

Abstract
The amino acid sequence of a protein governs its function. We used bulk competition and focused deep sequencing to investigate the effects of all ubiquitin point mutants on yeast growth rate. Many aspects of ubiquitin function have been carefully studied, which enabled interpretation of our growth analyses in light of a rich structural, biophysical and biochemical knowledge base. In one highly sensitive cluster on the surface of ubiquitin almost every amino acid substitution caused growth defects. In contrast, the opposite face tolerated virtually all possible substitutions. Surface locations between these two faces exhibited intermediate mutational tolerance. The sensitive face corresponds to the known interface for many binding partners. Across all surface positions, we observe a strong correlation between burial at structurally characterized interfaces and the number of amino acid substitutions compatible with robust growth. This result indicates that binding is a dominant determinant of ubiquitin function. In the solvent inaccessible core of ubiquitin all positions tolerated a limited number of substitutions, with hydrophobic amino acids especially interchangeable. Some mutations null for yeast growth were previously shown to populate folded conformations indicating that for these mutants subtle changes to conformation caused functional defects. The most sensitive region to mutation within the core was located near the C-terminus that is a focal binding site for many critical binding partners. These results indicate that core mutations may frequently cause functional defects through subtle disturbances to structure or dynamics.
Keywords: fitness, structure, function, thermodynamic stability, sequence
Introduction
Analyses of protein sequence-function relationships provide a powerful approach to understand mechanism. Mutational studies provide information on the functional impact of specific chemical changes to the protein. Systematic analyses of point mutations provide a detailed map of chemical space that can be mined to infer mechanism. While it has been possible to generate libraries of point mutants for many years1, until recently it had only been feasible to analyze the function of systematic mutations using amber suppresser strains of E. coli2. Functional analyses of mutant libraries in non-suppresser systems can now be performed in high-throughput by utilizing deep-sequencing to analyze mixtures of multiple mutants simultaneously. In this approach, sequence profiling, originally by microarray3 and now more commonly by deep sequencing is used to determine the relative abundance of mutants in response to selective pressures4; 5; 6; 7; 8; 9; 10; 11; 12.
To measure the fitness effects in cells of all possible point mutants for regions of genes, we developed an approach we refer to as EMPIRIC5; 6. This approach utilizes systematic site saturation libraries that incorporate a single degenerate codon (NNN) in an otherwise wild type (WT) coding sequence. Thus, all possible point mutants are included in the library design and the vast majority can be observed above background in deep sequencing analyses. We analyze libraries of point mutants in conditional yeast strains that contain a second copy of the gene whose activity can be tightly regulated. This enables the amplification of mutant libraries in yeast under permissive conditions where growth is not dependent on mutant function. Adjusting conditions to turn off the second copy of the gene then initiates growth competition based on mutant fitness. In previous work, we analyzed a nine amino acid loop in yeast Hsp906. Here, we report EMPIRIC fitness analyses for the entire yeast ubiquitin gene.
Ubiquitin is essential in all eukaryotes where it serves multiple functions via its ability to covalently attach to other proteins13. The covalent attachment of the C-terminus of ubiquitin to lysine side-chains is mediated by a series of enzymes referred to as E1, E2, and E314. Multiple ubiquitin molecules can be linked through covalent attachment between the C-terminus of one ubiquitin chain and a lysine from another ubiquitin. Lysine 48 in ubiquitin is the only lysine that is essential for yeast growth15. K48-linked poly-ubiquitin serves as a degradation signal16 with four K48-linked ubiquitins sufficient to target substrates for proteasome-mediated degradation17. Protein degradation by the ubiquitin-proteasome system is an important regulator of the composition of the proteome18. As such, the ubiquitin-proteasome system is required for homeostasis under constant conditions as well as rapid cellular responses to altered external conditions19. Protein degradation is often a critical signal in cells. For example, destruction of cyclins serves as the signal for progression through each step of the cell cycle20; and degradation of IκB serves as a key signal in many immune responses21. Disruptions to protein degradation pathways can lead to a variety of disorders including neurodegeneration22 and cancer19. Protein degradation pathways have emerged as promising targets for therapeutic drugs, including proteasome inhibitors that are currently in clinical use as anti-cancer agents23.
Because of its central role in mediating eukaryotic physiology, ubiquitin has been carefully analyzed by many approaches providing the opportunity to interpret fitness analyses in regards to a wealth of available structural and biochemical information. In particular, non-covalent binding interactions are critical to ubiquitin function. For example, the covalent attachment of ubiquitin depends on non-covalent interfaces between conjugating enzymes and ubiquitin24; 25; 26; 27. After covalent attachment to substrates most known functions of ubiquitin, including delivery of substrates to the proteasome, are mediated by non-covalent binding to ubiquitin-binding proteins28; 29; 30. There are many different ubiquitin-binding proteins in all eukaryotes and binding to ubiquitin is frequently mediated by a set of modular ubiquitin-binding domains (UBDs). The most common UBDs in both yeast and humans29 are the ubiquitin-interacting motif31 (UIM) that consists of a single α-helix32, and the ubiquitin-associated33 (UBA) domain that forms a three-helix bundle34; 35. Many UBDs bind to a hydrophobic patch on the surface of ubiquitin that includes residues L8, I44 and V7029; 36; 37.
Alanine scanning of the surface positions in ubiquitin successfully demarcated hotspots for ubiquitin binding partners by identifying 16 residues where substitutions prevented yeast growth, the majority of these positions located in the proximity of the L8, I44, V70 hydrophobic patch as well as the K48 and C-terminal sites of covalent linkage37. Of note, the alanine scan used a binary scoring of mutants (presence or absence of observed growth) and did not quantify potential intermediate growth defects nor did it sample the full diversity of possible mutations leaving many questions about the sensitivity of ubiquitin to surface mutations unknown. For example, are conservative mutations (e.g. Ile to Val) to the L8, I44, V70 hydrophobic patch functional, and at positions where alanine substitutions are functional are more severe mutations also tolerated (e.g. Asp to Lys charge reversals)?
In addition to binding, the thermodynamics of ubiquitin folding and unfolding have been subject to careful analysis. Ubiquitin is highly stable to temperature denaturation38 and predominantly populates a folded conformation even when subject to near boiling temperature (90 °C) at pH 4. Though it is a small protein of 76 amino acids, native conformations are sufficiently thermodynamically stabilized relative to unfolded conformations that folding is efficient even for many disruptive mutants in the solvent-inaccessible hydrophobic core including all individual alanine substitutions39, mutations that increase bulk40, and some hydrophobic to polar substitutions41; 42. Compared to their influence on protein folding, the impact of core mutations on ubiquitin function has not been thoroughly investigated. Of the few core mutants that have been studied functionally, we have previously analyzed Leu to Ser mutations at positions 67 and 69 near the C-terminus of ubiquitin. Both of these substitutions were capable of folding, but weakened binding affinity to proteasome receptors, resulted in increased accumulation of high molecular weight protein species in cells, and failed to support yeast growth41. These results indicated that small changes to the native structure or dynamics of ubiquitin can impair function.
To comprehensively examine both the sensitivity of the ubiquitin surface to mutation and the impact of core mutations on function, we analyzed the impacts of all ubiquitin point mutants on yeast growth rate. On the surface of ubiquitin, there were ten ultra-sensitive positions where only the wild type amino acid was observed to support robust growth. We also observed a cluster of ultra-tolerant positions on the α-helical face of ubiquitin where virtually all amino acid substitutions were compatible with robust growth. Structural analyses of 44 high-resolution co-crystal structures of ubiquitin bound to different partners indicated that burial at interfaces was a good predictor of sensitivity to mutation at surface positions. In the solvent-inaccessible core of ubiquitin hydrophobic substitutions were generally tolerated. Comparison of mutant effects on growth with previously determined effects on folding stability indicated that some mutants capable of folding were defective for growth. Functional sensitivity to mutation was asymmetrically distributed in the core. Core positions near the C-terminus where many critical binding interactions occur were the most sensitive to mutation. These findings indicate that binding interactions are a dominant contributor to ubiquitin function, which can be impacted by subtle conformational changes and/or dynamics in the folded state of ubiquitin.
Results and Discussion
Bulk competition of ubiquitin mutants in a shutoff strain
To facilitate the analyses of mutants with varied fitness, we used the Sub328 ubiquitin shutoff strain15. In Sub328, the only copy of the ubiquitin gene is expressed from a galactose regulated promoter that generates sufficient ubiquitin protein to support robust growth in galactose media, but that is effectively turned off in dextrose media. These properties enable Sub328 cells to host libraries of ubiquitin mutants in galactose media where growth does not require mutant function, and subsequently switch to dextrose media where growth is directly related to mutant function (Figure 1a). To characterize the timing of the shutoff process, we examined the growth of Sub328 cells harboring either a rescue plasmid constitutively expressing WT ubiquitin (utilizing a promoter and plasmid system previously developed to analyze ubiquitin mutants in yeast15), or a control plasmid lacking ubiquitin (Figure 1b). Cells with the rescue plasmid grow rapidly in dextrose media, but cells with the control plasmid stall in growth after about 10 hours in dextrose media. Based on these results, we decided to analyze selection on mutant libraries starting after 12 hours in dextrose so that most cells without functional mutants would have stalled in growth.
Figure 1.

Bulk competition analyses of the effect of ubiquitin mutants on yeast growth. (a) Experimental setup: systematic libraries of ubiquitin point mutants generated using saturation mutagenesis at sequential positions within a 9–10 amino acid window were generated on a plasmid with a constitutive promoter. These libraries were introduced into a ubiquitin strain whose only other source of ubiquitin was regulated by a galactose dependent promoter. Yeast libraries were amplified in galactose, and then competed in dextrose where growth relied on the mutant ubiquitin library. (b) Growth of the ubiquitin shutoff strain is rescued by constitutive expression of WT ubiquitin. (c) Positions 40–48 of ubiquitin were selected for initial method development. (d–e) Sequence based analyses of bulk competition of libraries of ubiquitin point mutants at positions 40–48. (d) Stop codons were rapidly depleted indicating that they were unable to support growth, while silent substitutions that change the nucleotide sequence without altering the protein sequence persisted in shutoff conditions. (e) Correlation between measured growth effects of mutants (selection coefficient) from full experimental repeats.
We examined the robustness of bulk competitions by analyzing a nine amino acid region (Figure 1c) of ubiquitin that included K48, the essential lysine involved in forming poly-ubiquitin chains that target substrates to the proteasome. The size of this region enabled it to be efficiently interrogated by Illumina short read (36 base) sequencing. We generated site saturation libraries for each position in the region, mixed libraries to create a combined library for the region and used focused deep sequencing5 to analyze the relative abundance of each point mutant in the combined library. The library was introduced into yeast, expanded in galactose media for 48 hours, and then switched to dextrose media for 50 hours. Samples from the library of yeast were saved at different time points in the competition and the relative abundance of mutants over time determined by sequencing, providing a direct measure of relative mutant fitness6. The fitness effects of WT synonyms (silent mutations) and stop codons (nonsense mutations) served as important internal positive and negative controls (Figure 1d). WT synonyms persisted in the bulk competition consistent with the near-neutral expectation for silent mutations. In contrast, stop codons rapidly decreased in relative abundance consistent with the critical function of the C-terminus of ubiquitin in conjugation to substrates 43. The slope of mutant to WT ratio versus time in WT generations was calculated and represents the selection coefficient (s) where s=0 indicates wild type growth and s=−1 indicates a null mutant. The rapid drop off in abundance of strongly deleterious mutants meant that this class of mutant could not be quantified as precisely as mutants that persisted in the culture. We performed a full experimental repeat to judge the reproducibility of our bulk fitness measurements. Excluding strongly deleterious mutants (s<−0.5), we observed a strong correlation between repeat measures of the effects of ubiquitin amino acid substitutions on yeast growth rate (Figure 1e). Compared to fit mutants, strongly deleterious mutants (s<−0.5) showed larger differences in the experimental repeat (Supplementary Figure 1A). Excluding strongly deleterious mutants, the correlation between repeat measurements indicates that we can accurately resolve growth differences of about 7%. This level of resolution is valuable for investigating the physical constraints on ubiquitin function. However, it is not sufficient to distinguish the full spectrum of selection that would act on natural populations where fitness effects on the order of the inverse of the effective population size (estimated at 10−7 in yeast44; 45) are subject to effective selection.
Analyzing mutants across the ubiquitin coding sequence
By investigating multiple different regions in parallel (Figure 2), we were able to analyze all positions in ubiquitin at the same time. We separated the ubiquitin gene into eight regions each encoding 9–10 amino acids that were amenable to our sequencing-based approach (Figure 2a and Supplementary Table S1). For each region, we generated site saturation libraries that we introduced into shutoff yeast and analyzed by bulk competition and sequencing. Utilizing this approach, we determined fitness effects across the ubiquitin coding sequence (Figure 2b). In order to assess reproducibility and selection in each region we analyzed both WT synonyms and stop codons (Supplementary Figure 1B). In all regions, WT synonyms were consistently highly fit (s 0) with narrow distributions (standard deviations ranging from 0.005 to 0.03). These observations indicate that highly fit mutants are accurately interrogated by our procedure as expected because they persist in the culture and are sampled throughout the competition experiment. The average fitness effects of stop codons is similar in each region (Supplementary Figure 1B), but with increased measurement variation. In all regions the average stop codon is highly deleterious (s<−0.65) indicating that selection is strong across all regions. Because highly deleterious mutants rapidly deplete from the culture, they are not sampled as extensively as other mutants and measurement accuracy has an increased dependence on the synchronization of selection pressure across the yeast culture and the number of sequence reads at the early selection time points. The variation in measurements of stop codons is approximately ten times greater than WT synonyms (standard deviation ranging from 0.07 to 0.1 for seven of the regions). Because of unintended variations in sequencing depth, one region (positions 49–58) had markedly lower number of reads for the early timepoints (Supplementary Table 1). In this region, the average stop codon was strongly deleterious (s=−0.75), but as expected measurement variation was large (standard deviation of 0.37). With the exception of this region, the experimental measure of highly deleterious mutants is reasonably precise across the dataset.
Figure 2.

Analyses of the growth effects of mutants across the ubiquitin gene. (a) The gene was subdivided into eight regions of 9–10 amino acids and each region was subject to saturation mutagenesis, bulk competition in yeast, and deep sequencing analyses. (b) Heat map representation of the effects of ubiquitin mutants on yeast growth. Mutants that were below a conservative detection limit at the beginning of the competition were omitted from fitness analyses. (c) Bi-modal distribution of observed mutant effects on yeast growth indicates that most mutants supported either WT-like or null growth in yeast (d) Distribution of growth effects for mutations that depleted by more than 2-fold during outgrowth in galactose media, but remained sufficiently abundant to quantify fitness. Most depleted mutants had null-like fitness and none were WT-like (s>−0.1). (e) Correlation between the growth rate of a panel of individually analyzed mutants relative to fitness measures from bulk competitions.
For amino acids encoded by multiple codons, we calculated fitness as the average over all synonyms (Supplementary Table S2). Across the entire data set, the fitness effects of all nucleotide changes that encode the same amino acid were similar indicating that protein sequence had a dominant impact on fitness compared to nucleotide sequence. We were able to directly analyze selection coefficients for the majority of possible amino acid substitutions (85% - colored in Fig 2b). These quantified mutants exhibited a bi-modal distribution of fitness effects (Figure 2c), which has been commonly observed in many different fitness studies 2; 46; 47; 48; 49.
At the first time point analyzed in shutoff selection, the relative abundance of some mutants was below our threshold for accurate analysis (colored grey in the heat map). We considered two potential explanations for this low mutant abundance: poor representation in the saturation mutagenesis, and/or depletion during growth in galactose where WT ubiquitin was co-expressed. We deep sequenced the plasmid pool and found that virtually all point mutants (99%) were represented in the plasmid library at relative abundances above our threshold for analysis (Supplementary Table S3). Sequencing of yeast samples obtained immediately following amplification in galactose media revealed that many mutations were depleted, indicating that they had a dominant negative growth defect. We observed greater than 2-fold depletion for 95% of mutants that were below the threshold level for fitness analysis (grey boxes in Figure 2b). We took advantage of mutants that were highly represented in the plasmid library to provide a dataset of mutants that depleted during co-expression with WT ubiquitin, but whose relative abundance after outgrowth was sufficient to enable accurate fitness measurements. These depletion-prone mutations are universally unfit (Figure 2D), indicating that depleted mutations that were not over-represented in the plasmid library (solid grey boxes in Figure 2b) are likely unfit. The dominant negative growth effects of ubiquitin mutants are both intriguing and mechanistically unclear as they occur at multiple different structural locations. Experiments focused on these mutants will likely be an exciting area of future research. As indicated in Figure 2b, a small number of mutations were low in abundance in the plasmid library or introduced an internal restriction site that interfered with sample processing (further described in the Methods section).
The bulk fitness measurements are in agreement with the known function of ubiquitin. For example positions 48 and 76 are sites of critical for covalent ubiquitin-ubiquitin linkages and are known to be sensitive to mutation13. In our EMPIRIC analyses, only the WT amino acids at positions 48 and 76 (outlined in dashed red lines in Figure 2b) are compatible with robust growth. There are three amino acid substitutions between the yeast and human versions of ubiquitin (outlined in maroon in Fig 2b). Consistent with the strong functional conservation in ubiquitin, all of these substitutions support robust yeast growth. To further probe the accuracy of our bulk competition measurements, we constructed 17 individual point mutations spread across the ubiquitin coding sequence and analyzed their growth rate in monoculture (Supplementary Figure S1). We find that monoculture growth rate was strongly correlated with EMPIRIC fitness measurements (Figure 2e).
Sensitivity to mutation on the surface of ubiquitin
We examined the tolerance of each position by quantifying the number of amino acids compatible with robust growth, defined here as having less than a 10% growth defect. This cutoff definition was chosen both because it is larger than our measurement precision and because it encompasses the main peak of near-WT fitness mutants (Fig 1c). This definition should include essentially all experimentally fit mutants in the robust class and minimize the number of mutants whose classification would switch with a small change in the cutoff. For amino acids located at or near the solvent-accessible surface, the observed tolerance was bimodal (Figure 3a). The majority of positions permitted either greater than 16 amino acids (and were classified as tolerant), or less than 5 amino acids (and were classified as sensitive). Of note, position 12 was classified as intermediate despite having only four fit amino acids because unusually poor mutant representation in the plasmid library hindered fitness quantification of 10 amino acids. Mapping to the mono-ubiquitin structure revealed that sensitive and tolerant positions clustered completely on opposite faces (Figure 3b). The fitness-sensitive positions are located on the β-sheet face, which contains the hydrophobic patch36 including L8, I44, and V70. This region is known to bind to many ubiquitin receptors.
Figure 3.

Effects of mutants on the solvent-accessible surface of ubiquitin on yeast growth. (a) Distribution of the number of amino acids observed to support growth within 90% or greater of wild type ubiquitin. Many positions in ubiquitin are either highly sensitive to mutation (4 or less amino acids support robust growth), or highly tolerant (17 or more amino acids support robust growth). (b) Space filling representations of ubiquitin structure (based on 1UBQ.PDB67) with sensitive positions colored blue, tolerant yellow, and intermediate green. (c) Heat map representations of sensitive, intermediate and tolerant positions on the ubiquitin surface. One-dimensional maps on the bottom compare our analyses with a previous alanine scan37. At positions where the WT amino acid is alanine, glycine substitutions are shown for both the EMPIRIC and previous alanine scan.
We compared the EMPIRIC fitness map for sensitive, intermediate and tolerant positions on the surface of ubiquitin to a previous alanine scan of the ubiquitin surface37 (Figure 3c). Overall, the EMPIRIC results correspond very well to the previous alanine scan. Of the 16 alanine scan mutants identified as growth defective, all have at least a 20% growth deficiency in our analyses. Of the 41 alanine scan mutants identified as supporting growth, 36 exhibited robust growth in our analyses while five exhibited growth defects ranging from 11% to 47%. The small number of discrepancies could be due to differences in experimental detail including the strains analyzed and the growth conditions. The alanine scan effectively identified sensitive sites, but did not distinguish tolerant sites from sites with intermediate sensitivity. By quantifying the effects of mutants and examining all possible substitutions, the EMPIRIC analyses presented here defines a continuous spectrum of mutational sensitivity from ultra-sensitive to ultra-tolerant.
The most tolerant positions in ubiquitin can accommodate almost any amino acid substitution without disturbing observed function (Figure 2c). These ultra tolerant positions cluster on one side of ubiquitin. The only mutations other than stop codons observed to reduce function were proline mutations within the α-helix that are known to disrupt helical structure, as well as minor defects for a small fraction of the possible hydrophobic substitutions (S28F, S28I, Q31I, P37W, P37F, P37Y, D39W, T55W) and a small number of charge reversals (D21K, D21R, D39R). Charge reversals are generally well tolerated on this face of ubiquitin (e.g. E16, E18, D39, D52, R54, and K63). The tolerance to charge reversal mutants, which dramatically change the interaction potential of a protein surface, suggests that this face of ubiquitin is not involved in critical binding interactions. The entire amino acid sequence of ubiquitin is highly conserved (there are only three substitutions between yeast and human ubiquitin), indicating that mutations throughout the protein impact fitness on a magnitude that is selectable in natural populations. Of note, the WT residues at the ultra tolerant positions are never aliphatic or aromatic. Based on these properties (polar and tolerant of individual mutations) we speculate that this region of ubiquitin may perform a solubility promoting role as similar properties have been observed in solubility promoting regions of the Hsp90 chaperone50; 51. These studies in Hsp90 support the idea that stringent natural selection for stability can result in highly optimized sequences that are so soluble that they are robust to individual mutations when measured with experimentally limited sensitivity. Ubiquitin is highly soluble (>100 mg/ml) and has been shown to impart solubility on genetically fused partner proteins52. In principle, ubiquitin solubility could provide a functional benefit by influencing the solubility of covalent complexes with substrates. The ultra-tolerance of this surface of ubiquitin to our experimental analyses is notable and motivated our discussion of solubility, which we acknowledge is highly speculative.
At the most sensitive positions to mutation (G10, R42, G47, K48, H68, R72, L73, R74, G75, G76), only the WT amino acid was observed to support robust growth. All ten ultra sensitive positions are located on the surface of the ubiquitin structure. Seven of these positions are either at or adjacent in primary sequence to the sites of critical covalent attachment (G47, K48, R72, L73, R74, G75, G76) consistent with the known role of these positions in conjugation to substrates and promoting recognition by the proteasome13. Of the other three ultra-sensitive positions, G10 is located in a β-turn in structural proximity to the C-terminus and has a positive main chain φ angle that is incompatible with other amino acids. The other two (R42 and H68) are both structurally adjacent to the hydrophobic patch formed by L8, I44, and V70 that is at the interface with many ubiquitin receptors. L8, I44, and V70 can all tolerate conservative substitutions (e.g. Leu to Ile) in our assay. These results are consistent with both the binding of partner proteins to this patch as well as the relatively low specificity of hydrophobic interactions relative to polar interactions such as hydrogen bonds that are highly directional. Of note, the contributions of H68 to function was unclear from alanine scanning as it has only a partial (20%) growth defect in our analyses (Figure 3c) and was positive for growth in plasmid swap experiments37.
Mapping fitness sensitivity to interfaces
Analyzing the effects of all ubiquitin mutants combined with the available structural information on interfaces with many different binding partners provides a unique opportunity to investigate how binding interactions influence sensitivity to mutation. Based on chemical intuition, it has long been posited that interaction surfaces will impose evolutionary constraints based on the prediction that many mutations located at interfaces will disrupt binding53. Consistent with previous observations, the most sensitive sites to mutation map to known binding interfaces including those with UBA and UIM domains (Figure 4a,b). To further examine the relationship between binding interfaces and sensitivity to mutation, we analyzed the surface area buried by each position in ubiquitin across 44 high resolution crystal structures (Supplementary Table S4). The average fraction of surface area buried was greater for positions that were sensitive to mutation in our screen compared to positions that were tolerant to mutation (Figure 4c). Across all surface positions, the fraction of surface area buried at structurally characterized interfaces predicted about 60% of the variance in observed tolerance to mutation (Figure 4d). Of note, the relationship between surface burial and tolerance to substation appears to have multiple phases. At low fraction surface burial many mutations are tolerated and at a threshold around 0.3, amino acid tolerance tends to decrease. Positions with a fraction of surface area buried greater than 0.4 are universally sensitive in our analyses. Consistent with these observations, the data fit well to a transition switching model related to those used in chemical denaturation of proteins (Figure 4d). These observations demonstrate that binding is a dominant determinant of sensitivity to mutation for ubiquitin and provide quantitative support for a long-standing intuition in molecular evolution.
Figure 4.

Relating fitness sensitivity on the surface of ubiquitin to binding interfaces. Structural representations of ubiquitin bound to common binding domains: (a) UBA domain (2OOB.PDB71), and (b) UIM domain (1QOW.PDB72). Top images show binding domains in magenta and ubiquitin as space-filled spheres with fitness sensitive positions in blue, fitness tolerant positions in yellow, and intermediate positions in green. Bottom images illustrate the underlying ubiquitin secondary structure. (c) Fraction of surface area buried per sensitive or tolerant residue on the surface of ubiquitin in 44 high-resolution co-crystal structures. Error bars represent standard deviations (N=18 and 19 respectively) (d) Correlation between fitness tolerance to amino acid substitution and burial at structurally characterized interfaces.
We further analyzed how sensitive and tolerant positions mapped to the structure of K48-linked tetra-ubiquitin because this is the minimal signal for proteasome-targeting17. The structure of tetra-ubiquitin in the proteasome-bound state is currently unavailable, as is the structure of poly-ubiquitin attached to a substrate. However, the structure of unanchored K48-linked tetra-ubiquitin representing the predominant conformation at physiological conditions is available54. Structural analyses of this “closed” conformation of tetra-ubiquitin showed that fitness-sensitive positions all cluster on the interior of tetra-ubiquitin, while fitness-tolerant positions all cluster on the exterior (Figure 5). This result suggests that the structural arrangement mediated by inter-ubiquitin contacts in closed tetra-ubiquitin may be biologically important. For example by presenting a molecular surface that is almost entirely polar, the closed conformation of tetra-ubiquitin may enhance solubility. In addition, the sequestration of binding sites for UBDs in the closed conformation may be important for modulating access to these interfaces by different effectors55. This potential mechanism is consistent with both an observed “open” conformation of K48-linked tetra-ubiquitin56 and from NMR studies demonstrating that binding by UBDs can require access to interfaces unavailable in the “closed” conformation57.
Figure 5.
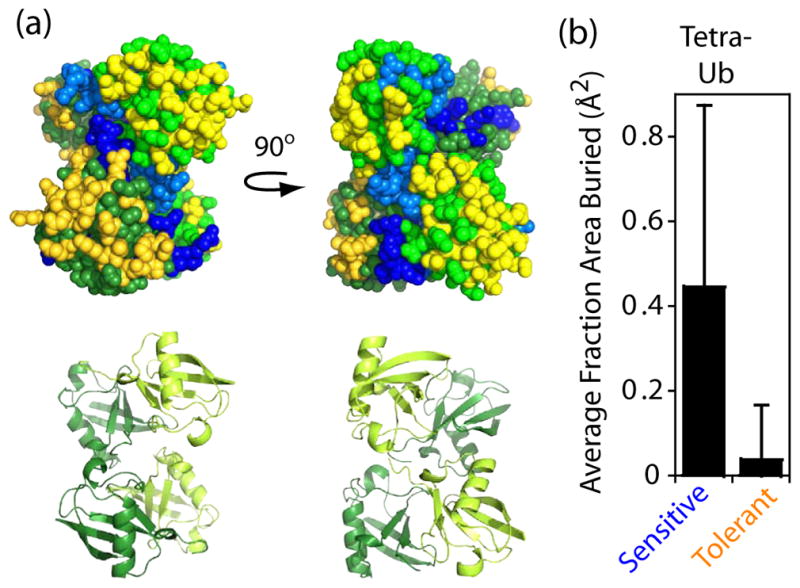
Relating fitness sensitivity to structure of tetra-ubiquitin (a) Structural image of K48-linked tetra-ubiquitin (2O6V.PDB54). Top images show space-filling representation with fitness sensitive positions colored blue, tolerant positions yellow, and intermediate positions grey. Different color shades were used to distinguish subunits. Bottom image illustrates the underlying secondary structure. (b) Fractoin surface area buried per sensitive or tolerant residue in tetra-ubiquitin. Error bars represent standard deviations (N=18 and 19 respectively)
Effects of Mutations in the Solvent-inaccessible Core
The majority of positions (10 of 16) located in the core of ubiquitin tolerated 3–6 mutations (Figure 6a,b) with substitutions between hydrophobic amino acids of different geometry frequently resulting in robust growth at all positions. This distribution of tolerated mutations is consistent with solvent-inaccessible residues contributing to a well-packed, largely hydrophobic core to energetically distinguish the native state from unfolded conformations. Aliphatic (Val, Leu, Ile) to aromatic (Phe, Tyr, Trp) substitutions that increase core bulk were generally poorly tolerated. For example, we did not observe any aliphatic to Trp substitutions that were compatible with robust growth. In the core mutations to Trp were only tolerated at positions where the WT amino acid was aromatic (F45, Y59). These observations indicate that large increases in core over packing, which are likely to alter the native conformation and dynamics, commonly result in functional defects to ubiquitin.
Figure 6.

Mutant effects in the solvent-inaccessible core of ubiquitin. (a) Heat map indicating the fitness of mutations at core positions indicates that substitutions among aliphatic amino acids are generally well-tolerated. (b) Positions in the core exhibit an intermediate tolerance to mutation with most positions having 3–6 different amino acids that support growth rates similar to the wild type sequence (s>−0.1). (c) Structural representation of ubiquitin showing the wild type side chains of core positions. Positions that tolerate more than eight amino acids (s>−0.1) are colored in yellow. (d) Relationship between core mutant impacts on folding stability39; 42 and yeast growth. Previously measured effects on ΔΔG of folding are plotted such that negative numbers represent destabilization. The amount of destabilization estimated to abolish folding is indicated as a dashed grey line on the left. Mutations to Q41, the only WT core polar amino acid, are shown in grey. All mutations in this panel are estimated to populate the unfolded state less than 1% in the absence of elevated temperature or denaturant based on the stability of wild type ubiquitin39. Mutants that are destabilized by more than 2 kcal/mol are shown in orange if they are highly fit (s>−0.12), blue if they are strongly deleterious (s<−0.49), or purple for those with intermediate fitness. (e) Structure of ubiquitin indicating the location of destabilized and highly fit (yellow) as well as destabilized and deleterious (cyan) mutations.
Polar amino acids in the core are generally incompatible with efficient ubiquitin function. Q41 is the only polar WT amino acid in the core. The side-chain of Q41 hydrogen bonds to a solvent-inaccessible and otherwise unsatisfied main-chain carbonyl oxygen. While most polar substitutions at position 41 exhibited a strong growth defect, multiple aliphatic substitutions (Leu, Ile, Met) were tolerated. These findings are consistent with the energetic penalty for burying unsatisfied hydrogen bonding atoms in the core of proteins as well as the energetic benefit from burial of hydrophobic atoms58; 59. The energetic penalty for burial of charged amino acids with unsatisfied hydrogen bonds in the core of proteins is especially severe and we observe growth defects at all core positions for substitutions to R, K, D, or E. The general trends that we observe in the core are consistent with a large body of work demonstrating that the interior of proteins is important for governing protein folding and dynamics60.
Within the core of ubiquitin, we observe that the sensitivity to mutation is unevenly distributed with the most tolerant positions (L15, V17, I23, F45) all clustered in one structural region (Figure 6c). We considered two potential explanations for this observation. First, protein folding may be a dominant determinant of the function of ubiquitin core mutants and the tolerant regions are less important for folding. Second, core mutations may impact ubiquitin function by subtle changes to the folded conformation and/or dynamics that affect critical binding interactions. The contribution of core positions to ubiquitin folding has been determined by φ analysis, which uses alanine mutations to identify residues that contribute to the folding transition state39. Of note, a broader folding transition state of ubiquitin was reported in studies using engineered double histidine substitutions and divalent metals as probes61. By φ analysis, the transition state for folding was found to occur in the same core region where we observe relatively high functional tolerance to mutation. In particular L15, V17, and I23 all have φ-values ≥ 0.5 (F45 was not analyzed) indicating that they have a large energetic contribution to the folding transition state. In contrast positions located near the C-terminal tail in the structure of ubiquitin all were observed to have φ-values close to zero indicating that they provide minimal energetic contributions to the folding transition state. These observations indicate that critical positions for ubiquitin folding can functionally tolerate more mutations than other core positions. Because protein folding to native conformations of ubiquitin should be required for function, this finding indicates either that the transition state for folding does not correlate with mutant effects on folding efficiency, or that ubiquitin core mutants may also have important impacts on protein behavior other than folding (e.g. binding).
We compared growth effects of core mutations to previously reported39; 42 observations of individual ubiquitin mutant impacts on the thermodynamic stability difference between folded and unfolded conformations (Figure 6d and Supplementary Table S5). All of the mutants in this panel were able to fold efficiently and populated native conformations under physiologically relevant conditions, but exhibited growth rates from WT to null in our assays. This observation indicates that some core ubiquitin mutants able to fold efficiently are functionally defective. Our previous work demonstrated that the L67S and L69S core mutations are capable of folding under physiological conditions, but are defective for binding to proteasome receptors and do not support yeast growth41. Thus core ubiquitin mutations capable of folding can be defective for binding to important receptors. In future studies, it will be interesting to determine the specific biochemical defects in core ubiquitin mutants that impair function. Of note, mutations that resulted in less than a 2 kcal/mol destabilization of the folded relative to unfolded states exhibited small to no observable growth defects. Within this stability region, mutations at Q41 had more pronounced functional defects than mutations at positions where the WT amino acid was hydrophobic. This observation is consistent with the observation that polar interactions in protein interiors can have a large influence on protein dynamics59.
Among the analyzed panel of core mutations that destabilized the folded state by 2 kcal/mol or greater there was a large variance in functional effects spanning from null to WT growth rates (Figure 6d). Within this stability regime, we mapped mutants with either minimal or severe growth effects onto structure (Figure 6e). The destabilized mutants that supported the most efficient growth included substitutions at three of the most functionally tolerant positions (L15A, V17N, I23A – mutants at F45 were not in the stability dataset). Mutations with a similar range of destabilization that exhibited severe growth defects were clustered near the β-sheet surface and C-terminus in the structure of ubiquitin (Figure 6e). Thus, core positions that are relatively sensitive to mutation were located adjacent to surface positions that make critical binding interfaces while tolerant core positions were located distant to known binding interfaces. Based on these observations, we speculate that many core ubiquitin mutations may impact function by affecting binding affinities. Interestingly, NMR studies have demonstrated that the C-terminal region of ubiquitin exhibits uncorrelated conformational dynamics62; 63 suggesting that it is capable of sampling many different conformations that could be important for binding to diverse UBDs. Indeed, recent reports indicate that distinct ubiquitin conformations mediated by core amino acids are important for affinity with different binding partners64. NMR studies on linked ubiquitin also observed conformational dynamics in the C-terminal region63, consistent with a potential role in mediating receptor binding to poly-ubiquitin.
Conclusion
The comprehensive analysis of ubiquitin mutants presented here provides a rigorous examination of the physical constraints at each position in the protein. Consistent with previous observations, our results strongly support binding as a dominant functional constraint for ubiquitin both for surface positions as well as for many core positions. Indeed, location at structurally characterized interfaces alone is a good predictor of the tolerance of surface positions to mutation. One face of ubiquitin that is not commonly at structurally characterized interfaces, tolerates almost all substitutions without causing detectable growth defects. The experimental tolerance that we observed for this surface indicates that its contribution to function is relatively insensitive to mutation. This mutational profile together with the polar composition of this region is consistent with a role in promoting solubility. We also find that the functional sensitivity to core mutations is asymmetrically distributed. Sites involved in the folding transition state are the most tolerant to mutation while sites in structural proximity to critical binding sites are the most sensitive. These results are consistent with an important role for ubiquitin conformational dynamics in mediating binding to critical partner molecules. The structural separation of folding centers and regions important for dynamics that influence binding events may be a design principle utilized by other proteins to balance the requirement for both folding and dynamics required for function.
Materials and Methods
High-throughput EMPIRIC Fitness Measurements
We constructed ubiquitin mutant libraries in a KanMX4-marked yeast high copy shuttle vector (p427) with ubiquitin expression driven by the GPD 1 promoter. To aid in cloning, the ampicillin-resistance gene was removed from the vector, and selection during bacterial cloning was performed with kanamycin (resistance provided by the KanMX4 marker). Of note, this high copy plasmid system is important for expressing ubiquitin at near-physiological levels because the ubiquitin gene is present at multiple chromosomal locations in wild type yeast. Libraries of saturated single codon substitutions in yeast ubiquitin were generated using a cassette ligation strategy in p427GPD as previously described6. To facilitate cloning and subsequent sequencing analyses, the ubiquitin gene was subdivided into eight regions of 9–10 amino acids. Pools of saturated point mutants within each region were generated that could be accurately and efficiently interrogated with short-read Illumina sequencing. To distinguish the growth properties of ubiquitin mutants, we utilized the Sub328 yeast strain15. The sole ubiquitin in these cells is supplied from a galactose regulated promoter.
Pooled plasmid libraries of mutants for each region of ubiquitin were transformed separately into the same batch of Sub328 cells as described65. In order to minimize doubly transformed cells66, a total of 1 μg of plasmid DNA was transformed per 100 μL competent yeast cells, and after recovery the cells were grown for 48 hours at 30 °C in 30 mL of liquid SRGal (synthetic 1% raffinose, 1% galactose) media supplemented with G418 (200 μg/mL). Cultures were diluted to maintain log-phase growth. After 48 hours of selection for G418 resistance in SRGal media, late-log cells were collected by centrifugation, then washed and resuspended in SD (synthetic 2% dextrose) media with G418 and ampicillin (50 μg/mL) to hinder bacterial contamination. Cultures of 100 mL were grown in a shaking incubator at 30 °C for 3 days with dilution to maintain the culture in logarithmic growth. Time point samples were collected throughout this period by centrifuging 2*108 cells, resuspending with 1 mL of water, transferring to a microfuge, pelleting, removing the supernatant and freezing the pellet at −80 °C. Fitness analyses were performed on samples isolated after 12, 15, 19, 23, 29, and 45 hours in dextrose. These fitness analyses were performed over 11 generations as the doubling time of the yeast harboring the WT rescue plasmid was 3 hours under these conditions. A full experimental repeat was performed for the ubiquitin region encompassing amino acids 40–48, including preparation of competent yeast from a separate colony, transformation, growth, and time point sampling.
Plasmid DNA was isolated from yeast pellets and processed for deep sequencing. Frozen pellets were thawed, lysed using zymolyase, and plasmid DNA isolated and prepared for sequencing as previously described 6. An initial PCR reaction was performed to amplify the library version of ubiquitin utilizing primers specific to the p427GPD vector. This PCR product was separated on an agarose gel, excised and purified using a silica column (Zymo Research). A second round of PCR was performed with region-specific primers that added a MmeI cut site immediately upstream of the randomized region and a universal primer binding site 250 bases downstream. The resulting PCR product was purified on a silica column, digested with MmeI and purified again on a silica column. Barcoded adapters were then ligated and samples processed and analyzed as previously described6.
Adapter cassettes with a sticky-end complementary to the resulting MmeI overhang were attached using T4 DNA ligase (New England Biolabs). Ligation adapters included a binding site for universal Illumina primers and a barcode to distinguish the time point and sample. Ligation reactions were separated on an agarose gel and the ligation product excised and purified. A final round of PCR was performed with Illumina primers. To minimize PCR errors, cycles were limited and the high-fidelity Pfusion enzyme (New England Biolabs) was utilized throughout. For each region, a processing control was included that started with a purified plasmid with wild type ubiquitin and was processed identical to time point samples (same number of PCR cycles, etc.). To distinguish mis-reads during sequencing, a sequencing control was also included in all deep sequencing samples. The sequencing control consisted of a region of the Sec61 gene cloned into a plasmid with flanking Illumina primer binding sites. Eight cycles of PCR from this plasmid generated a deep sequencing sample with minimal sequence heterogeneity.
Short-read (36 base) Illumina sequencing was utilized to analyze all time point samples. The resulting FastQ files were analyzed as previously described6 in order to determine the fitness effects of mutants. Processing and sequencing errors were directly estimated in each sequencing reaction from the number of apparent mutants observed in the internal processing and sequencing controls. The average per base error rate including PCR processing and sequencing was 0.005 per base. The majority of these errors (~90%) result in apparent double mutants that were filtered out of the data resulting in accurate abundance measurements of the underlying distribution of mutants in the library. Mutants with a relative abundance (mutant/WT) below 2−10 at the initial time point (12 hours in dextrose) under selection were deemed too noisy for accurate fitness measurements based on visual inspection of mutant versus time trajectories and were omitted from further analysis. In addition, mutants that created an internal MmeI site that would complicate processing were omitted from fitness analyses. The first three time points during library selection (corresponding to 12, 15, and 19 hours in dextrose) were utilized to analyze the rapid drop-off in stop codons and other null-like mutants. For mutations that persisted in the population, fitness effects were determined using all time points in selection. The residuals for each fit were determined to identify problematic mutants that were subsequently plotted, and eliminated from consideration if they showed multi-phasic behavior (< 1% of the data was eliminated using this approach). Depletion ratios were calculated from the relative abundance of mutants observed in sequencing of the plasmid library compared to cells collected at the end of growth in galactose. Mutants with a relative abundance (mutant/WT) below 2−8 in the plasmid library were deemed too noisy for depletion analyses based on visual inspection of trajectories.
Growth Rate of Individual Mutants in Monoculture
We measured the growth rate of 19 different point mutants in monoculture after shutoff in dextrose (Supplementary Figure S1). These mutants were chosen to span a range of EMPIRIC fitness values, and were from regions spanning the entire ubiquitin gene. Single mutants were generated in p427GPD and transformed into Sub328 yeast. Growth rates were determined by monitoring the OD600 during growth under identical conditions to the EMPIRIC bulk competitions (SD media with G418 and ampicillin at 30 °C).
Structural Analyses of Ubiquitin
Ubiquitin positions were characterized as at or near the solvent-accessible surface or in the solvent-inaccessible core based on the crystal structure of mono-ubiquitin (1UBQ.PDB67) and the classification algorithm of Mayo and colleagues68. We analyzed ubiquitin interfaces by quantifying the surface area buried69 by each amino acid in atomic resolution structures in the protein data bank. We searched the Protein Data Bank (PDB) for entries with high sequence identity to ubiquitin, at least 2 chains in the biological assembly, and X-ray resolution of 2.5 Å or better. We identified PDB files that had a BLAST E-value of less than 10−20, and length of alignment between 50 and 100 residues as the selection criteria. We manually curated this list to exclude structures of identical protein complexes (keeping only the highest resolution structure in each case) and structures of mono-ubiquitin, resulting in 117 ubiquitin chains in 44 PDB structures. We used areaimol from the CCP4 v.6.2 package70 to calculate ASA for each residue of every ubiquitin molecule both in the complex and in isolation. The resulting changes in ASA upon complex formation are provided in Supplementary Information (Table S4).
Supplementary Material
Highlights.
Mutations provide a powerful probe of protein mechanism
Bulk competition and deep sequencing was used to monitor ubiquitin mutants
Sensitivity to mutation correlated with binding interfaces at surface positions
In the core, positions near critical binding sites were most sensitive to mutation
Binding interactions impose dominant binding constraints throughout ubiquitin
Acknowledgments
We are appreciative to C.R. Matthews, F. Massi, O. Rando, R. Gilmore, E. Baehrecke, A. Ballesteros, and M. Moore for useful comments. The SUB328 yeast strain was kindly provided by D. Finley. This work was supported in part by grants from the National Institutes of Health (R01-GM083038) and the American Cancer Society (RSG-08-17301-GMC) to D.N.A.B., and by an NIH grant (R01-GM065334) to D.F.
Abbreviations
- UBD
Ubiquitin binding domain
- UIM
ubiquitin-interacting motif
- UBA
ubiquitin-associated domain
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchison CA., 3rd Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- 2.Rennell D, Bouvier SE, Hardy LW, Poteete AR. Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol. 1991;222:67–88. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- 3.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–91. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 4.Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S. High-resolution mapping of protein sequence-function relationships. Nat Methods. 2010;7:741–6. doi: 10.1038/nmeth.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hietpas R, Roscoe B, Jiang L, Bolon DN. Fitness analyses of all possible point mutations for regions of genes in yeast. Nat Protoc. 2012;7:1382–96. doi: 10.1038/nprot.2012.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hietpas RT, Jensen JD, Bolon DN. Experimental illumination of a fitness landscape. Proc Natl Acad Sci U S A. 2011;108:7896–901. doi: 10.1073/pnas.1016024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeBartolo J, Dutta S, Reich L, Keating AE. Predictive Bcl-2 family binding models rooted in experiment or structure. J Mol Biol. 2012;422:124–44. doi: 10.1016/j.jmb.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pitt JN, Ferre-D’Amare AR. Rapid construction of empirical RNA fitness landscapes. Science. 2010;330:376–9. doi: 10.1126/science.1192001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLaughlin RN, Jr, Poelwijk FJ, Raman A, Gosal WS, Ranganathan R. The spatial architecture of protein function and adaptation. Nature. 2012;491:138–42. doi: 10.1038/nature11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc Natl Acad Sci U S A. 2012;109:16858–63. doi: 10.1073/pnas.1209751109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA, Baker D. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol. 2012;30:543–8. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2012;332:816–21. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 15.Spence J, Sadis S, Haas AL, Finley D. A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol Cell Biol. 1995;15:1265–73. doi: 10.1128/mcb.15.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, Varshavsky A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 17.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. Embo J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–71. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 19.Goldberg AL. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans. 2007;35:12–7. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 20.Goebl MG, Yochem J, Jentsch S, McGrath JP, Varshavsky A, Byers B. The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science. 1988;241:1331–5. doi: 10.1126/science.2842867. [DOI] [PubMed] [Google Scholar]
- 21.Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1995;92:10599–603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dennissen FJ, Kholod N, van Leeuwen FW. The ubiquitin proteasome system in neurodegenerative diseases: Culprit, accomplice or victim? Prog Neurobiol. 2012;96:190–207. doi: 10.1016/j.pneurobio.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 23.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14:1649–57. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 24.Whitby FG, Xia G, Pickart CM, Hill CP. Crystal structure of the human ubiquitin-like protein NEDD8 and interactions with ubiquitin pathway enzymes. J Biol Chem. 1998;273:34983–91. doi: 10.1074/jbc.273.52.34983. [DOI] [PubMed] [Google Scholar]
- 25.Lee I, Schindelin H. Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell. 2008;134:268–78. doi: 10.1016/j.cell.2008.05.046. [DOI] [PubMed] [Google Scholar]
- 26.Burch TJ, Haas AL. Site-directed mutagenesis of ubiquitin. Differential roles for arginine in the interaction with ubiquitin-activating enzyme. Biochemistry. 1994;33:7300–8. doi: 10.1021/bi00189a035. [DOI] [PubMed] [Google Scholar]
- 27.Miura T, Klaus W, Gsell B, Miyamoto C, Senn H. Characterization of the binding interface between ubiquitin and class I human ubiquitin-conjugating enzyme 2b by multidimensional heteronuclear NMR spectroscopy in solution. J Mol Biol. 1999;290:213–28. doi: 10.1006/jmbi.1999.2859. [DOI] [PubMed] [Google Scholar]
- 28.Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem J. 2006;399:361–72. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hicke L, Schubert HL, Hill CP. Ubiquitin-binding domains. Nat Rev Mol Cell Biol. 2005;6:610–21. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- 30.Fushman D, Wilkinson KD. Structure and recognition of polyubiquitin chains of different lengths and linkage. F1000 Biol Rep. 2011;3:26. doi: 10.3410/B3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hofmann K, Falquet L. A ubiquitin-interacting motif conserved in components of the proteasomal and lysosomal protein degradation systems. Trends Biochem Sci. 2001;26:347–50. doi: 10.1016/s0968-0004(01)01835-7. [DOI] [PubMed] [Google Scholar]
- 32.Fisher RD, Wang B, Alam SL, Higginson DS, Robinson H, Sundquist WI, Hill CP. Structure and ubiquitin binding of the ubiquitin-interacting motif. J Biol Chem. 2003;278:28976–84. doi: 10.1074/jbc.M302596200. [DOI] [PubMed] [Google Scholar]
- 33.Hofmann K, Bucher P. The UBA domain: a sequence motif present in multiple enzyme classes of the ubiquitination pathway. Trends Biochem Sci. 1996;21:172–3. [PubMed] [Google Scholar]
- 34.Ohno A, Jee J, Fujiwara K, Tenno T, Goda N, Tochio H, Kobayashi H, Hiroaki H, Shirakawa M. Structure of the UBA domain of Dsk2p in complex with ubiquitin molecular determinants for ubiquitin recognition. Structure. 2005;13:521–32. doi: 10.1016/j.str.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Dieckmann T, Withers-Ward ES, Jarosinski MA, Liu CF, Chen IS, Feigon J. Structure of a human DNA repair protein UBA domain that interacts with HIV-1 Vpr. Nat Struct Biol. 1998;5:1042–7. doi: 10.1038/4220. [DOI] [PubMed] [Google Scholar]
- 36.Beal R, Deveraux Q, Xia G, Rechsteiner M, Pickart C. Surface hydrophobic residues of multiubiquitin chains essential for proteolytic targeting. Proc Natl Acad Sci U S A. 1996;93:861–6. doi: 10.1073/pnas.93.2.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sloper-Mould KE, Jemc JC, Pickart CM, Hicke L. Distinct functional surface regions on ubiquitin. J Biol Chem. 2001;276:30483–9. doi: 10.1074/jbc.M103248200. [DOI] [PubMed] [Google Scholar]
- 38.Wintrode PL, Makhatadze GI, Privalov PL. Thermodynamics of ubiquitin unfolding. Proteins. 1994;18:246–53. doi: 10.1002/prot.340180305. [DOI] [PubMed] [Google Scholar]
- 39.Went HM, Jackson SE. Ubiquitin folds through a highly polarized transition state. Protein Eng Des Sel. 2005;18:229–37. doi: 10.1093/protein/gzi025. [DOI] [PubMed] [Google Scholar]
- 40.Benitez-Cardoza CG, Stott K, Hirshberg M, Went HM, Woolfson DN, Jackson SE. Exploring sequence/folding space: folding studies on multiple hydrophobic core mutants of ubiquitin. Biochemistry. 2004;43:5195–203. doi: 10.1021/bi0361620. [DOI] [PubMed] [Google Scholar]
- 41.Haririnia A, Verma R, Purohit N, Twarog MZ, Deshaies RJ, Bolon D, Fushman D. Mutations in the hydrophobic core of ubiquitin differentially affect its recognition by receptor proteins. J Mol Biol. 2008;375:979–96. doi: 10.1016/j.jmb.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loladze VV, Ermolenko DN, Makhatadze GI. Thermodynamic consequences of burial of polar and non-polar amino acid residues in the protein interior. J Mol Biol. 2002;320:343–57. doi: 10.1016/S0022-2836(02)00465-5. [DOI] [PubMed] [Google Scholar]
- 43.Pickart CM, Kasperek EM, Beal R, Kim A. Substrate properties of site-specific mutant ubiquitin protein (G76A) reveal unexpected mechanistic features of ubiquitin-activating enzyme (E1) J Biol Chem. 1994;269:7115–23. [PubMed] [Google Scholar]
- 44.Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–4. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- 45.Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, Tsai IJ, Bergman CM, Bensasson D, O’Kelly MJ, van Oudenaarden A, Barton DB, Bailes E, Nguyen AN, Jones M, Quail MA, Goodhead I, Sims S, Smith F, Blomberg A, Durbin R, Louis EJ. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–41. doi: 10.1038/nature07743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanjuan R, Moya A, Elena SF. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc Natl Acad Sci U S A. 2004;101:8396–401. doi: 10.1073/pnas.0400146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carrasco P, de la Iglesia F, Elena SF. Distribution of fitness and virulence effects caused by single-nucleotide substitutions in Tobacco Etch virus. J Virol. 2007;81:12979–84. doi: 10.1128/JVI.00524-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Domingo-Calap P, Cuevas JM, Sanjuan R. The fitness effects of random mutations in single-stranded DNA and RNA bacteriophages. PLoS Genet. 2009;5:e1000742. doi: 10.1371/journal.pgen.1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perisic O, Xiao H, Lis JT. Stable binding of Drosophila heat shock factor to head-to-head and tail-to-tail repeats of a conserved 5 bp recognition unit. Cell. 1989;59:797–806. doi: 10.1016/0092-8674(89)90603-x. [DOI] [PubMed] [Google Scholar]
- 50.Pursell NW, Mishra P, Bolon DN. Solubility-promoting function of Hsp90 contributes to client maturation and robust cell growth. Eukaryot Cell. 2012;11:1033–41. doi: 10.1128/EC.00099-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wayne N, Bolon DN. Charge-rich regions modulate the anti-aggregation activity of Hsp90. J Mol Biol. 2011;401:931–9. doi: 10.1016/j.jmb.2010.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butt TR, Jonnalagadda S, Monia BP, Sternberg EJ, Marsh JA, Stadel JM, Ecker DJ, Crooke ST. Ubiquitin fusion augments the yield of cloned gene products in Escherichia coli. Proc Natl Acad Sci U S A. 1989;86:2540–4. doi: 10.1073/pnas.86.8.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King JL, Jukes TH. Non-Darwinian evolution. Science. 1969;164:788–98. doi: 10.1126/science.164.3881.788. [DOI] [PubMed] [Google Scholar]
- 54.Eddins MJ, Varadan R, Fushman D, Pickart CM, Wolberger C. Crystal structure and solution NMR studies of Lys48-linked tetraubiquitin at neutral pH. J Mol Biol. 2007;367:204–11. doi: 10.1016/j.jmb.2006.12.065. [DOI] [PubMed] [Google Scholar]
- 55.Varadan R, Walker O, Pickart C, Fushman D. Structural properties of polyubiquitin chains in solution. J Mol Biol. 2002;324:637–47. doi: 10.1016/s0022-2836(02)01198-1. [DOI] [PubMed] [Google Scholar]
- 56.Cook WJ, Jeffrey LC, Kasperek E, Pickart CM. Structure of tetraubiquitin shows how multiubiquitin chains can be formed. J Mol Biol. 1994;236:601–9. doi: 10.1006/jmbi.1994.1169. [DOI] [PubMed] [Google Scholar]
- 57.Varadan R, Assfalg M, Raasi S, Pickart C, Fushman D. Structural determinants for selective recognition of a Lys48-linked polyubiquitin chain by a UBA domain. Mol Cell. 2005;18:687–98. doi: 10.1016/j.molcel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 58.Bolon DN, Marcus JS, Ross SA, Mayo SL. Prudent modeling of core polar residues in computational protein design. J Mol Biol. 2003;329:611–22. doi: 10.1016/s0022-2836(03)00423-6. [DOI] [PubMed] [Google Scholar]
- 59.Bolon DN, Mayo SL. Polar residues in the protein core of Escherichia coli thioredoxin are important for fold specificity. Biochemistry. 2001;40:10047–53. doi: 10.1021/bi010427y. [DOI] [PubMed] [Google Scholar]
- 60.Cordes MH, Davidson AR, Sauer RT. Sequence space, folding and protein design. Curr Opin Struct Biol. 1996;6:3–10. doi: 10.1016/s0959-440x(96)80088-1. [DOI] [PubMed] [Google Scholar]
- 61.Krantz BA, Dothager RS, Sosnick TR. Discerning the structure and energy of multiple transition states in protein folding using psi-analysis. J Mol Biol. 2004;337:463–75. doi: 10.1016/j.jmb.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 62.Massi F, Grey MJ, Palmer AG., 3rd Microsecond timescale backbone conformational dynamics in ubiquitin studied with NMR R1rho relaxation experiments. Protein Sci. 2005;14:735–42. doi: 10.1110/ps.041139505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fushman D, Varadan R, Assfalg M, Walker O. Determining domain orientation in macromolecules by using spin-relaxation and residual dipolar coupling measurements. Progress in NMR Spectroscopy. 2004;44:189–214. [Google Scholar]
- 64.Zhang Y, Zhou L, Rouge L, Phillips AH, Lam C, Liu P, Sandoval W, Helgason E, Murray JM, Wertz IE, Corn JE. Conformational stabilization of ubiquitin yields potent and selective inhibitors of USP7. Nat Chem Biol. 2013;9:51–8. doi: 10.1038/nchembio.1134. [DOI] [PubMed] [Google Scholar]
- 65.Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 66.Scanlon TC, Gray EC, Griswold KE. Quantifying and resolving multiple vector transformants in S. cerevisiae plasmid libraries. BMC Biotechnol. 2009;9:95. doi: 10.1186/1472-6750-9-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vijay-Kumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 A resolution. J Mol Biol. 1987;194:531–44. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- 68.Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–7. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 69.Connolly ML. Solvent-accessible surfaces of proteins and nucleic acids. Science. 1983;221:709–13. doi: 10.1126/science.6879170. [DOI] [PubMed] [Google Scholar]
- 70.Collaborative-Computational-Project. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 71.Peschard P, Kozlov G, Lin T, Mirza IA, Berghuis AM, Lipkowitz S, Park M, Gehring K. Structural basis for ubiquitin-mediated dimerization and activation of the ubiquitin protein ligase Cbl-b. Mol Cell. 2007;27:474–85. doi: 10.1016/j.molcel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 72.Swanson KA, Kang RS, Stamenova SD, Hicke L, Radhakrishnan I. Solution structure of Vps27 UIM-ubiquitin complex important for endosomal sorting and receptor downregulation. Embo J. 2003;22:4597–606. doi: 10.1093/emboj/cdg471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.