

Abstract
Context:
Primary generalized glucocorticoid resistance is a rare genetic condition characterized by partial end-organ insensitivity to glucocorticoids. Most affected subjects present with clinical manifestations of mineralocorticoid and androgen excess. The condition has been associated with inactivating mutations in the human glucocorticoid receptor (hGR) gene, which impair the molecular mechanisms of hGRα action, thereby reducing tissue sensitivity to glucocorticoids.
Objective:
Τhe aim of our study was to investigate the molecular mechanisms through which one previously described natural heterozygous V423A mutation, the second mutation detected in the DNA-binding domain (DBD) of the hGRα, affects glucocorticoid signal transduction.
Design and Results:
Compared with the wild-type receptor, hGRαV423A demonstrated a 72% reduction in its ability to transactivate the glucocorticoid-inducible mouse mammary tumor virus promoter in response to dexamethasone. The hGRαV423A receptor showed a significant reduction in its ability to bind to glucocorticoid-response elements of glucocorticoid-responsive genes, owing to structural alterations of the DBD confirmed by computer-based structural analysis. In addition, hGRαV423A demonstrated a 2.6-fold delay in nuclear translocation following exposure to the ligand, although it did not exert a dominant negative effect on the wild-type hGRα, had a similar affinity to the ligand with the wild-type receptor, and displayed a normal interaction with the GRIP1 coactivator in vitro.
Conclusions:
The natural mutant receptor hGRαV423A causes primary generalized glucocorticoid resistance by affecting multiple steps in the cascade of glucocorticoid receptor action, which primarily involve decreased ability to bind to target glucocorticoid response elements and delayed translocation into the nucleus.
Primary generalized glucocorticoid resistance or Chrousos syndrome is a rare, familial, or sporadic condition characterized by generalized, partial, target-tissue insensitivity to glucocorticoids, compensatory elevations in circulating cortisol and ACTH concentrations, and resistance of the hypothalamic-pituitary-adrenal axis to dexamethasone suppression (1–5). The molecular basis of the condition has been ascribed to mutations in the human glucocorticoid receptor (hGR) gene, which impair the molecular mechanisms of hGR action (1–9). We have previously reported a new case of this disorder in a 9-year-old boy who presented with fatigue, anxiety, and refractory-to-treatment hypertension. Sequencing of the hGR gene revealed a novel heterozygous mutation that resulted in valine (V) to alanine (A) substitution at amino acid position 423 in the first zinc finger of the DNA-binding domain (DBD) of the protein (10). In the present study, we investigated the molecular mechanisms through which the natural mutant receptor hGRαV423A impairs glucocorticoid signal transduction.
Materials and Methods
Plasmids
The plasmids used in this study included pRShGRα, pF25GFP-hGRα, pBK/CMV-hGRα, pMMTV-luc, pGL4.73 [hRluc/SV40], pRSV-erbA-1, pGEX4T3-GRIP1(1–1462), pGEX4T3-GRIP1(596–774), and pGEX4T3-GRIP1(740–1217) (6–9). The plasmids pRShGRαV423A, pF25GFP-hGRαV423A, and pBK/CMV-hGRαV423A were constructed by introducing the V423A mutation into the pRShGRα, pF25GFP-hGRα, and pBK/CMV-hGRα plasmids, respectively, using PCR-assisted site-directed mutagenesis.
Transactivation assays
CV-1 cells were cotransfected with pRShGRα (0.0125 μg per well) or pRShGRαV423A (0.0125 μg per well), pMMTV-luc (0.125 μg per well), and pSV40-Renilla (0.02 μg per well) using lipofectin (Invitrogen, Carlsbad, California). Forty-eight hours after transfection, cells were exposed to dexamethasone or vehicle for 24 hours. Firefly and renilla luciferase activities were determined in the cell lysates (6–9).
Western blot analyses
CV-1 and COS-7 cells were transfected with pRShGRα or pRShGRαV423A (15 μg per flask) using lipofectin. Western blot analyses were performed as previously described (6–9).
Chromatin immunoprecipitation (ChIP) assays
HCT-116 cells were transiently transfected with pRShGRα-expressing or pRShGRαV423A-expressing plasmids using lipofectamine 2000 (Invitrogen). Sixteen hours after transfection, cells were treated with dexamethasone (10−6 M) or vehicle for 24 hours and then cross-linked in culture medium with 1% formaldehyde for 15 minutes at room temperature. ChIPs were performed according to Upstate protocol (www.upstate.com). Primers used for amplification of the promoter region of the glucocorticoid responsive glucocorticoid-induced leucine zipper (GILZ) and the glucose-6-phosphatase (G6Pase) genes were as follows: GILZ (promoter region: −1341 to −1209): F: 5′-CCTTAACTTCATCCAAACTG-3′, R: 5′-CACCAGAAGGAGCAAGAG-3′; G6Pase (promoter region: −257 to −39): F: 5′-CAGACCCTTGCACTGCCAAGAAGCATG-3′, R: 5′-TATCCAGTATTCAGGTCAACCCAGCCC-3′ (11). The calculation of specific enrichments (fold differences) was performed versus input using real-time PCR (ChIP/Input = 2Input Ct−ChIP Ct) (12).
Computer-based creation of the 3-dimenstional structure of the hGR DBD harboring the V423A mutation
Molecular dynamics simulations for the hGR DBD with and without the V423A mutation were performed in NAMD 2.7 (University of Illinois, Urbana-Champaign, Illinois) using the CHARMM force field (National Heart, Lung, and Blood Institute, Bethesda, Maryland) packaged with VMD 1.8.7 (University of Illinois) (13, 14). The simulations were performed in the presence of explicit water and counterions at constant temperature and pressure with periodic boundary conditions for 50 ns after minimization and equilibration. The α carbon root-mean-square deviation between the starting structure and the end points of the native and mutant simulations for both starting structures was less than 2 Å, whereas the root-mean-square deviation between the end points themselves was less than 0.7 Å. Snapshots of the hGR native and V423A trajectories were output every 100 ps and the molecular contacts of these 1000 structures were analyzed by VMD and LigPlot (University College, London, United Kingdom) (15).
Detection and localization of green fluorescent protein–fused hGRs
HeLa cells were transfected with pF25GFP-hGRα or pF25GFP-hGRαV423A (2 μg per dish) using FuGENE 6 according to the instructions of the manufacturer (Roche Diagnostics Corp, Indianapolis, Indiana). Nuclear translocation studies were performed as previously described (6–9).
Dexamethasone binding assays
COS-7 cells were transfected with pRShGRα or hGRαV423A (1.5 μg per well) using lipofectin. Confluent cells were incubated with 6 different concentrations of [3H]-dexamethasone at 37°C in the presence or absence of a 500-fold molar excess of nonradioactive dexamethasone for 1 hour. Dexamethasone-binding assays were performed as previously described (6–9).
Glutathione-S-transferase (GST) pull-down assays
GST fusion protein expression vectors (GST-fused GRIP1[1–1462], GRIP1[559–774], and GRIP1[740–1217]) were grown in Escherichia coli BL21 cells for 4 hours and induced with isopropyl β-d-thiogalactoside for an additional 2 hours. Glutathione-sepharose beads were added to the cell extracts and allowed to bind by gentle agitation for 1 hour at 4°C. The beads with the bound GST fusion protein were then pelleted, washed, and analyzed by SDS-PAGE for the amount of protein bound to the GST beads. COS-7 cells were transiently transfected with pRShGRα and pRShGRαV423A; 48 hours later, cells were washed and scraped, and cell extracts were prepared. The hGRα and hGRαV423A were incubated with equal amounts of GST fusion proteins bound to glutathione-sepharose beads overnight at 4°C, washed, eluted, and fractionated by SDS-PAGE as described above.
Results
hGRαV423A demonstrates decreased transactivational activity and does not exert a dominant negative effect on the wild-type hGRα
Compared with the wild-type receptor, the mutant receptor hGRV423A demonstrated a 72% reduction in its ability to transactivate the glucocorticoid-inducible mouse mammary tumor virus promoter in response to dexamethasone. The concentration of dexamethasone required to achieve 50% of transactivation was 10−9 M for the wild-type and 10−8 M for the mutant receptor (Figure 1A). Cotransfection with a constant amount of hGRα and progressively increasing concentrations of hGRV423A demonstrated that hGRV423A did not exert a dominant negative effect on the wild-type receptor. Western blot analyses demonstrated no differences in the expression of hGRα and hGRαV423A proteins.
Figure 1.

Transcriptional defects of hGRαV423A. A, hGRαV423A demonstrated a 72% reduction in the ability to transactivate the mouse mammary tumor virus promoter in response to dexamethasone. CV-1 cells were transfected with the wild-type (WT) or the mutant (V423A) hGRα-expressing plasmid together with reporter plasmids and were treated with indicated concentrations of dexamethasone. Circles indicate mean and SEM values of the firefly luciferase activity corrected with the renilla luciferase activity obtained in at least 5 independent experiments. **P < .01, compared to the wild-type hGRα. B and C, Ability of hGRαV423A to bind to DNA. The mutant receptor hGRαV423A demonstrates reduced binding to GREs of endogenous hGR target genes. HCT116 human colon carcinoma cells defective in endogenous GR proteins were transfected with WT hGRα-expressing or the mutant receptor hGRαV423A-expressing plasmid and were treated with 10−6 M of dexamethasone. Cross-linked chromatin from the cells was used in the ChIP assays employing anti-GRα antibody or control IgG. Primers specific for the amplification of GREs of the GILZ (B) and G6Pase (C) genes were used. Bars demonstrate mean and SEM values of specific enrichments (fold differences) vs input (ChIP/Input = 2Input Ct−ChIP Ct) obtained in at least 3 independent experiments. *P = .010 and **P = .046. Ct, threshold cycle.
hGRαV423A demonstrates impaired binding to DNA
Following induction with dexamethasone, the mutant receptor hGRαV423A bound to GREs of the endogenous GILZ and G6Pase genes less efficiently than the wild-type receptor (Figure 1, B and C). These findings indicate that the mutant receptor hGRαV423A demonstrates a statistically significant decrease in the ability to bind to GREs compared with the wild-type hGRα and is therefore less efficient for mediating the genomic actions of glucocorticoids.
The V423A substitution alters the hydrophobic nature of the zinc-binding site at the DBD of the receptor and reduces the intramolecular interactions with R477
The molecular dynamics simulations show striking differences in conformation around the mutation site and are illustrated in Figure 2A (the simulation through the entire time of analysis is shown in Supplemental Movie 1, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org). The hydrophobic V at position 423 shields the 4 zinc-binding cysteines (C421, C424, C438, and C441) from water. Replacement of V by A at position 423 in the hGR DBD permits water to diffuse into the ion-binding region of the protein, where it is captured by hydrogen bonds to C424 and C441 among others. In the mutant, there is a significant (P < .05) 12-fold increase in hydrogen bonding of water to C424 (P < .05). Water is never hydrogen bonded to C441 in the wild-type, but water is observed bound to C441 for more than 21% of the simulation for the mutant (Figure 2B).
Figure 2.
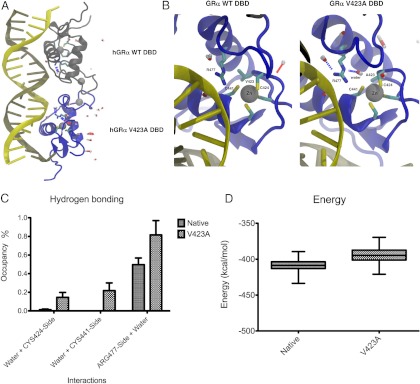
The V423A mutation alters the specific binding of hGR DBD to GREs. (A) Replacement of valine (V) by alanine (A) at position 423 in the hGR DBD reduces the hydrophobic nature of the site and permits water to diffuse into the ion-binding region of the protein, where it is captured by hydrogen bonds to C424 and C441 among others. Water is almost never observed in this area in the WT simulation. The structures in green indicate the WT receptor, whereas those in blue represent the mutant receptor. Water molecules are indicated by capsule-like molecules in gray and red. B, The most significant changes in hydrogen bonding in this area occur to C424, C441, and R477. C and D, The energy penalty in the V423A mutant for these nonnative interactions is greater than 15 kcal/mol. Given that R477 plays a key role for binding of the DBD of the receptor to GREs, the presence of the V423A mutation may explain the reduced binding of hGRαV423A to DNA and the subsequent reduction of its transcriptional activity.
The hydrophobic shielding by V423 extends to other nearby residues, including R477. In the wild-type, water hydrogen bonds to R477 for nearly half of the simulation, but in the V423A mutant, water hydrogen bonds to R477 for more than 80% of the simulation (P < .0001) and all hydrophobic interaction with A423 is lost (Figure 2B). The increased association of water with C424, C441, and R477 lead to a significant (P < .0001) gain of more than 15 kcal/mol of nonbonded energy for these residues and could therefore reduce the ability of R477 to specifically bind the phosphate backbone of DNA (Figure 2, C and D). R477 is critical for binding to GREs and a mutation at this position (R477H) abolishes DNA binding (8).
hGRαV423A demonstrates delayed nuclear translocation compared with the wild-type hGRα
In the absence of dexamethasone, both the wild-type and the mutant receptors were localized in the cytoplasm of cells. Addition of dexamethasone (10−6 M) resulted in translocation of hGRα and hGRαV423A into the nucleus within 13 minutes (12.6 ± 0.4 minutes) and 35 minutes (34.8 ± 1.4 minutes; P < .001), respectively. These findings suggest that the mutant receptor shows a 2.6-fold delay in translocating into the nucleus compared with the wild-type receptor.
hGRαV423A demonstrates similar affinity for the ligand compared with the wild-type hGRα
The apparent dissociation constant of hGRαV423A was similar to that of the wild-type receptor (6.7 ± 0.8 nM vs 6.0 ± 0.6 nM). No difference in the number of glucocorticoid receptor binding sites was noted between hGRα and hGRαV423A.
hGRαV423A displays normal interaction with the GRIP1 coactivator
Both the wild-type and the mutant receptor bound to full-length GRIP1, the carboxyl-terminal fragment of GRIP1 and the NRB fragment of GRIP1, indicating that hGRαV423A interacts with the GRIP1 coactivator in vitro through both its AF-1 and its AF-2 domains.
Discussion
In this study we systematically investigated the molecular mechanisms through which the V423A substitution impairs glucocorticoid signal transduction. Compared with the wild-type receptor, hGRαV423A demonstrated a 72% reduction in its ability to transactivate glucocorticoid-responsive genes, an impaired ability to bind to GREs, and a 2.6-fold delay in nuclear translocation. The mutant receptor did not exert a dominant negative effect on the wild-type hGRα, had a similar affinity for the ligand, and displayed a normal interaction with the GRIP1 coactivator in vitro. Therefore, the V423A causes generalized glucocorticoid insensitivity by affecting multiple steps in the cascade of hGR action.
In ChIP assays, hGRαV423A bound to the promoter regions of the endogenous glucocorticoid-induced GILZ and G6Pase genes less efficiently than the wild-type hGRα. Point mutations in the DBD of the hGR may abolish DNA binding, resulting in silencing of transcriptional activation, although they may not affect the ability of the mutant receptor to transrepress target genes, possibly through preserved protein-protein interactions and/or tethering of other cofactors to the transcriptional machinery (16–18).
Three-dimensional structural analysis demonstrated that the V423A mutation in the DBD of the receptor permits water to diffuse into the nearby zinc binding pocket, where it hydrogen bonds to the cysteines and reduces their affinity for zinc. The water also hydrogen bonds to R477 and reduces its affinity for the phosphate backbone of DNA. Because R477 plays a key role for binding of the DBD of the receptor to GREs, the presence of the V423A mutation may explain the reduced binding of hGRαV423A to DNA and the subsequent reduction of its transcriptional activity. Furthermore, the V423A mutation indirectly affects the function of NL-1 through R477, ultimately reducing the nuclear translocation of this mutant receptor (19, 20).
Finally, the normal affinity for the ligand and interaction with the GRIP1 coactivator of the mutant receptor concurs with previous observations (8) and reflects the integrity of the ligand-binding domain, given that the mutation is located in the DBD of the receptor.
We conclude that the mutant receptor hGRαV423A affects multiple steps in the cascade of the glucocorticoid receptor-signaling pathway, which primarily include decreased ability to bind to target GREs and delayed translocation into the nucleus. These findings expand on the previously described differential effects of hGR gene mutations on the glucocorticoid signal transduction pathway and may explain the differences observed in the clinical phenotype of affected subjects.
Supplementary Material
Acknowledgments
This work was supported in part by the University of Athens Medical School, Athens, Greece, the intramural program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ChIP
- chromatin immunoprecipitation
- Ct
- threshold cycle
- DBD
- DNA-binding domain
- G6Pase
- glucose-6-phosphatase
- GILZ
- glucocorticoid-induced leucine zipper
- GST
- glutathione-S-transferase
- hGR
- human glucocorticoid receptor.
References
- 1. Chrousos GP, Vingerhoeds A, Brandon D, et al. Primary cortisol resistance in man. A glucocorticoid receptor-mediated disease. J Clin Invest. 1982;69:1261–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chrousos GP, Detera-Wadleigh SD, Karl M. Syndromes of glucocorticoid resistance. Ann Intern Med. 1993;119:1113–1124 [DOI] [PubMed] [Google Scholar]
- 3. Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. 2008;93:1563–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Charmandari E, Kino T. Chrousos syndrome: a seminal report, a phylogenetic enigma and the clinical implications of glucocorticoid signalling changes. Eur J Clin Invest. 2010;40:932–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Charmandari E. Primary generalized glucocorticoid resistance and hypersensitivity. Horm Res Paediatr. 2011;76:145–155 [DOI] [PubMed] [Google Scholar]
- 6. Charmandari E, Kino T, Vottero A, Souvatzoglou E, Bhattacharyya N, Chrousos GP. Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: Molecular genotype, genetic transmission and clinical phenotype. J Clin Endocrinol Metab. 2004;89:1939–1949 [DOI] [PubMed] [Google Scholar]
- 7. Charmandari E, Raji A, Kino T, et al. A novel point mutation in the ligand-binding domain (LBD) of the human glucocorticoid receptor (hGR) causing generalized glucocorticoid resistance: the importance of the C terminus of hGR LBD in conferring transactivational activity. J Clin Endocrinol Metab. 2005;90:3696–3705 [DOI] [PubMed] [Google Scholar]
- 8. Charmandari E, Kino T, Ichijo T, Zachman K, Alatsatianos A, Chrousos GP. Functional characterization of the natural human glucocorticoid receptor (hGR) mutants hGRαR477H and hGRαG679S associated with generalized glucocorticoid resistance. J Clin Endocrinol Metab. 2006;91:1535–1543 [DOI] [PubMed] [Google Scholar]
- 9. Charmandari E, Kino T, Ichijo T, et al. A novel point mutation in helix 11 of the ligand-binding domain of the human glucocorticoid receptor gene causing generalized glucocorticoid resistance. J Clin Endocrinol Metab. 2007;92:3986–3990 [DOI] [PubMed] [Google Scholar]
- 10. Charmandari E, Kino T, Komianou E, Sertedaki A, Kassiou K, Chrousos GP. A novel point mutation in the DNA-binding domain of the human glucocorticoid receptor gene causing generalized glucocorticoid resistance. Proceedings of the 90th Endocrine Society Annual Meeting; San Francisco, CA, 2008:671 [Google Scholar]
- 11. Nader N, Chrousos GP, Kino T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB J. 2009;23:1572–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Litt MD, Simpson M, Recillas-Targa F, Prioleau MN, Felsenfeld G. Transitions in histone acetylation reveal boundaries of three separately regulated neighboring loci. EMBO J. 2001;20:2224–2235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. Comput Chem. 2005;26:1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38, 27–28 [DOI] [PubMed] [Google Scholar]
- 15. Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134 [DOI] [PubMed] [Google Scholar]
- 16. Liden J, Delaunay F, Rafter I, Gustafsson J, Okret S. A new function for the C-terminal zinc finger of the glucocorticoid receptor. Repression of RelA transactivation. J Biol Chem. 1997;272:21467–21472 [DOI] [PubMed] [Google Scholar]
- 17. Reichardt HM, Kaestner KH, Tuckermann J, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541 [DOI] [PubMed] [Google Scholar]
- 18. Tao Y, Williams-Skipp C, Scheinman RI. Mapping of glucocorticoid receptor DNA binding domain surfaces contributing to transrepression of NF-κB and induction of apoptosis. J Biol Chem. 2001;276:2329–2332 [DOI] [PubMed] [Google Scholar]
- 19. Picard D, Yamamoto KR. Two signals mediate hormone-dependent nuclear localization of the glucocorticoid receptor. EMBO J. 1987;6:3333–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Savory JG, Hsu B, Laquian IR, et al. Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol Cell Biol. 1999;19:1025–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.