

Abstract
Successful reverse engineering of mutants that have been obtained by nontargeted strain improvement has long presented a major challenge in yeast biotechnology. This paper reviews the use of genome-wide approaches for analysis of Saccharomyces cerevisiae strains originating from evolutionary engineering or random mutagenesis. On the basis of an evaluation of the strengths and weaknesses of different methods, we conclude that for the initial identification of relevant genetic changes, whole genome sequencing is superior to other analytical techniques, such as transcriptome, metabolome, proteome, or array-based genome analysis. Key advantages of this technique over gene expression analysis include the independency of genome sequences on experimental context and the possibility to directly and precisely reproduce the identified changes in naive strains. The predictive value of genome-wide analysis of strains with industrially relevant characteristics can be further improved by classical genetics or simultaneous analysis of strains derived from parallel, independent strain improvement lineages.
Keywords: inverse metabolic engineering, whole genome sequencing, transcriptomics, systems biology, yeast, evolutionary engineering
Introduction
Metabolic engineering, the targeted and knowledge-based modification of cellular processes by genetic modification with the aim to improve industrial performance (Bailey et al., 1990; Nielsen, 2001), is a key driver for progress in yeast biotechnology. Novel yeast-based processes for production of a wide range of chemical compounds, ranging from pharmaceuticals to bulk chemicals and biofuels, are intensively investigated and increasingly find their way toward industrial implementation (Nevoigt, 2008; Abbott et al., 2009; Tsuruta et al., 2009; Sauer et al., 2010; Zhang et al., 2011). Fifteen years after the first complete Saccharomyces cerevisiae genome sequence became available (Goffeau et al., 1996), functional genome analysis, quantitative physiology, and systems biology have advanced our understanding of yeast metabolic networks to such an extent that knowledge-based genetic intervention increasingly yields the intended positive impacts on industrial performance. Current developments in automated, high-throughput strain construction and analysis (Wang et al., 2009a; Anderson et al., 2010) and synthetic biology techniques for rapid synthesis and manipulation of DNA sequences (Gibson, 2011) further accelerate progress in knowledge-based metabolic engineering.
Despite the growing number of successes in yeast metabolic engineering, many cases remain in which the current level of understanding is insufficient to achieve the quantum leaps in performance demanded by industry. Knowledge-based engineering of traits such as pathway kinetics, cellular energetics, and robustness represent not only relevant and intellectually stimulating, but also painstaking and time-consuming challenges. Examples of such challenges include the long-running attempts to engineer industrially relevant aspects of yeast physiology such as glycolytic flux (Kern et al., 2007) and tolerance to ethanol and acetic acid (Mira et al., 2010; Stanley et al.,2010a, b). Consequently, there is a growing awareness in academia and industry that fast improvement of microbial strains requires integration of targeted metabolic engineering with modifications that do not a priori target specific genes (from here on referred to as nontargeted approaches) (Lee et al., 2011).
Nontargeted approaches for strain improvement have been a key driver in microbial biotechnology for over half a century. Even in the absence of detailed knowledge of genetics or physiology of the producing strain, their effectiveness is beyond dispute. The paradigm of such ‘classical’ strain development is the huge improvement, over a period of 60 years, of penicillin production by the filamentous fungus Penicillium chrysogenum (Nielsen, 1997; van den Berg et al., 2008). However, nontargeted strain improvement typically leads to a slow, incremental increase in performance, especially in the later stages of strain improvement. Moreover, its ‘black box’ character precludes the rapid transfer of relevant traits among strains or species. To address these limitations, it is essential to identify the genetic changes and mechanisms that underlie the improved performance of strains generated via nontargeted approaches.
In many technological disciplines, ranging from military to medical engineering, the process of elucidating the technological principles of a system via analysis and subsequent reconstruction of its structure and function is known as reverse engineering (Sutton, 1984; Rekoff, 1985). In a seminal paper by Bailey and co-workers (Bailey et al., 1996), this concept was introduced to the field of biotechnology as ‘inverse’ metabolic engineering. However, to maintain consistency with other engineering disciplines, ‘reverse’ metabolic engineering is used throughout this mini-review.
In contrast to the conventional ‘forward’ metabolic engineering cycle, which starts with a knowledge-based design that is subsequently tested by construction and analysis (Fig. 1), reverse metabolic engineering starts out with (an) existing microbial strain(s) with improved performance relative to (a) reference strain(s). High-performing strains can be either isolated from nature, obtained from culture collections, or created through nontargeted strain improvement efforts. Such nontargeted approaches for optimization of yeast strains include the following: (i) random mutagenesis combined with high-throughput selection, such as UV-C mutagenesis of the xylose-fermenting yeast Scheffersomyces stipitis for improved fermentation characteristics under anaerobic conditions (Hughes et al., 2011); (ii) laboratory evolution (‘evolutionary engineering’) under cultivation regimes that have been especially designed to convey a selective advantage to better performing strains (reviewed by Conrad et al., 2011). An example of this approach is the improvement of the fermentation kinetics of genetically engineered S. cerevisiae strains during growth on glucose-xylose-arabinose mixtures via evolutionary engineering (Wisselink et al., 2009); and (iii) the introduction of gene libraries, sometimes after mutagenizing the expressed genes. This has for instance been applied to transcription factor engineering of S. cerevisiae for improved ethanol tolerance (Alper et al., 2006).
Fig. 1.

The ‘forward’ metabolic engineering and ‘reverse’ metabolic engineering cycles and their interaction. In forward metabolic engineering, analysis of strains constructed based on rational design often results in scientific questions that need to be addressed by further analysis and consultation of the rapidly expanding knowledge on microbial metabolism and its regulation. In reverse metabolic engineering, generation and analysis of biodiversity – obviously, with special attention for strains that show improved performance – contributes to accelerated strain improvement and knowledge development (After Nielsen, 2001; Bailey et al., 1996; Bro & Nielsen, 2004).
The next step of the reverse metabolic engineering cycle is the elucidation of the genetic basis for improved performance (Fig. 1). This elucidation should be rigorous and move beyond merely establishing genotype–phenotype correlations. Instead, it should be unambiguously demonstrated that reintroduction of a defined set of genetic changes can wholly or partially reconstruct the improved performance. Reverse metabolic engineering has the added benefit that it enables the extraction of productive mutations, thereby avoiding the accumulation of nonproductive mutations that may occur in prolonged nontargeted strain improvement programmes.
Integration of the ‘forward’ and reverse metabolic engineering cycles can be accomplished in several ways. Once the molecular basis for improved performance, preferably including understanding of the underlying biochemical mechanism, has been elucidated by a reverse engineering approach, this knowledge can be implemented in ‘forward’ metabolic engineering of the same strain lineage or of other strains (Fig. 1). For example, although not trivial, the identified relevant mechanisms can be investigated for their potential in robust industrial strains. Additionally, secondary effects of mutations that not only give a selective benefit, but also have a much broader impact, such as mutations in regulatory networks, can be prevented by only engineering the relevant trait. Strains constructed via ‘forward’ metabolic engineering can, after additional nontargeted modification of their genomes, be re-entered into the reverse metabolic engineering cycle. This can, for example, accelerate the evaluation of optimal gene sequences or pathway configurations (Fig. 1).
The unequivocal and fast identification of the genetic basis of improved performance remains the key challenge in reverse metabolic engineering of yeasts. In the 15 years since the first S. cerevisiae genome sequence was published, the toolbox for integral analysis of yeasts at different information levels (genome, proteome and metabolome) has rapidly expanded. In addition, the decreasing costs of several key analytical technologies are making them increasingly accessible for application in industrial and academic yeast research. This fast progress in tool development brings about a new challenge: How to make informed choices from a wide range of expensive analytical approaches? The goal of the present paper is not to exhaustively review the literature on reverse metabolic engineering of yeast. Instead, by discussing published examples on reverse metabolic engineering of yeast, we will identify advantages and limitations of the genome-wide analytical approaches that are currently available. Emphasis will be on analysis of yeast strains generated in ‘linear’ strain improvement programmes, for example, via classical mutagenesis or evolutionary engineering, rather than on the systematic exploration of yeast biodiversity.
Genome expression analysis
Until recently, the costs of whole genome sequencing precluded its use as a routine laboratory technique in reverse metabolic engineering. Therefore, analysis of the molecular basis of industrially relevant traits has, for the past decade, strongly depended on genome expression studies. The goal of genome expression analysis in the context of reverse metabolic engineering is to correlate expression levels of individual genes with an industrially relevant performance parameter, such as productivity, yield, or robustness. These correlations form the basis for identification of lead genes and/or cellular processes, whose contribution to the phenotype of high-performing strains should subsequently be assessed by targeted genetic modification. As the coverage of state-of-the art proteomics platforms is still incomplete, transcriptome analysis is currently the only widely available means of truly genome-wide analysis of gene expression in yeast. We will therefore mainly focus our evaluation of genome expression for reverse metabolic engineering on transcriptome analysis. The potential added value of a few examples of proteomics and metabolomics in yeast reverse metabolic engineering is briefly discussed in separate paragraphs.
Experimental design for genome expression analysis
DNA microarray analyses (DeRisi, 1997 Daran-Lapujade et al., 2009) and RNA sequencing (Wang et al., 2009b; Ozsolak & Milos, 2011) enable the rapid, quantitative, and inclusive correlation of the yeast transcriptome to environmental or genetic contexts. Furthermore, powerful algorithms enable analysis of the overrepresentation of functional categories (Kresnowati et al., 2006; Huang et al., 2009a, b) and transcription factor-binding sequences (Harbison et al., 2004; MacIsaac et al., 2006) that show a transcriptional up- or downregulation in a given context. Additionally, transcriptome data can be correlated with a vast body of information on the transcriptional responses of S. cerevisiae to a range of environmental parameters and genetic interventions. In the interpretation of transcriptome data, it is important to consider the context dependency of transcriptional regulation in yeast (Knijnenburg et al., 2008).
Cultivation conditions and specific growth rate have a substantial impact on yeast genome expression, which has been especially well documented for transcriptome analysis (Boer et al., 2003; Tai et al., 2005; Regenberg et al., 2006; Abbott et al., 2007; Castrillo et al., 2007; De Nicola et al., 2007; Fazio et al., 2008; Daran-Lapujade et al., 2009). This has important implications for the use of transcriptome data in reverse metabolic engineering. When experimental conditions and/or specific growth rates differ for the strains that are compared, this may generate nonproductive leads, that is, gene expression differences that do not reflect a positive contribution to the industrially relevant phenotype under study. Controlled cultivation in chemostat cultures avoids the impact of the changing environmental conditions that occur in batch cultivation, and the fixed dilution rate eliminates the impact of specific growth rate on transcriptome analyses of different yeast strains and environmental conditions (Daran-Lapujade et al., 2009).
An additional experimental design challenge, which is specifically associated with reverse metabolic engineering, arises when mutagenesis or laboratory evolution leads to ‘gain of function’ phenotypes. Ideally, transcriptome analysis should be performed under conditions where the trait of interest is expressed, but these do not always allow growth of the reference strain. For example, after laboratory evolution of strains for anaerobic growth on pentose sugars or for strongly induced ethanol or acetic acid tolerance (Sonderegger & Sauer, 2003; Stanley et al., 2010a; Wisselink et al., 2010; Wright et al., 2011), the reference strain cannot be grown under the conditions in which the selected phenotype becomes apparent. Consequently, the use of identical cultivation conditions that are permissive for both strains can only yield genes whose transcriptional up- or downregulation in the evolved strain does not depend on the conditions that led to its selection. An example of such ‘constitutive’ expression is the upregulation of transaldolase and transketolase-encoding genes in yeast strains selected for growth on xylose or arabinose (Wahlbom et al., 2003; Wisselink et al., 2010), which could already be observed in a comparison of glucose-grown cultures of the evolved and parental strains. Additional, important transcriptional changes may only be observable under conditions that are nonpermissive for the reference strain. For example, in an evolved l-arabinose-fermenting strain grown on glucose, transcript levels of the GAL regulon were the same as those in the nonevolved strain, presumably as a result of glucose repression (Wisselink et al., 2010). However, very high transcript levels of this regulon were observed during growth of the evolved strain on l-arabinose and, subsequently, linked to the deregulation of the GAL2-encoded transporter, which is responsible for l-arabinose transport in engineered S. cerevisiae strains (Becker & Boles, 2003; Wisselink et al., 2010). Similarly, S. cerevisiae strains whose acetic acid tolerance had been strongly increased by evolutionary engineering required induction by acetic acid to express the acquired hyper tolerance (Wright et al., 2011), thereby precluding a meaningful analysis of gene expression in cultures grown without acetic acid. In such cases, a three-way comparison can be applied by comparing genome expression of both the evolved and reference strains under conditions that are permissive for the reference strain with the evolved strain under the relevant condition (Wahlbom et al., 2003; van Maris et al., 2007; Wisselink et al., 2010). However, the inevitable consequence of such a comparison is that nonproductive leads are likely to be generated as a result of the different cultivation conditions.
Interpretation of transcriptome data: sources of nonproductive leads
Studies in which analysis of genome-wide transcriptional responses were followed up by systematic analysis of the fitness of null mutants suggest that, generally, only a small fraction of transcriptionally responsive genes positively contribute to fitness under the conditions to which they showed a transcriptional response (Winzeler et al., 1999b; Birrell et al., 2002; Giaever et al., 2002, 2004; Tai et al., 2007). Also in transcriptome-based reverse metabolic engineering, the number of responsive genes usually far exceeds the number of productive leads, even when cultivation conditions are rigorously controlled and standardized. Several causes for nonproductive leads will be discussed below.
Nonproductive leads can occur when productive mutations directly or indirectly affect expression of other genes. A direct influence occurs, for example, when the productive mutation affects the in vivo activity of a transcriptional regulator that, in addition to genes that positively affect an industrially relevant phenotype, controls the expression of genes that do not. An example is provided by a study on a S. cerevisiae strain in which the sucrose-hydrolyzing enzyme invertase was relocated to the cytosol to improve ethanol yields on sucrose. Prolonged cultivation in sucrose-limited chemostat cultures led to a drastic improvement of the affinity for sucrose. Analysis of two independently evolved strains revealed increased transcript levels of many genes involved in maltose metabolism, whereas the improved affinity for sucrose could be entirely attributed to upregulation of a single maltose transporter gene (MAL11; Basso et al., 2011). Although the specific mutation responsible for the deregulation of MAL genes in the evolved strain was not identified, it seems plausible that it affected a transcriptional regulator. Similarly, the contribution of the very high expression of the entire GAL regulon in an S. cerevisiae strain evolved for fast l-arabinose fermentation (Wisselink et al., 2010) could be entirely explained from the essential role of Gal2p in l-arabinose transport (see above). The high expression levels of other GAL genes in the evolved l-arabinose-fermenting strain were most probably a side effect of a mutation in a regulatory protein whose primary evolutionary significance was the deregulation of GAL2. Although, in these cases, engineering of the regulator may reproduce the selected phenotype, identification and targeted engineering of the responsible reaction or transport step may be desirable, for example, to minimize protein burden (Snoep et al., 1995).
Another cause for nonproductive leads from transcriptome analysis is the deletion or amplification of multigene DNA fragments. Even in cases where the evolutionary distance between strains is small, as in laboratory evolution experiments with S. cerevisiae, which generally do not involve more than a few hundred generations of selective growth, deletion or amplification of multigene DNA fragments is frequently observed (Brown et al., 1998; Dunham et al., 2002; Jansen et al., 2005; Wisselink et al., 2010; Basso et al., 2011). In such cases, only a single gene on an amplified or deleted fragment may contribute to the phenotype of interest. By plotting transcript levels on a physical map of the yeast genome, amplified or deleted regions larger than a couple of genes stand out from the experimental background noise and can be identified directly. For example, in a transcriptome analysis of an S. cerevisiae strain evolved for fast anaerobic fermentation of l-arabinose, a 250-kb fragment of chromosome VII appeared to be duplicated (Wisselink et al., 2010; Fig. 2). One of the genes on this fragment (YGR043C) encodes a transaldolase isoenzyme and was subsequently shown to contribute to faster arabinose fermentation rates. Although the impact of other genes on the duplicated fragment was not studied, it seems plausible that their increased expression levels reflect ‘collateral damage’ of the duplication event that led to increased expression level of YGR043C.
Fig. 2.
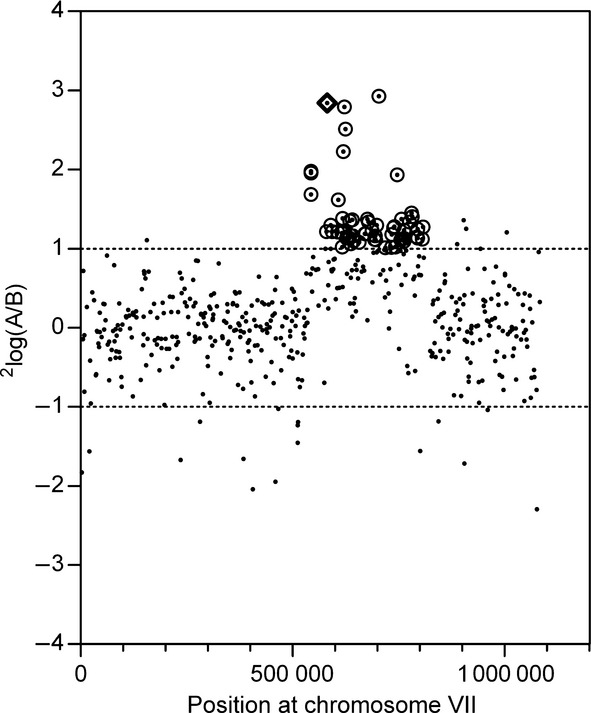
Transcriptome comparison of a genetically engineered Saccharomyces cerevisiae strain and an arabinose-fermenting derivative strain obtained by laboratory evolution. The dotted lines indicate a twofold difference. The circled dots represent genes located between positions 543 555 and 807 659 with an at least twofold increased transcript level in the evolved strain, indicating a duplication of a 250-kb region on chromosome VII. The increased expression level of YGR043C (diamond), encoding a transaldolase, was subsequently shown to contribute to enhanced arabinose fermentation rates in the evolved strain (reproduced with permission from Wisselink et al., 2010).
Genetic differences that are related to the parameter of interest can sometimes be enriched by comparison of different independent strains that share the same industrially relevant phenotype. Of course, this does not provide leads if the evolved phenotype is caused by different mechanisms in the independent evolutions. A successful example of this approach is an early transcriptome-based reverse metabolic engineering study on strains with an evolved freeze resistance (Tanghe et al., 2002). A transcriptome comparison of three freeze-resistant strains with a freeze-sensitive reference strain enabled the demonstration that overexpression of a specific allele of the aquaporin-encoding AQY2 gene led to increased freeze resistance in a naive strain. Recently, transcriptomes of three independently evolved S. cerevisiae strains with increased rates of galactose metabolism were compared with those of the nonevolved ancestor strain (Hong et al., 2011). Expression of genes controlled by the RAS/PKA pathway was found to be affected in all three evolved strains. When a specific point mutation in RAS2 was introduced in the ancestor strain, this led to a significant increase in the specific growth rate on galactose (Hong et al., 2011).
In evolutionary engineering and in classical mutagenesis and selection, the relevant yeast strains generally share a common genetic background. Interpretation of transcriptome data becomes progressively more complicated when the genetic background of strains becomes more diverse. In addition to technical issues (e.g. the design of microarrays), the different ‘wiring’ of transcriptional regulation networks complicates interpretation of transcriptome data. However, even when different strain backgrounds are compared, excellent results are occasionally obtained when analysis is focused on subsets of genes that have been selected based on prior knowledge. A successful example is a transcriptome-based study on the molecular basis of tolerance to hydroxymethylfurfural (HMF), an important inhibitor of yeast fermentation in lignocellulosic hydrolysates (Petersson et al., 2006). On the basis of the knowledge that reduction of HMF to the corresponding alcohol is a key detoxification mechanism, a microarray-based transcriptome analysis of two nonrelated S. cerevisiae strains with different degrees of HMF tolerance focused on oxido-reductase-encoding genes. A set of 15 such genes were expressed at a higher level in the tolerant strain. Individual overexpression of these genes in an HMF-sensitive laboratory strain led to the identification of Adh6p as a major HMF reductase, whose overexpression led to increased rates of in vivo HMF reduction (Petersson et al., 2006). Similarly, a recent example is the identification of ILV6 as a target to reduce diacetyl formation in lager brewers’ yeast through a combination of microarray-based comparative genome hybridization and a transcriptome analysis (Duong et al., 2011).
The potential of proteome analysis in reverse metabolic engineering
A large and growing body of evidence shows that in S. cerevisiae, the correlation between transcript and protein levels is not perfect (de Groot et al., 2007; Wu et al., 2008; Rossignol et al., 2009; Olivares-Hernández et al., 2010; Rossouw et al., 2010), which indicates that, in many cases, regulation occurs at the level of translation and/or protein turnover. Consequently, analysis of gene expression at the level of transcription may overlook important changes, which might be detected by a thorough proteome analysis (Kolkman et al., 2005; Beck et al., 2011). However, we are aware of only very few studies in which proteome analysis has been performed in a reverse metabolic engineering context. A relevant study is a proteome analysis of a S. cerevisiae strain evolved for improved fermentation of xylose (Karhumaa et al., 2009). Because the same strain and its parental strain had previously been studied at the transcriptome level (Wahlbom et al., 2003), this investigation enabled a clear view on the additional information that can be gained from proteome analysis. Strikingly, the leads generated from the transcriptome and proteome comparisons showed very little correlation. Firstly, major increases were observed in the protein levels of the heterologous xylose reductase and xylitol dehydrogenase that were introduced into the ancestor strain via targeted genetic modification. No information on expression levels of their structural genes was obtained in the earlier transcriptome analysis, because they were not represented on the commercial S. cerevisiae microarrays. Secondly, six- to eightfold changes in the levels of several proteins – some of which were involved in key pathways of sugar metabolism – were not accompanied by significant changes in the corresponding mRNA (Karhumaa et al., 2009). This study reinforces the warning that, especially for central metabolic pathways, transcript levels cannot be considered as reliable indicators of either in vivo metabolic activity or protein levels (Daran-Lapujade et al., 2009). Clearly, proteomics analysis has high potential for use in reverse metabolic engineering, but requires further developments, such as increased coverage of the proteome and low-labor, high-throughput methodologies.
Metabolite analysis to support lead generation
Quantitative measurements of intracellular metabolite levels can contribute to the identification of pathways whose capacity controls the rate of substrate consumption or product formation. Even though developments in (intracellular) metabolite analysis progress rapidly (Roessner & Bowne, 2009; Christen & Sauer, 2011), there are currently no methods available that enable the complete and accurate analysis of the yeast metabolome, and interpretation of intracellular metabolite data is complicated by the metabolic compartmentation of yeast cells. Moreover, metabolite analysis shares the challenges in experimental design that are inherent to all gene expression studies, such as context dependency or defining a reference situation for ‘gain of function’ phenotypes. Additionally, changes at the metabolite level alone are never sufficient to identify the underlying molecular mechanism. These limitations notwithstanding, in some studies metabolite analysis successfully resulted in the generation of leads for reverse metabolic engineering.
In genetically engineered strains of S. cerevisiae that were evolved for faster metabolism of xylose, analysis of intracellular metabolite levels indicated that the capacity of the nonoxidative pentose phosphate pathway is a key factor in engineering of pentose-fermenting strains (Zaldivar et al., 2002; Pitkänen et al., 2005). This indication is in line with metabolic engineering studies, which showed that overexpression of key enzymes of the pentose phosphate pathway is indeed essential to achieve high rates of pentose fermentation in S. cerevisiae (Hahn-Hägerdal et al., 2007; van Maris et al., 2007). However, as the metabolite studies cited above were not linked to transcriptome analysis or genome sequencing, improved performance of the evolved strains could not be linked to mutations or altered expression of specific genes.
A recent study on S. cerevisiae strains evolved for faster growth on galactose integrated different analytical approaches (Hong et al., 2011). Decreased intracellular concentrations of glucose-1-phosphate and galactose-1-phosphate, key metabolites of the Leloir pathway for galactose fermentation, coincided with an increased expression of PGM2, which encodes phosphoglucomutase. Identification of PGM2 as a reverse engineering target was validated by previous work, which showed that its overexpression leads to increased rates of galactose metabolism in S. cerevisiae (Bro et al., 2005). In this case, metabolite analyses basically led to the confirmation of targets that would also have been identified by transcriptome analysis.
One of the major challenges in intracellular metabolite analysis is to assess whether altered metabolite concentrations are cause or consequence of an increased flux through a pathway. Integration of metabolite data with, for example, gene expression studies and a thermodynamic analysis can increase their predictive value for reverse metabolic engineering. For example, Wisselink et al. (2010) analyzed an engineered S. cerevisiae strain that was evolved for fast fermentation of l-arabinose via integrated analysis of the transcriptome and intracellular concentrations of intermediates of central carbon metabolism. A thermodynamic analysis based on measured intracellular metabolite concentrations of glycolysis and the pentose phosphate pathway indicated that the driving force for the transaldolase and transketolase reactions was much higher in arabinose-grown cultures of the evolved strain than in glucose-grown cultures of the evolved and parental strains. This suggested a limiting capacity of these two reactions. The two major genes for transaldolase and transketolase (TAL1 and TKL1, respectively) were already strongly overexpressed in the evolved strain owing to previous targeted metabolic engineering (Wisselink et al., 2007). However, transcriptome analysis showed an increased expression in the evolved strain of two genes encoding ‘minor’ isoenzymes of transaldolase (YGR043C, Fig. 2) and transketolase (TKL2). Subsequent knockout studies confirmed the involvement of these genes in the improved arabinose fermentation kinetics of the evolved strain (Wisselink et al., 2010). Although these genes might also have been identified as targets for reverse engineering based on a transcriptome analysis only, their expression level was low relative to those of the ‘major’ TAL1 and TKL1 genes (Wisselink et al., 2010). In the examples discussed above, metabolite analysis led to improved understanding of the impact of various mutations on the biochemistry. Moreover, metabolite analysis provided additional, strong incentives to prioritize mutations for follow-up studies.
Analysis of gene and genome sequences
In contrast to gene expression data, genome sequences of genetically homogeneous (‘pure’) cultures are context independent and offer a direct view on molecular changes at the DNA level. Furthermore, whereas a change in a single transcript, protein, or metabolite often has a drastic impact on the complete transcriptome, proteome, or metabolome, individual mutations will generally have little impact on the likelihood of mutations elsewhere on the genome. One notable exception to this are mutator phenotypes (Thompson et al., 2006; Raynes et al., 2011), in which a genetic change in one gene leads to an increased mutation frequency elsewhere on the genome and which may well be enriched for in classical strain improvement and evolutionary engineering.
Classical methods, such as genomic libraries and transposon mutagenesis, have been instrumental in identifying genotype–phenotype relations (Ross-Macdonald et al., 1999; de Jesus Ferreira et al., 2001; Jin et al., 2005; Ni et al., 2007; Hong et al., 2010). However, these techniques are often laboreous and can only identify dominant mutations. DNA sequencing is a powerful alternative, but the associated costs and the large size of the yeast genome in comparison with prokaryotes have long been prohibitive for its routine use in reverse metabolic engineering studies. Although the number of sequence- or hybridization-based reverse metabolic engineering studies with yeast is small, interesting insights into the potential of a sequence-based approach for the identification of reverse engineering targets that these studies provide is discussed below.
(Re)sequencing of selected genes or plasmids
When available knowledge, models, flux analysis, or expression studies strongly point toward a certain gene or sequence, partial sequencing can sometimes economize the discovery of relevant mutations. This is exemplified by a recent study on the introduction of a heterologous phospho-enol-pyruvate carboxykinase (PCK) into S. cerevisiae as an alternative, ATP-efficient C3→C4 carboxylating pathway. Laboratory evolution was required to enable the heterologous PCK to functionally replace the S. cerevisiae pyruvate carboxylases. Based on a physiological analysis, it was hypothesized that the high activities of pyruvate kinase in S. cerevisiae might compete for phospho-enol-pyruvate with the heterologous PCK. Resequencing of the PYK1 gene in the evolved strain revealed a point mutation, whose introduction in the nonevolved strain led to a reduced pyruvate kinase activity and enabled growth via the heterologous pathway (Zelle et al., 2010).
When heterologous enzymes or pathways are expressed from episomal vectors, it is straightforward to first establish, by plasmid curing and plasmid reintroduction into a naive strain, whether a mutation is chromosomal or plasmid borne. In the latter case, sequencing of the plasmid can be a fast, cost-effective alternative to whole genome analysis. In a recent study, a pyruvate carboxylase–negative mutant of S. cerevisiae, expressing an Escherichia coli malic enzyme gene, was evolved for growth on glucose as the sole carbon source. As in the previous example, the goal of this study was to explore energy-efficient pathways for production of C4-dicarboxylic acids (Zelle et al., 2011). After establishing that, in two independent mutants, the relevant mutations were plasmid borne, two different point mutations were identified in the E. coli gene. These were subsequently shown to drastically affect their redox cofactor preference, thereby enabling them to function in the carboxylating direction and to replace the yeast pyruvate carboxylase (Zelle et al., 2011). Scientific curiosity, time-to-results, and the disproportionality of the cost per base pair of sequencing specific genes or plasmids vs. whole genome analyses in the end determine the choice between these techniques.
Hybridization-based microarray genome analysis
Comparative genome hybridization and oligonucleotide microarrays have proven to be a powerful tool in the discovery of variations between yeast strains (Winzeler et al., 1999a; Daran-Lapujade et al., 2003; Gresham et al., 2006; Schacherer et al., 2007, 2009), ranging from single-nucleotide polymorphisms to structural variations (Dunham et al., 2002; Gresham et al., 2010). DNA hybridization experiments assay the presence of complementary DNA that is present in a sample, usually on a DNA array. All array types, from BAC arrays to tiling arrays, can be used to detect structural variations between the sample and the reference on the chip. The density of DNA probes on the array determines the resolution of the analysis, with resequencing and tiling arrays having the ability to discover single-nucleotide variations (SNVs) (reviewed by Gresham et al., 2008a). Array-based genotyping has long had the advantage over whole genome resequencing that it is faster. Moreover, especially in dynamic experiments and in large comparative studies, the lower costs of array-based techniques provided an advantage (reviewed by Gresham et al., 2008a; but see next paragraph). However, these hybridization-based techniques have the inherent disadvantage that in quality as well as in quantity, they only allow a comparison with sequences that are represented on the microarray. Therefore, when used to analyze strains resulting from mutagenesis or evolutionary engineering, arrays need to be representative for the ancestor strains used in strain improvement. Additionally, while some types of microarray allow for the accurate mapping of the position of SNVs, determination of the exact identity of the sequence variation requires the use of either resequencing arrays or resequencing (see review Gresham et al., 2008a).
Whole genome (re)sequencing of yeast strains for reverse metabolic engineering
The 1000-dollar genome (Mardis, 2006), an iconic target in human genomics, is rapidly becoming a reality for S. cerevisiae. Currently (September 2011), the costs for custom resequencing of a 12-Mb genome with short-read paired-end technology at 40+-fold coverage is about €850 (E. Zeinstra, B., Leiden, the Netherlands; personal communication). In principle, (re)sequencing of S. cerevisiae strains that have been obtained via evolutionary engineering or mutagenesis offers huge opportunities for reverse engineering.
The advantage of whole genome resequencing over array-based techniques with respect to the identification of SNVs was indicated by the discovery of additional mutations of yeast strains (Araya et al., 2010; Kvitek & Sherlock, 2011) that had previously been analyzed by tiling array-based genotyping (Gresham et al., 2008b; Kao & Sherlock, 2008). On the other hand, identification of structural variation with short next-generation sequence reads is challenging with current alignment techniques, but is likely to be solved with further technology improvements (reviewed by Alkan et al., 2011). For the reliable and comprehensive detection of relevant mutations, including structural rearrangements such as indels (insertions and deletions), inversions and duplications, it is not always sufficient to align and compare sequence data of a strain of interest to a reference genome, such as the first published S. cerevisiae genome (strain S288C, Goffeau et al., 1996).
To gain the full benefit of whole genome or whole transcriptome sequencing, it is important to have access to a well assembled and annotated genome sequence of the reference strain that is used as the ancestor in mutagenesis or evolutionary engineering experiments. Although new algorithms enable the de novo assembly of entire S. cerevisiae genomes from short-read sequence information only (see e.g. Nijkamp et al., 2010), reliable ‘gold standard’ genome sequences for reference strains will generally require sequencing of additional libraries. This will include, preferably, longer-read sequencing (classical Sanger sequencing; Roche 454 or new Pacific Biosystems) and mate-paired libraries of several insert sizes (ranging from 400 bp to 10 kbp) (Table 1) to close gaps in the assembled sequence and to obtain reliable assemblies of repetitive sequences (Argueso et al., 2009; Novo et al., 2009). The costs of fully assembling and annotating such a reference genome exceed the costs of routine resequencing, which limits the true availability of fully de novo assembled ‘gold standard’ genome sequences.
Table 1.
Comparison of ‘next generation’ methods for whole genome sequence. Costs per megabase are estimated based on price quotes (September 2011) from companies that offer commercial sequencing services
| Company (sequencing platform) | Chemistry | Read length (bp) | Total base count per run (Gb) | Accuracy (%) | Cost per Mb (€) |
|---|---|---|---|---|---|
| Illumina (Hiseq2000, GAIIX) | Reversible dye terminators | 50–150 | 5 | 99.0 | 2.5 |
| Life Technologies (ABI SOLiD, SOLiD4) | Oligonucleotide probe ligation | 35–50 | 10 | 99.9 | 3 |
| Roche (454 GS FLX Titanium) | Pyrosequencing | 350–450 | 0.4 | 99.5 | 20 |
| Pacific Biosystems (SMRT) | Phospho-linked fluorescent nucleotides | 600–1400 | 100 | 85.0 | 15 |
| Helicos Biosciences (HeliScope) | Reversible dye terminators | 35 | 25 | 99.9 | – |
There are, hitherto, only few cases in which whole genome sequencing of S. cerevisiae has been performed in a reverse metabolic engineering context (Timmermann et al. 2010; Dhar et al., 2011; Hong et al., 2011; Kvitek & Sherlock, 2011). Timmermann et al. (2010) used whole genome sequencing to analyze the molecular basis for oxidative stress tolerance in a S. cerevisiae mutant that was obtained by mutagenesis with ethyl methanosulfonate. Comparison of raw sequence data, followed by a manual inspection of results for ambiguous sequence calls, yielded only four mutations that were predicted to cause amino acid changes in proteins. A mutation in the peroxiredoxin protein Tsa1 was subsequently shown to be responsible for the improved oxidative stress tolerance. The discovery of only four lead genes represents a marked contrast with many microarray-based transcriptome analyses, which typically yields dozens, if not hundreds of differentially expressed genes. This contrast was even clearer in a direct comparison of transcriptome analysis and whole genome sequencing in a study on S. cerevisiae strains evolved for faster growth on galactose (Hong et al., 2011). After selecting strains by 400 generations of growth on galactose as sole carbon source, hundreds of genes were differentially expressed relative to the parental strain. However, systematic analysis of whole genome sequencing data revealed only small numbers of nonconservative SNVs and insertions/deletions within genes (fewer than 20 in two of three strains). On the basis of the observation that mutations in genes involved in the RAS/PKA pathway occurred in all three strains, a specific point mutation in RAS2 was reverse engineered in the ancestor strain and was shown to explain about half of the observed increase in growth rate on galactose (Hong et al., 2011).
Prolonged glucose-limited growth of S. cerevisiae in chemostat cultivation is a popular model for laboratory evolution and its molecular analysis. Transcriptome-based studies (Ferea et al., 1999; Jansen et al., 2005) identified hundreds of genes whose transcript levels changed as the yeast adapted to this nutrient limitation. In contrast, in a similar evolution experiment, resequencing of an adaptive clone revealed mutations in only six genes (Kvitek & Sherlock, 2011). Interestingly, a long terminal repeat insertion in GPB2, previously predicted based on tiling array analysis of this clone (Kao & Sherlock, 2008), was not identified during resequencing (Fig. 3). Mutations in HXT6/7 and GPB2 were shown to confer a statistically significant (P < 0.05) advantage over the ancestor strain during competitive glucose-limited cultivation (Kvitek & Sherlock, 2011). Unpublished results from our laboratory confirm that whole genome sequencing of parallel evolution lines can contribute to the rapid identification of key mutations in yeast strains generated in laboratory evolution experiments. For example, two suppressor mutants were isolated from independent laboratory evolution experiments with a jen1 null mutant, which encodes the S. cerevisiae lactate transporter (Casal et al., 1999). These strains regained the ability to grow on lactate as sole carbon source through different point mutations in the same membrane transporter gene. Reverse engineering of these mutations confirmed that each of them enabled the transporter to act as an efficient lactate transporter. Interestingly, this gene was not identified as a target in a parallel transcriptome analysis (S. de Kok et al., in press).
Fig. 3.
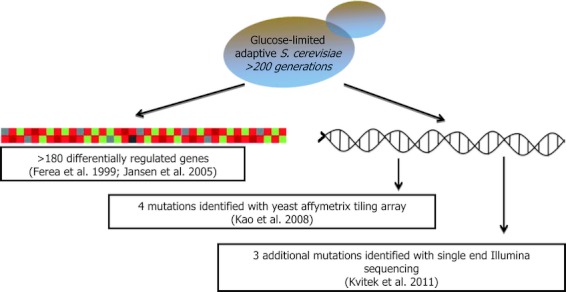
Analyses of evolved Saccharomyces cerevisiae strains adapted for glucose-limited cultivation conditions illustrate the advantages of whole genome sequencing over microarray-based transcriptome analysis in reverse metabolic engineering. Transcriptome analysis of four independently evolved strains consistently yielded more than 180 differentially expressed genes. Genotyping of a single cell line using tiling arrays and whole genome sequencing showed a much smaller number of underlying nonconservative mutations. Some mutations identified by whole genome sequencing went unnoticed by a previous analysis using tiling arrays.
Discussion
An overview of the relevant literature enables two clear recommendations on experimental design of reverse metabolic engineering experiments of S. cerevisiae. Firstly, although the number of studies in which whole genome sequencing has been applied for the reverse metabolic engineering of yeasts is still small, the available information consistently indicates that this technique is a real game changer. In ‘linear’ strain improvement studies (e.g. chemical mutagenesis and laboratory evolution), whole genome sequencing typically yields many fewer lead genes than transcriptome analysis. Moreover, the changes that are identified at the DNA level can be immediately and exactly reconstructed in naive strain backgrounds. Also in view of its rapidly decreasing costs, whole genome sequencing should now be the first-choice analytical approach in reverse metabolic engineering of yeast strains. Metabolomics, proteomics, and transcriptomics can subsequently be used for further interpretation of genome sequencing data and to elucidate the biochemical impact of the mutation, but, in general, are less suitable as first-line analytical approaches than genome sequencing. An interesting development in this respect is the sequencing of mRNA (RNA-seq), because both genetic changes in coding sequences and the transcriptional responses are measured in one step (Wang et al., 2009b).
Secondly, prioritization of mutations is greatly facilitated by the use of parallel strain improvement experiments. Focusing on functional analysis of mutations that affect the same gene, pathway or cellular process in multiple independent evolution or mutagenesis experiments has been repeatedly shown to facilitate the fast identification of relevant targets (Zelle et al., 2010; Hong et al., 2011; S. de Kok et al., in press). Especially when the number of parallel strain improvement experiments is small, an exclusive focus on the ‘overlap’ of a small number of selected genotypes can potentially lead to loss of valuable information. Such a loss can result from mutations in different genes or processes that have a similar positive effect on the phenotype but which do not occur in all strains selected for analysis. Moreover, the phenotypic effect of a mutation, be it on evolutionary fitness or on industrial performance, can be strongly dependent on the genetic context. The relevance of this context dependency, which in genetics is known as epistasis, is illustrated by an elegant study by Kvitek & Sherlock (2011), who monitored the occurrence of mutations during laboratory evolution experiments with S. cerevisiae in glucose-limited chemostat cultures. The authors convincingly demonstrated that mutations in the hexose transporter gene HXT6/7 and in the regulator gene MTH1 exhibited negative epistasis: individual introduction of the mutations in a naive strain background led to an improved fitness, while their combined introduction had a negative effect. In addition to monitoring the incidence of mutations during evolution, increasing the number of parallel strain improvement experiments should facilitate identification of negatively epistatic mutations.
Wherever whole genome sequencing results in too many candidate leads, the power of molecular techniques should be amplified by their integration with classical yeast genetics. Analysis of segregation patterns after mating with a reference strain and systematic backcrossing can rapidly provide insight into the complexity of acquired genotypes and reduce the number of nonproductive mutations (Timmermann et al. 2010). Moreover, segregation of mutations in the offspring of a backcross with the strain of interest is a powerful technique in discovering positive epistasis (i.e. multigenic traits) through the identification of quantitative trait loci (Kearsey, 1998; Liti et al., 2009b; Ehrenreich et al., 2010; Cubillos et al., 2011; Parts et al., 2011). When knowledge on gene or protein function is limited, parallel strain improvement and identification of quantitative trait loci give information for target prioritization without requiring a priori knowledge on gene or protein function.
We anticipate that further technology developments and decreases in sequencing costs, combined with the automation of the parallel and combinatorial reconstruction of different genetic variations, will make reverse metabolic engineering one of the major driving forces in yeast biotechnology in the coming decade.
Acknowledgments
The PhD project of B.O. is funded by the Netherlands Genomics Initiative. This work is supported by the European Commission (project SYSINBIO-Systems Biology as a Driver for Industrial Biotechnology). We thank our colleagues Jens Nielsen and Greg Stephanopoulos for an inspiring discussion on terminology (reverse vs. inverse metabolic engineering).
References
- Abbott DA, Knijnenburg TA, de Poorter LMI, Reinders MJT, Pronk JT, van Maris AJA. Generic and specific transcriptional responses to different weak organic acids in anaerobic chemostat cultures of Saccharomyces cerevisiae. FEMS Yeast Res. 2007;7:819–833. doi: 10.1111/j.1567-1364.2007.00242.x. [DOI] [PubMed] [Google Scholar]
- Abbott DA, Zelle RM, Pronk JT, van Maris AJA. Metabolic engineering of Saccharomyces cerevisiae for production of carboxylic acids: current status and challenges. FEMS Yeast Res. 2009;9:1123–1136. doi: 10.1111/j.1567-1364.2009.00537.x. [DOI] [PubMed] [Google Scholar]
- Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper H, Moxley J, Nevoigt E, Fink GR, Stephanopoulos G. Engineering yeast transcription machinery for improved ethanol tolerance and production. Science. 2006;314:1565–1568. doi: 10.1126/science.1131969. [DOI] [PubMed] [Google Scholar]
- Anderson JC, Dueber JE, Leguia M, Wu GC, Goler JA, Arkin AP, Keasling JD. BglBricks: a flexible standard for biological part assembly. J Biol Eng. 2010;4:1. doi: 10.1186/1754-1611-4-1. DOI: 10.1186/1754-1611-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya CL, Payen C, Dunham MJ, Fields S. Whole-genome sequencing of a laboratory-evolved yeast strain. BMC Genomics. 2010;11:88. doi: 10.1186/1471-2164-11-88. DOI: 10.1186/1471-2164-11-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argueso JL, Carazzolle MF, Mieczkowski PA, et al. Genome structure of a Saccharomyces cerevisiae strain widely used in bioethanol production. Genome Res. 2009;19:2258–2270. doi: 10.1101/gr.091777.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JE, Birnbaum S, Galazzo JL, Khosla C, Shanks JV. Strategies and challenges in metabolic engineering. Ann N Y Acad Sci. 1990;589:1–15. doi: 10.1111/j.1749-6632.1990.tb24230.x. [DOI] [PubMed] [Google Scholar]
- Bailey JE, Sburlati A, Hatzimanikatis V, Lee K, Renner WA, Tsai PS. Inverse metabolic engineering: a strategy for directed genetic engineering of useful phenotypes. Biotechnol Bioeng. 1996;52:109–121. doi: 10.1002/(SICI)1097-0290(19961005)52:1<109::AID-BIT11>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Basso TO, de Kok S, Dario M, et al. Engineering topology and kinetics of sucrose metabolism in Saccharomyces cerevisiae for improved ethanol yield. Metab Eng. 2011;13:694–703. doi: 10.1016/j.ymben.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Beck M, Claassen M, Aebersold R. Comprehensive proteomics. Curr Opin Biotechnol. 2011;22:3–8. doi: 10.1016/j.copbio.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Becker J, Boles E. A modified Saccharomyces cerevisiae strain that consumes l-arabinose and produces ethanol. Appl Environ Microbiol. 2003;69:4144–4150. doi: 10.1128/AEM.69.7.4144-4150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell GW, Brown JA, Wu HI, Giaever G, Chu AM, Davis RW, Brown JM. Transcriptional response of Saccharomyces cerevisiae to DNA-damaging agents does not identify the genes that protect against these agents. P Natl Acad Sci USA. 2002;99:8778–8783. doi: 10.1073/pnas.132275199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer VM, de Winde JH, Pronk JT, Piper MDW. The genome-wide transcriptional responses of Saccharomyces cerevisiae grown on glucose in aerobic chemostat cultures limited for carbon, nitrogen, phosphorus, or sulfur. J Biol Chem. 2003;278:3265–3274. doi: 10.1074/jbc.M209759200. [DOI] [PubMed] [Google Scholar]
- Bro C, Nielsen J. Impact of “ome” analyses on inverse metabolic engineering. Metab Eng. 2004;6:204–211. doi: 10.1016/j.ymben.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Bro C, Knudsen S, Regenberg B, Olsson L, Nielsen J. Improvement of galactose uptake in Saccharomyces cerevisiae through overexpression of phosphoglucomutase: example of transcript analysis as a tool in inverse metabolic engineering. Appl Environ Microbiol. 2005;71:6465–6472. doi: 10.1128/AEM.71.11.6465-6472.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Todd KM, Rosenzweig RF. Multiple duplications of yeast hexose transport genes in response to selection in a glucose-limited environment. Mol Biol Evol. 1998;15:931–942. doi: 10.1093/oxfordjournals.molbev.a026009. [DOI] [PubMed] [Google Scholar]
- Casal M, Paiva S, Andrade RP, Gancedo C, Leão C. The lactate-proton symport of Saccharomyces cerevisiae is encoded by JEN1. J Bacteriol. 1999;181:2620–2623. doi: 10.1128/jb.181.8.2620-2623.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillo JI, Zeef LA, Hoyle DC, et al. Growth control of the eukaryote cell: a systems biology study in yeast. J Biol. 2007;6:4. doi: 10.1186/jbiol54. DOI: 10.1186/jbiol54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen S, Sauer U. Intracellular characterization of aerobic glucose metabolism in seven yeast species by 13C flux analysis and metabolomics. FEMS Yeast Res. 2011;11:263–272. doi: 10.1111/j.1567-1364.2010.00713.x. [DOI] [PubMed] [Google Scholar]
- Conrad TM, Lewis NE, Palsson BØ. Microbial laboratory evolution in the era of genome-scale science. Mol Syst Biol. 2011;7:509. doi: 10.1038/msb.2011.42. DOI: 10.1038/msb.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos FA, Billi E, Zörgö E, Parts L, Fargier P, Omholt S, Blomberg A, Warringer J, Louis EJ, Liti G. Assessing the complex architecture of polygenic traits in diverged yeast populations. Mol Ecol. 2011;20:1401–1413. doi: 10.1111/j.1365-294X.2011.05005.x. [DOI] [PubMed] [Google Scholar]
- Daran-Lapujade P, Daran J, Kotter P, Petit T, Piper M, Pronk JT. Comparative genotyping of the laboratory strains S288C and CEN.PK113-7D using oligonucleotide microarrays. FEMS Yeast Res. 2003;4:259–269. doi: 10.1016/S1567-1356(03)00156-9. [DOI] [PubMed] [Google Scholar]
- Daran-Lapujade P, Daran JM, van Maris AJA, de Winde JH, Pronk JT. Chemostat-based micro-array analysis in baker's yeast. Adv Microb Physiol. 2009;54:257–311. doi: 10.1016/S0065-2911(08)00004-0. [DOI] [PubMed] [Google Scholar]
- de Groot MJL, Daran-Lapujade P, van Breukelen B, Knijnenburg TA, de Hulster EAF, Reinders MJT, Pronk JT, Heck AJR, Slijper M. Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals post-transcriptional regulation of key cellular processes. Microbiology. 2007;153:3864–3878. doi: 10.1099/mic.0.2007/009969-0. [DOI] [PubMed] [Google Scholar]
- de Jesus Ferreira M, Bao X, Laizé V, Hohmann S. Transposon mutagenesis reveals novel loci affecting tolerance to salt stress and growth at low temperature. Curr Genet. 2001;40:27–39. doi: 10.1007/s002940100237. [DOI] [PubMed] [Google Scholar]
- de Kok S, Nijkamp JF, Oud B, Roque FC, De Ridder D, Daran J-M, Pronk JT, van Maris AJA. Laboratory evolution of new lactate transporter genes in a jen1 mutant of Saccharomyces cerevisiae and their identification as ADY2 alleles by whole-genome resequencing and transcriptome analysis. FEMS Yeast Res. doi: 10.1111/j.1567-1364.2012.00787.x. (in press), in press. [DOI] [PubMed] [Google Scholar]
- De Nicola R, Hazelwood LA, De Hulster EAF, Walsh MC, Knijnenburg TA, Reinders MJT, Walker GM, Pronk JT, Daran JM, Daran-Lapujade P. Physiological and transcriptional responses of Saccharomyces cerevisiae to zinc limitation in chemostat cultures. Appl Environ Microbiol. 2007;73:7680–7692. doi: 10.1128/AEM.01445-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi JL. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Dhar R, Sägesser R, Weikert C, Yuan J, Wagner A. Adaptation of Saccharomyces cerevisiae to saline stress through laboratory evolution. J Evol Biol. 2011;24:1135–1153. doi: 10.1111/j.1420-9101.2011.02249.x. [DOI] [PubMed] [Google Scholar]
- Dunham MJ, Badrane H, Ferea T, Adams J, Brown PO, Rosenzweig F, Botstein D. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. P Natl Acad Sci USA. 2002;99:16144–16149. doi: 10.1073/pnas.242624799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong CT, Strack L, Futschik M, Katou Y, Nakao Y, Fujimura T, Shirahige K, Kodama Y, Nevoigt E. Identification of Sc-type ILV6 as a target to reduce diacetyl formation in lager brewers’ yeast. Metab Eng. 2011;13:638–647. doi: 10.1016/j.ymben.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Ehrenreich IM, Torabi N, Jia Y, Kent J, Martis S, Shapiro JA, Gresham D, Caudy AA, Kruglyak L. Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature. 2010;464:1039–1042. doi: 10.1038/nature08923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazio A, Jewett MC, Daran-Lapujade P, Mustacchi R, Usaite R, Pronk JT, Workman CT, Nielsen J. Transcription factor control of growth rate dependent genes in Saccharomyces cerevisiae: a three factor design. BMC Genomics. 2008;9:341. doi: 10.1186/1471-2164-9-341. DOI: 10.1186/1471-2164-9-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferea TL, Botstein D, Brown PO, Rosenzweig RF. Systematic changes in gene expression patterns following adaptive evolution in yeast. P Natl Acad Sci USA. 1999;96:9721–9726. doi: 10.1073/pnas.96.17.9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW. Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. P Natl Acad Sci USA. 2004;101:793–798. doi: 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG. Gene and genome construction in yeast. Curr Protoc Mol Biol. 2011;Chapter 3:Unit 3.22. doi: 10.1002/0471142727.mb0322s94. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, et al. Life with 6000 genes. Science. 1996;274:546. doi: 10.1126/science.274.5287.546. 563–567. [DOI] [PubMed] [Google Scholar]
- Gresham D, Ruderfer DM, Pratt SC, Schacherer J, Dunham MJ, Botstein D, Kruglyak L. Genome-wide detection of polymorphisms at nucleotide resolution with a single DNA microarray. Science. 2006;311:1932–1936. doi: 10.1126/science.1123726. [DOI] [PubMed] [Google Scholar]
- Gresham D, Dunham M, Botstein D. Comparing whole genomes using DNA microarrays. Nat Rev Genet. 2008a;9:291–302. doi: 10.1038/nrg2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, Desai MM, Tucker CM, Jenq HT, Pai DA, Ward A, DeSevo CG, Botstein D, Dunham MJ. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet. 2008b;4:e1000303. doi: 10.1371/journal.pgen.1000303. DOI: 10.1371/journal.pgen.1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, Usaite R, Germann SM, Lisby M, Botstein D, Regenberg B. Adaptation to diverse nitrogen-limited environments by deletion or extrachromosomal element formation of the GAP1 locus. P Natl Acad Sci USA. 2010;107:18551–18556. doi: 10.1073/pnas.1014023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn-Hägerdal B, Karhumaa K, Fonseca C, Spencer-Martins I, Gorwa-Grauslund MF. Towards industrial pentose-fermenting yeast strains. Appl Environ Microbiol. 2007;74:937–953. doi: 10.1007/s00253-006-0827-2. [DOI] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong M-E, Lee K-S, Yu BJ, Sung Y-J, Park SM, Koo HM, Kweon D-H, Park JC, Jin Y-S. Identification of gene targets eliciting improved alcohol tolerance in Saccharomyces cerevisiae through inverse metabolic engineering. J Biotechnol. 2010;149:52–59. doi: 10.1016/j.jbiotec.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Hong K-K, Vongsangnak W, Vemuri GN, Nielsen J. Unravelling evolutionary strategies of yeast for improving galactose utilization through integrated systems level analysis. P Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1103219108. DOI: 10.1073/pnas.1103219108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hughes SR, Gibbons WR, Bang SS, et al. Random UV-C mutagenesis of Scheffersomyces (formerly Pichiastipitis NRRL Y-7124 to improve anaerobic growth on lignocellulosic sugars. J Ind Microbiol Biotechnol. 2011 doi: 10.1007/s10295-011-1012-x. DOI: 10.1007/s10295-011-1012-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen MLA, Diderich JA, Mashego M, Hassane A, de Winde JH, Daran-Lapujade P, Pronk JT. Prolonged selection in aerobic, glucose-limited chemostat cultures of Saccharomyces cerevisiae causes a partial loss of glycolytic capacity. Microbiology. 2005;151:1657–1669. doi: 10.1099/mic.0.27577-0. [DOI] [PubMed] [Google Scholar]
- Jin Y-S, Alper H, Yang YT, Stephanopoulos G. Improvement of xylose uptake and ethanol production in recombinant Saccharomyces cerevisiae through an inverse metabolic engineering approach. Society. 2005;71:8249–8256. doi: 10.1128/AEM.71.12.8249-8256.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao KC, Sherlock G. Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae. Nat Genet. 2008;40:1499–1504. doi: 10.1038/ng.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karhumaa K, Påhlman AK, Hahn-Hägerdal B, Levander F, Gorwa-Grauslund MF. Proteome analysis of the xylose-fermenting mutant yeast strain TMB 3400. Yeast. 2009;26:371–382. doi: 10.1002/yea.1673. [DOI] [PubMed] [Google Scholar]
- Kearsey M. The principles of QTL analysis (a minimal mathematics approach) J Exp Bot. 1998;49:1619–1623. [Google Scholar]
- Kern A, Tilley E, Hunter IS, Legisa M, Glieder A. Engineering primary metabolic pathways of industrial micro-organisms. J Biotechnol. 2007;129:6–29. doi: 10.1016/j.jbiotec.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Knijnenburg TA, Wessels LFA, Reinders MJT. Combinatorial influence of environmental parameters on transcription factor activity. Bioinformatics. 2008;24:i172–i181. doi: 10.1093/bioinformatics/btn155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolkman A, Slijper M, Heck AJR. Development and application of proteomics technologies in Saccharomyces cerevisiae. Trends Biotechnol. 2005;23:598–604. doi: 10.1016/j.tibtech.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Kresnowati MTAP, van Winden WA, Almering MJH, ten Pierick A, Ras C, Knijnenburg TA, Daran-Lapujade P, Pronk JT, Heijnen JJ, Daran JM. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Mol Syst Biol. 2006;2:49. doi: 10.1038/msb4100083. DOI: 10.1038/msb4100083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitek DJ, Sherlock G. Zhang J, editor. Reciprocal sign epistasis between frequently experimentally evolved adaptive mutations causes a rugged fitness landscape. PLoS Genet. 2011;7:e1002056. doi: 10.1371/journal.pgen.1002056. DOI: 10.1371/journal.pgen.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Kim TY, Jang Y-S, Choi S, Lee SY. Systems metabolic engineering for chemicals and materials. Trends Biotechnol. 2011;29:370–378. doi: 10.1016/j.tibtech.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Liti G, Haricharan S, Cubillos FA, Tierney AL, Sharp S, Bertuch AA, Parts L, Bailes E, Louis EJ. Segregating YKU80 and TLC1 alleles underlying natural variation in telomere properties in wild yeast. PLoS Genet. 2009b;5:e1000659. doi: 10.1371/journal.pgen.1000659. DOI: 10.1371/journal.pgen.1000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac KD, Wang T, Gordon DB, Gifford DK, Stormo GD, Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. DOI: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. Anticipating the 1,000 dollar genome. Genome Biol. 2006;7:112. doi: 10.1186/gb-2006-7-7-112. DOI: 10.1186/gb-2006-7-7-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira NP, Palma M, Guerreiro JF, Sá-Correia I. Genome-wide identification of Saccharomyces cerevisiae genes required for tolerance to acetic acid. Microb Cell Fact. 2010;9:79. doi: 10.1186/1475-2859-9-79. DOI: 10.1186/1475-2859-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevoigt E. Progress in metabolic engineering of Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2008;72:379–412. doi: 10.1128/MMBR.00025-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H, Laplaza JM, Jeffries TW. Transposon mutagenesis to improve the growth of recombinant Saccharomyces cerevisiae on d-xylose. Appl Environ Microbiol. 2007;73:2061–2066. doi: 10.1128/AEM.02564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J. Singapore: World Scientific Publishing Co; 1997. Physiological engineering aspects of Penicillium chrysogenum. [Google Scholar]
- Nielsen J. Metabolic engineering. Appl Environ Microbiol. 2001;55:263–283. doi: 10.1007/s002530000511. [DOI] [PubMed] [Google Scholar]
- Nijkamp J, Winterbach W, van den Broek M, Daran JM, Reinders M, de Ridder D. Integrating genome assemblies with MAIA. Bioinformatics. 2010;26:i433–i439. doi: 10.1093/bioinformatics/btq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novo M, Bigey F, Beyne E, et al. Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. P Natl Acad Sci USA. 2009;106:16333–16338. doi: 10.1073/pnas.0904673106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares-Hernández R, Usaite R, Nielsen J. Integrative analysis using proteome and transcriptome data from yeast to unravel regulatory patterns at post-transcriptional level. Biotechnol Bioeng. 2010;107:865–875. doi: 10.1002/bit.22868. [DOI] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12:87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts L, Cubillos FA, Warringer J, et al. Revealing the genetic structure of a trait by sequencing a population under selection. Genome Res. 2011;21:1131–1138. doi: 10.1101/gr.116731.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson A, Almeida JRM, Modig T, Karhumaa K, Hahn-Hägerdal B, Gorwa-Grauslund MF, Lidén G. A 5-hydroxymethyl furfural reducing enzyme encoded by the Saccharomyces cerevisiae ADH6 gene conveys HMF tolerance. Yeast. 2006;23:455–464. doi: 10.1002/yea.1370. [DOI] [PubMed] [Google Scholar]
- Pitkänen JP, Rintala E, Aristidou A, Ruohonen L, Penttilä M. Xylose chemostat isolates of Saccharomyces cerevisiae show altered metabolite and enzyme levels compared with xylose, glucose, and ethanol metabolism of the original strain. Appl Environ Microbiol. 2005;67:827–837. doi: 10.1007/s00253-004-1798-9. [DOI] [PubMed] [Google Scholar]
- Raynes Y, Gazzara MR, Sniegowski PD. Mutator dynamics in sexual and asexual experimental populations of yeast. BMC Evol Biol. 2011;11:158. doi: 10.1186/1471-2148-11-158. DOI: 10.1186/1471-2148-11-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regenberg B, Grotkjaer T, Winther O, Fausbøll A, Akesson M, Bro C, Hansen LK, Brunak S, Nielsen J. Growth-rate regulated genes have profound impact on interpretation of transcriptome profiling in Saccharomyces cerevisiae. Genome Biol. 2006;7:R107. doi: 10.1186/gb-2006-7-11-r107. DOI: 10.1186/gb-2006-7-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekoff M. On reverse engineering. IEEE T Syst Man Cyb. 1985;15:244–252. [Google Scholar]
- Roessner U, Bowne J. What is metabolomics all about? BioTechniques. 2009;46:363–365. doi: 10.2144/000113133. [DOI] [PubMed] [Google Scholar]
- Rossignol T, Kobi D, Jacquet-Gutfreund L, Blondin B. The proteome of a wine yeast strain during fermentation, correlation with the transcriptome. J Appl Microbiol. 2009;107:47–55. doi: 10.1111/j.1365-2672.2009.04156.x. [DOI] [PubMed] [Google Scholar]
- Ross-Macdonald P, Coelho PS, Roemer T, et al. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature. 1999;402:413–418. doi: 10.1038/46558. [DOI] [PubMed] [Google Scholar]
- Rossouw D, van den Dool AH, Jacobson D, Bauer FF. Comparative transcriptomic and proteomic profiling of industrial wine yeast strains. Appl Environ Microbiol. 2010;76:3911–3923. doi: 10.1128/AEM.00586-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer M, Porro D, Mattanovich D, Branduardi P. 16 years research on lactic acid production with yeast – ready for the market? Biotechnol Genet Eng Rev. 2010;27:229–256. doi: 10.1080/02648725.2010.10648152. [DOI] [PubMed] [Google Scholar]
- Schacherer J, Ruderfer DM, Gresham D, Dolinski K, Botstein D, Kruglyak L. Genome-wide analysis of nucleotide-level variation in commonly used Saccharomyces cerevisiae strains. PLoS ONE. 2007;2:e322. doi: 10.1371/journal.pone.0000322. DOI: 10.1371/journal.pone.0000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacherer J, Shapiro JA, Ruderfer DM, Kruglyak L. Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature. 2009;458:342–345. doi: 10.1038/nature07670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoep JL, Yomano LP, Westerhoff HV, Ingram LO. Protein burden in Zymomonas mobilis: negative flux and growth control due to overproduction of glycolytic enzymes. Microbiology. 1995;141:2329–2337. [Google Scholar]
- Sonderegger M, Sauer U. Evolutionary engineering of Saccharomyces cerevisiae for anaerobic growth on xylose. Appl Environ Microbiol. 2003;69:1990–1998. doi: 10.1128/AEM.69.4.1990-1998.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley D, Chambers PJ, Stanley GA, Borneman A, Fraser S. Transcriptional changes associated with ethanol tolerance in Saccharomyces cerevisiae. Appl Environ Microbiol. 2010a;88:231–239. doi: 10.1007/s00253-010-2760-7. [DOI] [PubMed] [Google Scholar]
- Stanley D, Fraser S, Chambers PJ, Rogers P, Stanley GA. Generation and characterisation of stable ethanol-tolerant mutants of Saccharomyces cerevisiae. J Ind Microbiol Biotechnol. 2010b;37:139–149. doi: 10.1007/s10295-009-0655-3. [DOI] [PubMed] [Google Scholar]
- Sutton J. Reverse engineering the 80C86. Electron Eng. 1984;56:51–54. 58. [Google Scholar]
- Tai SL, Boer VM, Daran-Lapujade P, Walsh MC, de Winde JH, Daran JM, Pronk JT. Two-dimensional transcriptome analysis in chemostat cultures. Combinatorial effects of oxygen availability and macronutrient limitation in Saccharomyces cerevisiae. J Biol Chem. 2005;280:437–447. doi: 10.1074/jbc.M410573200. [DOI] [PubMed] [Google Scholar]
- Tai SL, Snoek I, Luttik MAH, Almering MJH, Walsh MC, Pronk JT, Daran JM. Correlation between transcript profiles and fitness of deletion mutants in anaerobic chemostat cultures of Saccharomyces cerevisiae. Microbiology. 2007;153:877–886. doi: 10.1099/mic.0.2006/002873-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanghe A, Van Dijck P, Dumortier F, Teunissen A, Hohmann S, Thevelein JM. Aquaporin expression correlates with freeze tolerance in baker's yeast, and overexpression improves freeze tolerance in industrial strains. Appl Environ Microbiol. 2002;68:5981–5989. doi: 10.1128/AEM.68.12.5981-5989.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, Desai MM, Murray AW. Ploidy controls the success of mutators and nature of mutations during budding yeast evolution. Curr Biol. 2006;16:1581–1590. doi: 10.1016/j.cub.2006.06.070. [DOI] [PubMed] [Google Scholar]
- Timmermann B, Jarolim S, Russmayer H, Kerick M, Michel S, Krüger A, Bluemlein K, Laun P, Grillari J, Lehrach H, Breitenbach M, Ralser M. A new dominant peroxiredoxin allele identified by whole-genome re-sequencing of random mutagenized yeast causes oxidant-resistance and premature aging. Aging. 2010;2:475–486. doi: 10.18632/aging.100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruta H, Paddon CJ, Eng D, Lenihan JR, Horning T, Anthony LC, Regentin R, Keasling JD, Renninger NS, Newman JD. High-level production of amorpha-4,11-diene, a precursor of the antimalarial agent artemisinin, in Escherichia coli. PLoS ONE. 2009;4:e4489. doi: 10.1371/journal.pone.0004489. DOI: 10.1371/journal.pone.0004489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg MA, Albang R, Albermann K, et al. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat Biotechnol. 2008;26:1161–1168. doi: 10.1038/nbt.1498. [DOI] [PubMed] [Google Scholar]
- van Maris AJA, Winkler AA, Kuyper M, de Laat WTAM, van Dijken JP, Pronk JT. Development of efficient xylose fermentation in Saccharomyces cerevisiae: xylose isomerase as a key component. Adv Biochem Eng Biotechnol. 2007;108:179–204. doi: 10.1007/10_2007_057. [DOI] [PubMed] [Google Scholar]
- Wahlbom CF, Cordero Otero RR, van Zyl WH, Hahn-Hägerdal B, Jönsson LJ. Molecular analysis of a Saccharomyces cerevisiae mutant with improved ability to utilize xylose shows enhanced expression of proteins involved in transport, initial xylose metabolism, and the pentose phosphate pathway. Appl Environ Microbiol. 2003;69:740–746. doi: 10.1128/AEM.69.2.740-746.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009a;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009b;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Lee B, McCusker JH, Davis RW. Whole genome genetic-typing in yeast using high-density oligonucleotide arrays. Parasitology. 1999a;118:S73–S80. doi: 10.1017/s0031182099004047. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, et al. Functional characterization of the Saccharomyces cerevisiae genome by gene deletion and parallel analysis. Science. 1999b;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Wisselink HW, Toirkens MJ, del Rosario Franco Berriel M, Winkler AA, van Dijken JP, Pronk JT, van Maris AJA. Engineering of Saccharomyces cerevisiae for efficient anaerobic alcoholic fermentation of l-arabinose. Appl Environ Microbiol. 2007;73:4881–4891. doi: 10.1128/AEM.00177-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisselink HW, Toirkens MJ, Wu Q, Pronk JT, van Maris AJA. Novel evolutionary engineering approach for accelerated utilization of glucose, xylose, and arabinose mixtures by engineered Saccharomyces cerevisiae strains. Appl Environ Microbiol. 2009;75:907–914. doi: 10.1128/AEM.02268-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisselink HW, Cipollina C, Oud B, Crimi B, Heijnen JJ, Pronk JT, van Maris AJA. Metabolome, transcriptome and metabolic flux analysis of arabinose fermentation by engineered Saccharomyces cerevisiae. Metab Eng. 2010;12:537–551. doi: 10.1016/j.ymben.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Wright J, Bellissimi E, de Hulster E, Wagner A, Pronk JT, van Maris AJA. Batch and continuous culture-based selection strategies for acetic acid tolerance in xylose-fermenting Saccharomyces cerevisiae. FEMS Yeast Res. 2011;11:299–306. doi: 10.1111/j.1567-1364.2011.00719.x. [DOI] [PubMed] [Google Scholar]
- Wu G, Nie L, Zhang W. Integrative analyses of posttranscriptional regulation in the yeast Saccharomyces cerevisiae using transcriptomic and proteomic data. Curr Microbiol. 2008;57:18–22. doi: 10.1007/s00284-008-9145-5. [DOI] [PubMed] [Google Scholar]
- Zaldivar J, Borges A, Johansson B, Smits HP, Villas-Bôas SG, Nielsen J, Olsson L. Fermentation performance and intracellular metabolite patterns in laboratory and industrial xylose-fermenting Saccharomyces cerevisiae. Appl Environ Microbiol. 2002;59:436–442. doi: 10.1007/s00253-002-1056-y. [DOI] [PubMed] [Google Scholar]
- Zelle RM, Trueheart J, Harrison JC, Pronk JT, van Maris AJA. Phosphoenolpyruvate carboxykinase as the sole anaplerotic enzyme in Saccharomyces cerevisiae. Appl Environ Microbiol. 2010;76:5383–5389. doi: 10.1128/AEM.01077-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelle RM, Harrison JC, Pronk JT, van Maris AJA. Anaplerotic role for cytosolic malic enzyme in engineered Saccharomyces cerevisiae strains. Appl Environ Microbiol. 2011;77:732–738. doi: 10.1128/AEM.02132-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Rodriguez S, Keasling JD. Metabolic engineering of microbial pathways for advanced biofuels production. Curr Opin Biotechnol. 2011;22:1–9. doi: 10.1016/j.copbio.2011.04.024. [DOI] [PubMed] [Google Scholar]