. Local/regional invasion of vital structures in the neck is also a cause of substantial mortality, since it can be difficult or impossible to obtain clear margins around the tumor in a surgical resection. Therefore there is great interest in developing strategies to mitigate the invasive and metastatic potential of HNSCC.
TAFs are reported to produce a number of pro-invasive molecules, including brain-derived neurotrophic factor (BDNF), hepatocyte growth factor (HGF), insulin-like growth factor 2 (IGF2), bone morphogenic protein 4 (BMP4), and chemokine ligand 7 (CCL7) [7, 9, 10, 24, 25]. In addition, TAFs are capable of directly facilitating invasion/metastasis through the production of proteases that can help digest the extracellular matrix including matrix metalloproteases [1–3, 26–28]. Production of these factors by the TAFs appears to be induced by signals from the cancer cells, especially tumor necrosis factor alpha (TNFα) and interleukin-1 alpha (IL-1α) [9, 10]. Furthermore, TAFs produce factors that encourage production of TNFα and IL-1α by the HNSCC cells [29]; it appears that the TAFs and the HNSCC cells communicate with each other in a mutually reinforcing cycle. Table 3 summarizes the molecules reported to be involved in tumor-TAF cross-talk.
Table 3.
Pro-invasive signaling – molecules are categorized by which compartment secretes them and which compartment reacts to them.
Brain derived neurotrophic factor (BDNF) is one of the invasion promoting signals produced by HNSCC-TAFs but not normal oral fibroblasts [7]. BDNF exerts its effects by promoting an epithelial-mesenchymal transition (EMT), thereby facilitating metastasis [7]. Cancer cells exposed to BDNF changed their gene expression profile to one characteristic of cells undergoing EMT, specifically an increase in vimentin and a decrease in E-cadherin expression [7]. BDNF production by TAFs is theorized to be driven by exposure to HNSCC-produced TNFα [7]. Although this has not yet been proved rigorously, it is suspected because TNFα up-regulation in the tumor accompanies BDNF up-regulation in the stroma [7]. Furthermore, anti-TNFα antibody infliximab significantly inhibits TAF-induced invasion by HNSCC cells [28]. TNFα blockers such as infliximab and etanercept have met with success in clinical trials [30, 31], but they have not yet been tested in head and neck cancer. Given the significant role of TNFα in HNSCC invasion, infliximab and etanercept may be useful in preventing HNSCC metastasis.
In co-cultured TAFs and HNSCC cells, BDNF production is increased by the TAFs, while TrkB (a receptor for BDNF) is up-regulated by the cancer cells [7]. In addition, IL-1β was found to be increased in the fibroblasts, and TNFα in the cancer cells. These cytokines were suggested as possible regulators of BDNF and TrkB, but the exact mechanism remains unclear. TrkB and BDNF are being investigated as potential drug targets. Inhibitors of TrkB have already been developed and met with some success in cell culture models [32]. Furthermore, down-regulation of TrkB has been linked to reduced tumor growth in a murine xenograft model of HNSCC [33]. Blocking the interaction of BDNF and TrkB could potentially reduce rates of metastasis.
Hepatocyte growth factor (HGF) is a pro-invasive molecule produced by activated TAFs and binds to the MET receptor on the cancer cells, triggering enhanced invasion through the basement membrane [9, 21, 25, 34]. The HGF/MET signaling pathway is already being extensively targeted in anti-cancer research [25, 35]. Production of HGF by TAFs may be regulated by exposure to HNSCC-secreted IL-1α [9, 10]. Stromal-derived factor 1 (SDF-1) also induces invasiveness, and like HGF, it is upregulated in fibroblasts exposed to IL-1α [9]. A recombinant IL-1 receptor antagonist called anakinra has been used successfully to treat inflammatory conditions, such as rheumatoid arthritis [31]. Anakinra could potentially be used to inhibit IL-1 signaling between tumors and fibroblasts. Inhibitors of SDF-1 signaling have been found useful in preventing metastasis in a mouse model of breast cancer [36] and they also extended survival in a mouse model of leukemia [37], but these inhibitors have not yet been tested in HNSCC.
TAF-secreted insulin-like growth factor 2 (IGF2), bone morphogenic protein 4 (BMP4), and CCL7 have also been found to promote HNSCC invasion [10, 24]. Neutralizing antibodies to these molecules abrogated the pro-invasive effect, which confirms the involvement of these mediators [10, 24]. CCL7’s role in metastasis was confirmed by the fact that its receptor, CCR7, was found to be substantially upregulated in HNSCC cells with higher metastatic tendencies [38]. It has not yet been determined what factors the tumor cells secrete to encourage IGF2 and BMP4 production; however some inducers of CCL7 expression are known. IL-1α, but not vascular endothelial growth factor (VEGF), produced by the cancer cells was determined to be specifically responsible for stimulating CCL7 secretion from TAFs [10]. However, exposure to both VEGF and IL-1α further increased CCL7 secretion compared to IL-1α alone.
Other chemokines of the CXCL and CCL families are also overproduced when fibroblasts are exposed to tumor cell factors and some of these, such as CCL2 (monocyte chemotactic protein-1) have also been found to enhance invasion by HNSCC cells [9, 26, 34, 39]. SDF-1 (CXCL12), which was mentioned before, is also a member of this family [26]. So far only CXCL12 has been targeted for pharmacotherapy, but the success of these experiments should encourage efforts at blocking CCL7, CCL2, and other molecules in the CCL and CXCL families.
Matrix metalloproteases (MMPs) degrade components of the ECM including collagens, fibronectin and laminin [40]. Collagen I produced by TAFs interacts with the α2β1 integrin on the HNSCC cells, inducing production of MMP 2 [26]. Expression of membrane type 1 matrix metalloproteinase by TAFs has been reported in HNSCC [26–28]. Western blotting of TAF lysates showed elevated expression of membrane type 1 MMP (MT1-MMP) as compared to normal mucosal fibroblasts [28]. Further, HNSCC cells promote MMP secretion from TAFs via expression of extracellular matrix metalloprotease inducer (EMMPRIN) [41]. EMPIRIN is a glycoprotein reported to function as a cell adhesion molecule and a paracrine inducer of MMP expression. MT1-MMP cleaves EMPIRIN from the cell surface releasing a 22 KD fragment that mediates expression of MMP-2 from fibroblasts [42]. Normal gingival fibroblasts activated by exposure to HNSCC cells showed up-regulation of MMPs 1–3, along with tissue inhibitors of metalloproteinases 1 and 3 (TIMPs 1 and 3) [27]. Expression of TIMP 1 and 3 in HNSCC tissue was observed in TAF’s via immunohistochemistry [43]. Inhibition of specific MMPs produced in the tumor and in the stroma has the potential to achieve antitumor effects.
Interactions between TAFs and Immune Cells
Less work has been done in this area, but there is evidence that TAFs secrete arachidonic acid derivatives that can impair immune surveillance [11, 12]. IL-1β secretion by tumor cells induces production of COX2 [11, 12] and microsomal PGE synthase 1 by TAFs, both of which are involved in synthesis of prostaglandin E2 (PGE2) [11]. Increased PGE2 in the tumor microenvironment functions to suppress immune attack [11]. It is thought that PGE2 accomplishes this through its effects on CD4 (helper) and CD8 (killer) T cells; PGE2 reduces the proliferation of both types of T cells and alters their production of cytokines [44]. In addition to its role in inflammation, PGE2 promotes angiogenesis by inducing endothelial cell growth, tumor migration and proliferation [11]. As a result of these properties, COX2 activity in the stroma is correlated with worse tumor grade in various cancers, but specifically in HNSCC [45]. These results suggest that COX2 inhibitors should have value as anti-cancer agents, but more specific inhibition of microsomal PGE synthase might be an even better line of attack [11]. These studies help to explain the long-established anti-cancer activity of COX2 inhibitor celecoxib [46], which is known to be active against many tumors [46] including oral squamous cell carcinoma [47]. Inhibitors of microsomal PGE synthase have been created [48], but have not yet been tested for efficacy against HNSCC.
Interestingly, TAFs also produce cytokines which are known to have a pro-immune effect. CCL7 and SDF-1 (CXCL12) have been found to enhance the activity and migration of T cells [49, 50], which are a pivotal cell type for immune defense against malignancies. It seems counterintuitive that TAFs would secrete both immune stimulating and immune suppressing mediators, but since fibroblast activation is a normal part of the wound repair process, it makes sense that activated fibroblasts would produce mediators to stimulate an immune response at the site of damage.
TAFs also secrete Monocyte Chemotactic Protein 1 (MCP1) [39], and thus may play a role in attracting tumor associated macrophages (TAMs) to head and neck cancers. In several types of cancer, TAMs have been found to mature predominantly into type 2 macrophages, which tend to support tumor growth by favoring angiogenesis, remodeling, and repair [34, 51]. Furthermore, a high level of macrophage infiltration into HNSCC tumors is associated with more aggressive disease [34, 52]. Therefore TAFs may play an additional role in facilitating tumor progression by attracting monocytes, which the tumor cells then co-opt.
Angiogenesis
TAFs are reported to induce angiogenesis in HNSCC via COX2-mediated production of PGE2 [11, 12]. In addition, HGF [53], CXCL12/SDF-1 [54], and BMP4 [55] secreted by TAFs have documented angiogenic properties. Although it has not been confirmed in HNSCC, TAFs have also been shown to express vascular endothelial growth factor (VEGF), a well-known angiogenic signal in transgenic models and human breast carcinoma [56, 57]. Therefore, TAFs may be involved in neovascularization through an array of molecules that promote angiogenesis. Since TAFs produce pro-angiogenic factors besides VEGF, they may be also involved in “evasive resistance”, a phenomenon where tumors develop resistance to VEGF inhibitors by exploiting alternative angiogenic pathways [58]. Targeting pro-angiogenic factors produced by TAFs could complement existing anti-angiogenic therapies based on inhibiting VEGF signaling.
Proliferation/Mitotic Activity
Several TAF-secreted factors have been linked with increased proliferation of HNSCC cells including PGE2, keratinocyte growth factor (KGF), and activin A [11, 13, 14]. Oral cancer TAFs have been found to produce substantially higher levels of KGF than normal fibroblasts [13]. Furthermore, inhibition of KGF with blocking antibodies abrogated TAF-induced HNSCC mitosis and viability [13]. Although inhibitors targeting the KGF receptor were developed a few years ago and have shown promise against breast cancer in in vitro studies [59], these agents have not yet been tested in HNSCC. The utility of blocking KGF is somewhat dubious, however, since KGF seems to stimulate the growth of normal keratinocytes just as strongly as it stimulates the growth of malignant keratinocytes [60].
Activin A, which plays a role in regulating the HNSCC cell-cycle through its effects on SMAD-family proteins [61], is produced at a much higher levels by TAFs than normal resting fibroblasts [14]. Further, activin A knockdown with siRNA reduced TAF-induced tumor growth [14]. Inhibitors of activin A have been developed and have met with some success in certain cancers, such as multiple myeloma [62]. Further studies on the effects of targeting activin A in HNSCC are warranted.
Conclusions and Future Directions
TAFs and HNSCC cells engage in molecular communications facilitating tumor progression through a variety of mechanisms. TAFs secrete factors including CCL7 and HGF [9,10, 21,25], that induce HNSCC invasion. TAFs also encourage tumor cell proliferation via KGF, activin A, and PGE [11, 13, 14]. Although the role of TAFs in angiogenesis and immune escape are less-well studied, TAFs produce prostaglandin E2, which encourages tumor angiogenesis and interferes with T cell mediated cytotoxicity [11]. Molecular networks involved in tumor-stroma communication will likely be areas of active research in the near future. As the communication between tumor and reactive stroma is more fully elucidated, more potential drug targets will become apparent.
For the present, however, some of these targets look very promising in the near term, while others might take years of research to reach clinical application. The molecular targets which may be most useful are those for which clinically tested inhibitors already exist. TNFα inhibitors such as adalimumab and etanercept have already been developed and tested in cancers including head and neck [30, 31]. Further, inhibitors of prostaglandin synthesis (COX2 inhibitors), c-Met, IL-1, and activin A are also currently available and could be used to target the tumor associated stroma [25, 31, 46, 62]. In all likelihood, several of these approaches will need to be combined with more traditional medical and surgical management in order to achieve maximum anti-tumor effects.
Acknowledgements
This work was supported by the University of Pittsburgh Medical School Dean’s Summer Research Award to GL, a career development award (P50 CA097190), Competitive Medical Research Fund award, University of Pittsburgh Medical Center and Department of Otolaryngology, University of Pittsburgh funds to SMT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Franco OE, Shaw AK, Strand DW, Hayward SW. Cancer associated fibroblasts in cancer pathogenesis. Semin Cell Dev Biol. 2010;21:33–39. doi: 10.1016/j.semcdb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Döbróssy L. Epidemiology of head and neck cancer: Magnitude of the problem. Cancer and Metastasis Reviews. 2005;24:9–17. doi: 10.1007/s10555-005-5044-4. [DOI] [PubMed] [Google Scholar]
- 3.Chin D, Boyle GM, Williams RM, Ferguson K, Pandeya N, Pedley J, et al. Novel markers for poor prognosis in head and neck cancer. International Journal of Cancer. 2005;113:789–797. doi: 10.1002/ijc.20608. [DOI] [PubMed] [Google Scholar]
- 4.Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. 2008;359:1143–1154. doi: 10.1056/NEJMra0707975. [DOI] [PubMed] [Google Scholar]
- 5.Lim KP, Cirillo N, Hassona Y, Wei W, Thurlow JK, Cheong SC, et al. Fibroblast gene expression profile reflects the stage of tumour progression in oral squamous cell carcinoma. The Journal of Pathology. 2011;223:459–469. doi: 10.1002/path.2841. [DOI] [PubMed] [Google Scholar]
- 6.Vered M, Dayan D, Yahalom R, Dobriyan A, Barshack I, Bello IO, et al. Cancer associated fibroblasts and epithelial mesenchymal transition in metastatic oral tongue squamous cell carcinoma. International journal of cancer. 2010;127:1356–1362. doi: 10.1002/ijc.25358. [DOI] [PubMed] [Google Scholar]
- 7.Dudas J, Bitsche M, Schartinger V, Falkeis C, Sprinzl GM, Riechelmann H. Fibroblasts produce brain-derived neurotrophic factor and induce mesenchymal transition of oral tumor cells. Oral Oncology. 2011;47:98–103. doi: 10.1016/j.oraloncology.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudas J, Fullar A, Bitsche M, Schartinger V, Kovalszky I, Sprinzl GM, et al. Tumor-produced, active interleukin-1beta regulates gene expression in carcinomaassociated fibroblasts. Exp Cell Res. 2011;317:2222–2229. doi: 10.1016/j.yexcr.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daly AJ, McIlreavey L, Irwin CR. Regulation of HGF and SDF-1 expression by oral fibroblasts, implications for invasion of oral cancer. Oral oncology. 2008;44:646–651. doi: 10.1016/j.oraloncology.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 10.Jung DW, Che ZM, Kim J, Kim K, Kim KY, Williams D. Tumor stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. International Journal of Cancer. 2010;127:332–344. doi: 10.1002/ijc.25060. [DOI] [PubMed] [Google Scholar]
- 11.Alcolea S, Anton R, Camacho M, Soler M, Alfranca A, Aviles-Jurado FX, et al. Interaction between head and neck squamous cell carcinoma (HNSCC) cells and fibroblasts in the biosynthesis of PGE2. Journal of Lipid Research. 2012;53:630–642. doi: 10.1194/jlr.M019695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dudas J, Fullar A, Bitsche M, Schartinger V, Kovalszky I, Sprinzl GM, et al. Tumor-produced, active Interleukin-1 [beta] regulates gene expression in carcinomaassociated fibroblasts. Experimental Cell Research. 2011;317:2222–2229. doi: 10.1016/j.yexcr.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin J, Liu C, Ge L, Gao Q, He X, Liu Y, et al. Carcinoma-associated fibroblasts promotes the proliferation of a lingual carcinoma cell line by secreting keratinocyte growth factor. Tumor Biology. 2011;32:597–602. doi: 10.1007/s13277-011-0158-5. [DOI] [PubMed] [Google Scholar]
- 14.Sobral LM, Bufalino A, Lopes MA, Graner E, Salo T, Coletta RD. Myofibroblasts in the stroma of oral cancer promote tumorigenesis via secretion of activin A. Oral oncology. 2011;47:840–846. doi: 10.1016/j.oraloncology.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 15.Sweeny L, Liu Z, Lancaster W, Hart J, Hartman YE, Rosenthal EL. Inhibition of fibroblasts reduced head and neck cancer growth by targeting fibroblast growth factor receptor. The Laryngoscope. 2012;122:1539–1544. doi: 10.1002/lary.23266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature Reviews Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 17.Hall B, Dembinski J, Sasser AK, Studeny M, Andreeff M, Marini F. Mesenchymal stem cells in cancer: tumor-associated fibroblasts and cell-based delivery vehicles. International journal of hematology. 2007;86:8–16. doi: 10.1532/IJH97.06230. [DOI] [PubMed] [Google Scholar]
- 18.Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer research. 2004;64:8492–8495. doi: 10.1158/0008-5472.CAN-04-1708. [DOI] [PubMed] [Google Scholar]
- 19.Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, et al. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PloS one. 2009;4:e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emura M, Ochiaai A, Horino M, Arndt W, Kamino K, Hirohashi S. Development of myofibroblasts from human bone marrow mesenchymal stem cells cocultured with human colon carcinoma cells and TGF beta 1. In Vitro Cellular & Developmental Biology-Animal. 2000;36:77–80. doi: 10.1290/1071-2690(2000)036<0077:domfhb>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 21.Lewis M, Lygoe K, Nystrom M, Anderson W, Speight P, Marshall J, et al. Tumour-derived TGFβ1 modulates myofibroblast differentiation and promotes HGF/SF-dependent invasion of squamous carcinoma cells. British Journal of Cancer. 2004;90:822–832. doi: 10.1038/sj.bjc.6601611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenthal E, McCrory A, Talbert M, Young G, Murphy, Ullrich J, et al. Elevated expression of TGFβ1 in head and neck cancer associated fibroblasts. Molecular carcinogenesis. 2004;40:116–121. doi: 10.1002/mc.20024. [DOI] [PubMed] [Google Scholar]
- 23.Bhave SL, Teknos TN, Pan Q. Molecular Parameters of Head and Neck Cancer Metastasis. Critical Reviews Eukaryotic Gene Expression. 2011;21:143–153. doi: 10.1615/critreveukargeneexpr.v21.i2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strnad H, Lacina L, Kolar M, Cada Z, Vlçek C, Dvorankova B, et al. Head and neck squamous cancer stromal fibroblasts produce growth factors influencing phenotype of normal human keratinocytes. Histochemistry and cell biology. 2010;133:201–211. doi: 10.1007/s00418-009-0661-6. [DOI] [PubMed] [Google Scholar]
- 25.Knowles LM, Stabile LP, Egloff AM, Rothstein ME, Thomas SM, Gubish CT, et al. HGF and c-Met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clinical Cancer Research. 2009;15:3740–3750. doi: 10.1158/1078-0432.CCR-08-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koontongkaew S, Amornphimoltham P, Monthanpisut P, Saensuk T, Leelakriangsak M. Fibroblasts and extracellular matrix differently modulate MMP activation by primary and metastatic head and neck cancer cells. Medical Oncology. 2011;29:690–703. doi: 10.1007/s12032-011-9871-6. [DOI] [PubMed] [Google Scholar]
- 27.Fullar A, Kovalszky I, Bitsche M, Romani A, Schartinger VH, Sprinzl GM, et al. Tumor cells and carcinoma-associated fibroblasts interaction regulates matrix metalloproteinases and their inhibitors in oral squamous cell carcinoma. Experimental Cell Research. 2012;318:1517–1527. doi: 10.1016/j.yexcr.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenthal EL, McCrory A, Talbert M, Carroll W, Magnuson JS, Peters GE. Expression of proteolytic enzymes in head and neck cancer-associated fibroblasts. Archives of Otolaryngology -- Head and Neck Surgery. 2004;130:943–947. doi: 10.1001/archotol.130.8.943. [DOI] [PubMed] [Google Scholar]
- 29.Koontongkaew S, Amornphimoltham P, Yapong B. Tumor-stroma interactions influence cytokine expression and matrix metalloproteinase activities in paired primary and metastatic head and neck cancer cells. Cell biology international. 2009;33:165–173. doi: 10.1016/j.cellbi.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 30.Brown E, Charles K, Hoare S, Rye R, Jodrell D, Aird R, et al. A clinical study assessing the tolerability and biological effects of infliximab, a TNFα inhibitor, in patients with advanced cancer. Annals of oncology. 2008;19:1340–1346. doi: 10.1093/annonc/mdn054. [DOI] [PubMed] [Google Scholar]
- 31.Wang F, Arun P, Friedman J, Chen Z, Van Waes C. Current and potential inflammation targeted therapies in head and neck cancer. Current opinion in pharmacology. 2009;9:389–395. doi: 10.1016/j.coph.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Desmet C, Peeper D. The neurotrophic receptor TrkB: a drug target in anti cancer therapy. Cellular and molecular life sciences. 2006;63:755–759. doi: 10.1007/s00018-005-5490-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kupferman M, Jiffar T, El-Naggar A, Yilmaz T, Zhou G, Xie T, et al. TrkB induces EMT and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene. 2010;29:2047–2059. doi: 10.1038/onc.2009.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathew M, Thomas SM. The Cellular Microenvironment of Head and Neck Squamous Cell Carcinoma. In: Rijeka Li X., editor. Squamous Cell Carcinoma. Croatia: InTech; 2012. pp. 163–174. [Google Scholar]
- 35.Gherardi E, Birchmeier W, Birchmeier C, Woude GV. Targeting MET in cancer: rationale and progress. Nature Reviews Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 36.Liang Z, Wu T, Lou H, Yu X, Taichman RS, Lau SK, et al. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer research. 2004;64:4302. doi: 10.1158/0008-5472.CAN-03-3958. [DOI] [PubMed] [Google Scholar]
- 37.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clinical Cancer Research. 2010;16:2927. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 38.Wang J, Xi L, Hunt JL, Gooding W, Whiteside TL, Chen Z, et al. Expression Pattern of Chemokine Receptor 6 (CCR6) and CCR7 in Squamous Cell Carcinoma of the Head and Neck Identifies a Novel Metastatic Phenotype. Cancer Research. 2004;64:1861–1866. doi: 10.1158/0008-5472.can-03-2968. [DOI] [PubMed] [Google Scholar]
- 39.Wu MH, Hong HC, Hong TM, Chiang WF, Jin YT, Chen YL. Targeting galectin-1 in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis by downregulating MCP-1/CCL2 expression. Clinical Cancer Research. 2011;17:1306. doi: 10.1158/1078-0432.CCR-10-1824. [DOI] [PubMed] [Google Scholar]
- 40.Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor-host cell communication. Differentiation. 2002;70:561–573. doi: 10.1046/j.1432-0436.2002.700909.x. [DOI] [PubMed] [Google Scholar]
- 41.Rosenthal EL, Zhang W, Talbert M, Raisch KP, Peters GE. Extracellular matrix metalloprotease inducer-expressing head and neck squamous cell carcinoma cells promote fibroblast-mediated type 1 collagen degreadation in vitro. Mol Cancer Res. 2005;3:195–202. doi: 10.1158/1541-7786.MCR-04-0203. [DOI] [PubMed] [Google Scholar]
- 42.Egawa N, Koshikawa N, Tomari T, Nabeshima K, Isobe T, Seiki M. Membrane type 1 matrix metalloproteinase (MT1-MMP/MMP-14) cleaves and releases a 22-kDa extracellular matrix metalloproteinase inducer (EMMPRIN) fragment from tumro cells. Journal of Biological Chemistry. 2006;281:37576–37585. doi: 10.1074/jbc.M606993200. [DOI] [PubMed] [Google Scholar]
- 43.Sutinen M, Kainulainen T, Hurskainen T, Vesterlund E, Alexander JP, Overall CM, et al. Expression of matrix metalloproteinases (MMP-1 and-2) and their inhibitors (TIMP-1, -2, and -3) in oral lichen planus, dysplasia, squamous cell carcinoma, and lymph node metastasis. British Journal of Cancer. 1998;77:2239–2245. doi: 10.1038/bjc.1998.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends in immunology. 2002;23:144–150. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 45.Kourelis K, Vandoros G, Kourelis T, Papadas T, Goumas P, Sotiropoulou-Bonikou G. Low COX2 in tumor and upregulation in stroma mark laryngeal squamous cell carcinoma progression. The Laryngoscope. 2009;119:1723–1729. doi: 10.1002/lary.20569. [DOI] [PubMed] [Google Scholar]
- 46.Koki AT, Masferrer JL. Celecoxib: a specific COX-2 inhibitor with anticancer properties. Cancer Control. 2002;9:28–35. doi: 10.1177/107327480200902S04. [DOI] [PubMed] [Google Scholar]
- 47.Li N, Sood S, Wang S, Fang M, Wang P, Sun Z, et al. Overexpression of 5-lipoxygenase and cyclooxygenase 2 in hamster and human oral cancer and chemopreventive effects of zileuton and celecoxib. Clinical cancer research. 2005;11:2089–2096. doi: 10.1158/1078-0432.CCR-04-1684. [DOI] [PubMed] [Google Scholar]
- 48.Riendeau D, Aspiotis R, Ethier D, Gareau Y, Grimm EL, Guay J, et al. Inhibitors of the inducible microsomal prostaglandin E2 synthase (mPGES-1) derived from MK-886. Bioorganic & medicinal chemistry letters. 2005;15:3352–3355. doi: 10.1016/j.bmcl.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 49.Qiu Y, Zeltzer S, Zhang Y, Wang F, Chen G-H, Dayrit J, et al. Early Induction of CCL7 Downstream of TLR9 Signaling Promotes the Development of Robust Immunity to Cryptococcal Infection. The Journal of Immunology. 2012;188:3940–3948. doi: 10.4049/jimmunol.1103053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savino W, Smaniotto S, Mendes-da-Cruz DA, Dardenne M. Growth hormone modulates migration of thymocytes and peripheral T cells. Annals of the New York Academy of Sciences. 2012;1261:49–54. doi: 10.1111/j.1749-6632.2012.06637.x. [DOI] [PubMed] [Google Scholar]
- 51.Sica A, Larghi P, Mancino A, Rubino L, Porta C, Tortaro M, et al. Macrophage Polarization in Tumor Progression. Semin Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 52.Marcus B, Arenberg D, Lee J, Kleer C, Chepeha D, Schmalbach C, et al. Prognostic factors in oral cavity and oropharyngeal squamous cell carcinoma. Cancer. 2004;101(12):2779–2787. doi: 10.1002/cncr.20701. [DOI] [PubMed] [Google Scholar]
- 53.Ding S, Merkulova-Rainon T, Han ZC, Tobelem G. HGF receptor upregulation contributes to the angiogenic phenotype of human endothelial cells and promotes angiogenesis in vitro. Blood. 2003;101:4816–4822. doi: 10.1182/blood-2002-06-1731. [DOI] [PubMed] [Google Scholar]
- 54.Streiter RM, Belperio JA, Phillips RJ, Keane MP. CXC chemokines in angiogenesis of cancer. Sem Cancer Bio. 2004;14:195–200. doi: 10.1016/j.semcancer.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 55.Rothhammer T, Bataille F, Spruss T, Eissner G, Bosserhoff AK. Functional implication of BMP4 expression on angiogenesis in malignant melanoma. Oncogene. 2007;26:4158–4170. doi: 10.1038/sj.onc.1210182. [DOI] [PubMed] [Google Scholar]
- 56.Fukumura D, Xavier R, Sugiura T, Chen Y, Park E, Lu N, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–725. doi: 10.1016/s0092-8674(00)81731-6. [DOI] [PubMed] [Google Scholar]
- 57.Hlatky L, Tsionou C, Hahnfeldt, Coleman CN. Mammary Fibroblasts may influence breast tumor angiogenesis via hypoxia-induced vascular endothelial growth factor up-regulation and protein expression. Cances Res. 1994;54:6083–6086. [PubMed] [Google Scholar]
- 58.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nature Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hackett J, Xiao Z, Zang XP, Lerner ML, Brackett DJ, Brueggemeier RW, et al. Development of keratinocyte growth factor receptor tyrosine kinase inhibitors for the treatment of cancer. Anticancer research. 2007;27:3801–3806. [PubMed] [Google Scholar]
- 60.Drugan CS, Stone A, Game SM, Prime SS. The mitogenic effect of KGF and the expression of its cell surface receptor on cultured normal and malignant human oral keratinocytes and on contiguous fibroblasts. J Oral Path Med. 1997;26:327–333. doi: 10.1111/j.1600-0714.1997.tb00224.x. [DOI] [PubMed] [Google Scholar]
- 61.Chen YG, Wang Q, Lin SL, Chang CD, Chung J, Ying SY. Activin signaling and its role in regulation of cell proliferation, apoptosis, and carcinogenesis. Experimental biology and medicine. 2006;231:534–544. doi: 10.1177/153537020623100507. [DOI] [PubMed] [Google Scholar]
- 62.Vallet S, Smith MR, Raje N. Novel bone-targeted strategies in oncology. Clinical Cancer Research. 2010;16:4084–4093. doi: 10.1158/1078-0432.CCR-10-0600. [DOI] [PubMed] [Google Scholar]