

Abstract
We report a rare case of paraganglioma that developed in the mesentery of terminal ileum. A 78-year-old woman complained of right-sided abdominal pain. Abdominal computed tomography revealed a solid heterogeneously enhanced mass in the right lower abdomen. The tumor was laparoscopically excised. The mesenteric tumor was well circumscribed, ovoid, and encapsulated and measured 3 cm × 1.5 cm × 1.5 cm. Histological examination showed a cellular neoplasm comprised of nests and groups of tumor cells separated by fibrovascular connective tissue, giving a characteristic nested Zellballen pattern. Immunohistochemically, the tumor cells were positive for chromogranin, synaptophysin, CD56, and vimentin and negative for cytokeratins, SMA, CD34, CD117/c-kit and S100. On the basis of histologic and immunohistochemical features, a diagnosis of mesenteric paraganglioma was made. The operative and postoperative courses were unremarkable, and the patient was discharged on postoperative day 7. She was doing well 1 year after the surgery with no signs of recurrence. Extra-adrenal paragangliomas most commonly develop adjacent to the aorta, particularly the area corresponding to the organ of Zuckerkandl. Mesenteric paraganglioma, as in our case, is extremely rare; only 11 cases have been reported in the literature. We herein discuss the clinical findings of these cases.
Keywords: Mesenteric tumor, Extra-adrenal paraganglioma, Pheochromocytoma, Surgical management, Preoperative diagnosis
INTRODUCTION
Paraganglia are groups of morphologically and cytochemically similar cells derived from the neural crest. They include such tissues as the adrenal medulla, carotid and aortic bodies, organs of Zuckerkandl, and other unnamed paraganglia in the distribution of sympathetic and parasympathetic nerves.
Paragangliomas are uncommon tumors arising from the neuroendocrine elements (chief cells) of the paraganglia. However, they have been described in virtually every site in which normal paraganglia are known to occur; only 5%-10% of sporadic paragangliomas are extra-adrenal[1-3]. Paraganglioma as a mesenteric mass is extremely rare, and only occasional reports have been published. The present case report describes a quite rare mesenteric paraganglioma, including its imaging features and histopathological characteristics. In addition, a review of the current literature summarizes the clinical findings associated with mesenteric paragangliomas.
CASE REPORT
A 78-year-old woman, who underwent distal gastrectomy for early gastric cancer in 1994 and total thyroidectomy for papillary thyroid carcinoma in 2000 was followed up at our hospital. In June 2010, she complained of right-sided abdominal pain. Abdominal computed tomography (CT) revealed a solid mass, 16 mm × 22 mm × 25 mm in size, in the right lower abdomen. Contrast-enhanced CT showed a smoothly marginated, heterogeneously enhanced hypervascular tumor adjacent to the right major psoas muscle (Figure 1A and B). Magnetic resonance imaging (MRI) showed that the lesion was hypointense on T1-weighted images and hyperintense on T2-weighted images. After the bolus infusion of gadolinium chelate, the lesion had marked contrast enhancement on T1-weighted images (Figure 1C-E). Whole-body 18F-fluorodeoxyglucose (FDG)-positron emission tomography (PET) was negative (Figure 1F), and subsequent upper gastrointestinal endoscopy and colonoscopy were not remarkable. Laboratory studies yielded normal blood chemistry and hematology results. The carcinoembryonic antigen and carbohydrate antigen 19-9 levels were both within normal limits. In retrospect, follow-up CT after gastrectomy in 2002 already showed the tumor, which was 16 mm × 13 mm × 15 mm in size and was not pointed out at that time. For 8 years, the tumor had been slowly but definitely growing.
Figure 1.
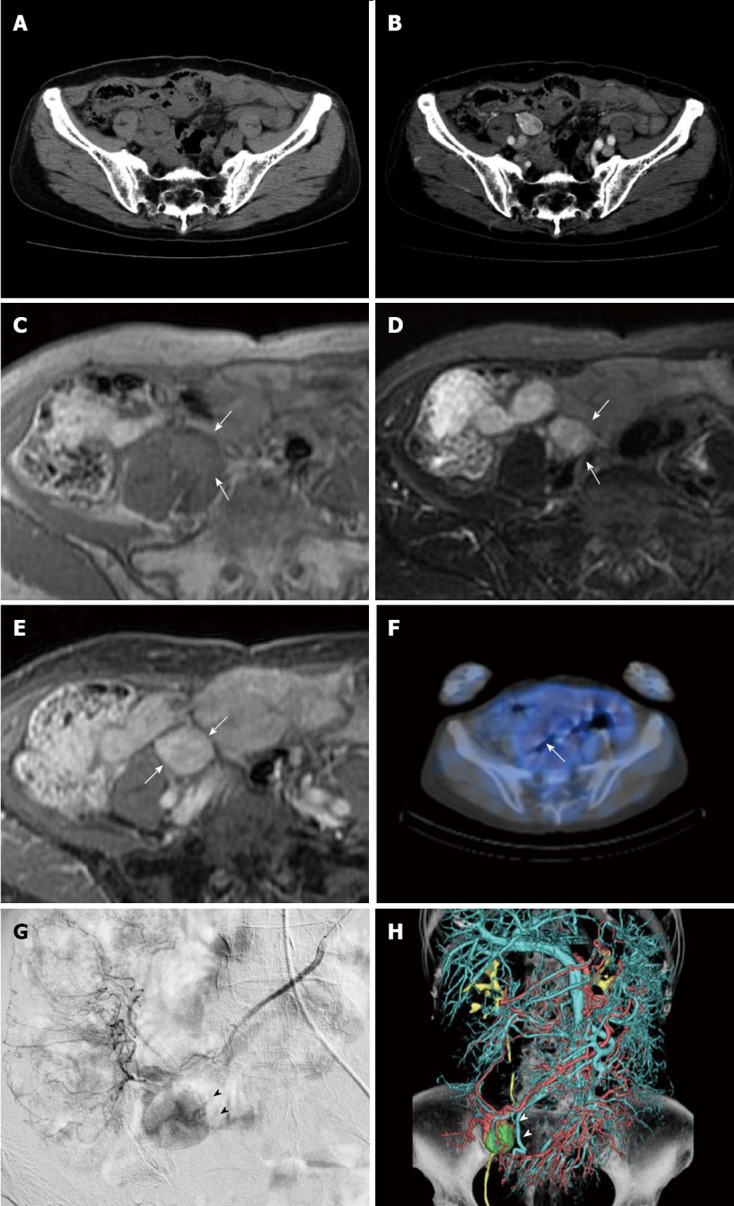
Imaging features of tumor (white arrows) before treatment. A: Axial plain; B: contrast-enhanced computed tomography (CT), CT shows a smoothly marginated, heterogeneously enhanced tumor adjacent to the right major psoas muscle, 16 mm × 22 mm × 25 mm in size; C: T1-weighted magnetic resonance image shows a well defined, isointense mass; D: On T2-weighted images, the mass shows heterogeneous high intensity; E: On T1-weighted images after a bolus infusion of gadolinium chelate, the mass had marked contrast enhancement; F: Positron emission tomography-CT scan was negative; G: Superior mesenteric arteriography displays a markedly hypervascular mass (black arrow heads) adjacent to the terminal ileum; H: Volume rendering image acquired from angio-CT (white arrow heads).
For a definitive diagnosis, surgical resection was recommended to the patient, and she was admitted to our hospital. Physical examination showed a blood pressure of 118/80 mmHg and a regular pulse of 68 bpm. On angiography, the tumor appeared as a hypervascular lesion fed by the superior mesenteric artery (Figure 1G and H). Before surgery, although the differential diagnosis included gastrointestinal stromal tumors, leiomyoma and Castleman’s disease, we could not definitively diagnose this tumor.
In March 2011, exploratory laparoscopy confirmed a solid, brownish-red mass in the mesentery of the terminal ileum. There was no lymph node swelling or ascites. Throughout the exploration, there was no remarkable fall or rise in blood pressure. The mass was excised under laparoscopy without ileum resection. Grossly, the mesenteric mass was well circumscribed, ovoid, and encapsulated and measured 3 cm × 1.5 cm × 1.5 cm (Figure 2A). Histological examination showed a cellular neoplasm comprised of nests and groups of tumor cells separated by fibrovascular connective tissue, giving a characteristic nested Zellballen pattern (Figure 2B). Immunohistochemically, the tumor cells were positive for chromogranin, synaptophysin, CD56, and vimentin and negative for cytokeratins, SMA, CD34, CD117/c-kit, and S100. The proportion of Ki-67-positive cells was low (Figure 2C-E).
Figure 2.
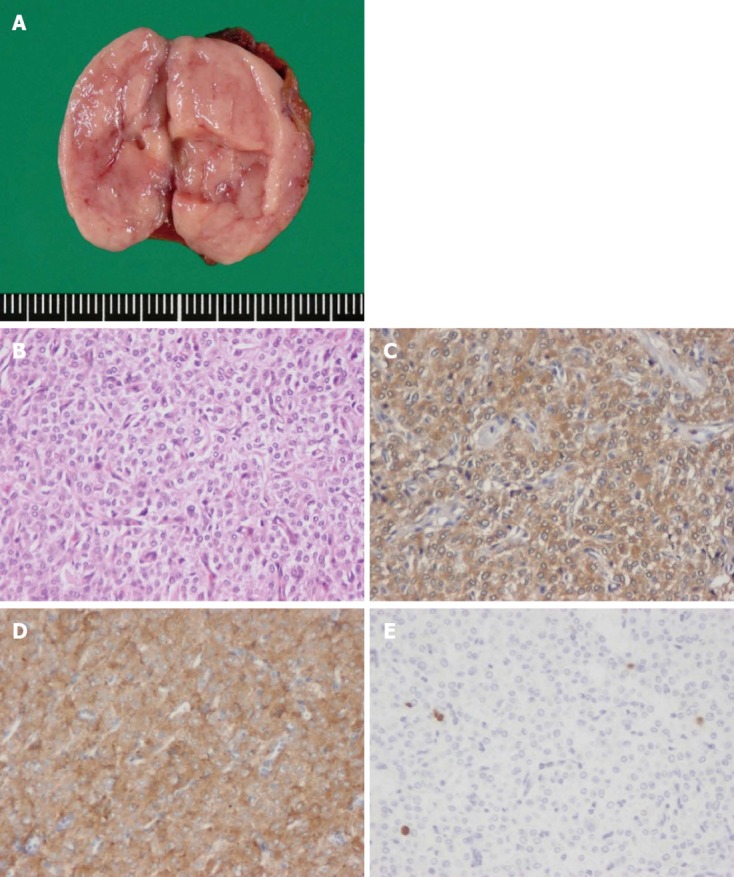
Macroscopic findings and pathological features of the resected tumor. A: Gross findings of the resected specimen. The tumor was encapsulated and measured 3 cm × 1.5 cm × 1.5 cm; B: The paraganglioma comprised a dual cell population arranged in a characteristic nested Zellballen pattern (HE stain, × 400); C: Immunohistochemistory of Chromogranin A, × 400; D: Synaptophysin were strongly positive and confirmed a neuroendocrine origin, supporting the diagnosis of paraganglioma, × 400; E: The MIB-1 labeling index, × 400.
On the basis of histologic and immunohistochemical features, a diagnosis of mesenteric paraganglioma was made. The operative and postoperative courses were unremarkable, and the patient was discharged on postoperative day 7. She was doing well 1year after the surgery with no signs of recurrence.
DISCUSSION
Paraganglioma is a rare tumor of neural crest cell origin that arises from sympathetic or parasympathetic neural paraganglia. While the most common location of paragangliomas is the adrenal medulla, where they give rise to pheochromocytomas, approximately 5%-10% of sporadic paragangliomas occur in extra-adrenal sites[1-4]. Although extra-adrenal paragangliomas may develop in every site in which normal paraganglia exist, 70%-85% of cases actually occur intra-abdominally, most commonly adjacent to the aorta and particularly the area corresponding to the organ of Zuckerkandl[3,4]. Paragangliomas that develop in the mesentery, as in our case, are extremely rare, with only 11 cases in the literature[3] (Table 1).
Table 1.
Clinical characteristics of the 12 reported cases of mesenteric paraganglioma
| No. of cases | Ref. | Age (yr) | Sex | Location | Symptoms | Size (cm) | Hypertension | Preoperative diagnosis | Surgical procedures | Prognosis |
| 1 | Arean et al[18] | 32 | M | Mesentery of the small intestine | Nausea, vomiting, diarrhea | 10 × 7 × 6 | - | Abdominal mass | Resection of the intestine and its mesentery along with mass | 8 mo: Alive without recurrence |
| 2 | Carmichael et al[20] | 62 | F | Mesentery of the small intestine | Nausea, vomiting, back pain | 3.2 | + | Abdominal mass | Resection of the intestine and its mesentery along with mass | Not documented |
| 3 | Tanaka et al[20] | 29 | F | Descending colon | Nausea, vomiting | 10 × 9 × 7 | - | Retroperitoneal mass | Resection of themass | 32 mo: Alive without recurrence |
| 4 | Ishikura et al[21] | 33 | F | Sigmoid colon | Lower abdominal pain, dysuria | 15 × 15 × 15 | - | Ovarian tumor | Resection of the sigmoid colon and its mesentery along with mass | Not documented |
| 5 | Onoue et al[22] | 38 | F | Mesentery of the small intestine | None | 4.5 × 3.2 | - | Mesenteric tumor | Resection of the intestine and its mesentery along with mass | 24 mo: Alive without recurrence |
| 6 | Jaffer et al[3] | 76 | M | Mesentery of the small intestine | Abdominal mass, vomiting, diarrhea | 8.5 × 8 | + | Abdominal mass | Resection of the intestine and its mesentery along with mass | Not documented |
| 7 | Muzaffar et al[23] | 76 | F | Mesentery of the small intestine | Abdominal mass | 20 × 15 | - | Abdominal mass | Not documented | 15 mo: Alive without recurrence |
| 8 | Ponsky et al[24] | 35 | F | Mesentery of the small intestine | Abdominal mass, headache | 5.5 | + | Abdominal mass | Resection of the intestine and its mesentery along with mass | 24 mo: Alive without recurrence |
| 9 | Kudoh et al[25] | 72 | F | Mesentery of the small intestine (ileum) | Abdominal pain and mass | 10 × 9 × 9 | - | Mesenteric tumor | Resection of segment of ileum and mesentery containing mass | 12 mo: Alive without recurrence |
| 10 | Nobeyama et al[26] | 53 | M | Mesentery of the small intestine (ileum) | Abdominal mass | 15 × 10 × 7 | - | Abdominal mass | Resection of segment of ileum and mesentery containing mass | Not documented |
| 11 | Matsumoto et al[27] | 77 | F | Mesentery of the small intestine (near Bauhin's valve) | Abdominal mass | 7 × 5.5 | - | Mesenteric tumor | Resection of segment of ileum and mesentery containing mass | 9 mo: Alive without recurrence |
| 12 | Present case | 78 | F | Mesentery of the small intestine (near Bauhin's valve) | None | 3 × 1.5 × 1.5 | - | Mesenteric tumor | Resection of themass | 8 mo: Alive without recurrence |
M: Male; F: Female.
As shown in Table 1, there appears to be a marked predilection for females (9:3), which contrasts with the slight male predominance (1.3:1) reported for retroperitoneal paraganglioma[5,6]. At the time of diagnosis, most patients are older (median, 57.5 years of age) than those with retroperitoneal paraganglioma (median, 39-43 years of age[4-6]). No significant difference was noted in the size of mesenteric (average, 9.3 cm) and retroperitoneal tumors (average, 7.4-10.5 cm[4-6]).
The pathogenesis of paragangliomas is not fully understood. They may be either sporadic or hereditary. Overall, as many as 10%-50% of paragangliomas are considered to be hereditary[7]. Hereditary paragangliomas are multicentric in 20%-50% of cases[8,9], whereas sporadic paragangliomas are multicentric in 10% of cases. In hereditary cases, they may be associated with multiple endocrine neoplasia type 2, von Hippel-Lindau disease, familial paraganglioma, Carney triad and neurofibromatosis type 1[10]. For this reason, especially in patients diagnosed before 50 years of age and in those who present with bilateral, multifocal, and malignant paragangliomas, genetic testing may be beneficial[11]. In the present case, the tumor was solitary and the patient was a 78-year-old woman with no history of genetic disorders; thus, genetic screening was not performed.
From a diagnostic viewpoint, functional tumors are easier to diagnose. Most patients undergo paroxysmal episodic hypertension and the typical triad of symptoms associated with pheochromocytoma: palpitations, headache, and profuse sweating. When functional paraganglioma is suspected, biochemical analysis of catecholamine hypersecretion should precede any form of imaging.
However, a majority of extra-adrenal paraganglioma is nonfunctional[11], as in our case. A large proportion of these tumors are incidentally discovered in normotensive patients during imaging evaluation for other reasons. In addition, the CT features of extra-adrenal paraganglioma include a nonspecific soft tissue density and overlap those of other neoplasms. Specifically, tumors of neural or mesodermal origin and those of metastatic disease must be considered[1]. Thus, because of their clinical manifestation and the overlap with other tumors in terms of medical imaging findings, the preoperative diagnosis of extra-adrenal paraganglioma is usually difficult. Especially when extra-adrenal paragangliomas arise from unusual sites, as in the present case, accurate diagnosis is seldom made preoperatively (Table 1).
The MRI characteristics of our case are quite typical for paraganglioma. Paragangliomas have low signal intensity on T1-weighted images and enhance strongly after administration of contrast material. On T2-weighted images, they appear hyper intense. In addition, a speckled appearance with multiple flow voids is typical in tumors > 2 cm in diameter[12]. Angiography was thus useful to outline the location and vascular supply of the tumor in our case; theoretically, however, clinically silent functional tumors should be ruled out by urine analysis before manipulation.
In functional paraganglioma, 131I-metaiodobenzylguanidine (MIBG) scintigraphy is the best imaging study for a preoperative diagnosis. MIBG scintigraphy may also be helpful to rule out clinically silent cases, but the specificity for diagnosis of nonfunctional paraganglioma is unclear[13]. In certain cases, FDG-PET may be indicated to investigate metastatic disease[7]. It was recently reported that the newest technique using fluorine-18-dihydroxyphenylalanine-PET imaging offers even higher accuracy than MIBG scintigraphy in the localization of paragangliomas[14].
In the case described here, diagnostic imaging played a very important role preoperatively to determine tumor localization, vascularity, and extent of disease. Differential diagnosis including gastrointestinal stromal tumors, leiomyoma, malignant lymphoma, Castleman’s disease and other metastatic tumor could be made preoperatively. However, pitfall for misdiagnosis in our case was tumor location. Because of the tumor location away from the para-aortic area, a preoperative diagnosis of paraganglioma could not be made. Although rare, paraganglioma should be included in the preoperative differential diagnosis of solid hypervascular mesenteric tumors.
The treatment of choice for paraganglioma is surgical resection. As shown in Table 1, most tumors were excised along with a segment of small bowel, probably because of the large tumor size and intestinal vascularity. From the viewpoint of lymph node dissection, however, recurrence in cervical lymph node was reported for retroperitoneal paraganglioma[5], neither local nor distant lymph node metastasis was reported for mesenteric paragangliomas.
With regard to malignant potential, the incidence of malignant change reportedly ranges from 14% to 50%[15,16]. In these reports, the clinical and histological distinction between benign and malignant tumors was unclear, and the definitive diagnosis of malignancy was based solely on the presence of metastases. The distinction of endocrine tumors was recently well defined according to the World Health Organization classification[17]. In particular, mitotic counts and the Ki-67 labeling index are of considerable significance in grading its malignant potential.
In the present case, the Ki-67 labeling index was low and mitoses were rare. The tumor presented as a well circumscribed mass with no metastases. The patient was considered to be at low risk of malignancy. However, in retroperitoneal paraganglioma, the 5- and 10-year disease-free survival rates were 75% and 45% even after successful resection, indicating that more than half of these patients will experience a relapse if followed long enough after resection[5]. Although recurrence of mesenteric paraganglioma has not been reported, long-term follow-up after surgical excision is likely to be necessary.
In conclusion, mesenteric paraganglioma is a very rare entity with a limited number of cases reported. Preoperative diagnosis of extra-adrenal paraganglioma in asymptomatic patients is usually difficult. Although rare, paraganglioma should be included in the preoperative differential diagnosis of solid mesenteric tumors. Even after complete resection, patients should continue to be followed up carefully.
Footnotes
P- Reviewers Karmazanovsky GG, Guan YS S- Editor Wen LL L- Editor A E- Editor Xiong L
References
- 1.Hayes WS, Davidson AJ, Grimley PM, Hartman DS. Extraadrenal retroperitoneal paraganglioma: clinical, pathologic, and CT findings. AJR Am J Roentgenol. 1990;155:1247–1250. doi: 10.2214/ajr.155.6.2173385. [DOI] [PubMed] [Google Scholar]
- 2.Vázquez-Quintana E, Vargas R, Pérez M, Porro R, Gómez Duarte C, Tellado M, Marcial M. Pheocromocytoma and gastrointestinal bleeding. Am Surg. 1995;61:937–939. [PubMed] [Google Scholar]
- 3.Jaffer S, Harpaz N. Mesenteric paraganglioma: a case report and review of the literature. Arch Pathol Lab Med. 2002;126:362–364. doi: 10.5858/2002-126-0362-MP. [DOI] [PubMed] [Google Scholar]
- 4.Lack EE, Cubilla AL, Woodruff JM, Lieberman PH. Extra-adrenal paragangliomas of the retroperitoneum: A clinicopathologic study of 12 tumors. Am J Surg Pathol. 1980;4:109–120. doi: 10.1097/00000478-198004000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Sclafani LM, Woodruff JM, Brennan MF. Extraadrenal retroperitoneal paragangliomas: natural history and response to treatment. Surgery. 1990;108:1124–1129; discussion 1129-1130. [PubMed] [Google Scholar]
- 6.Cunningham SC, Suh HS, Winter JM, Montgomery E, Schulick RD, Cameron JL, Yeo CJ. Retroperitoneal paraganglioma: single-institution experience and review of the literature. J Gastrointest Surg. 2006;10:1156–1163. doi: 10.1016/j.gassur.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Young WF. Paragangliomas: clinical overview. Ann N Y Acad Sci. 2006;1073:21–29. doi: 10.1196/annals.1353.002. [DOI] [PubMed] [Google Scholar]
- 8.Robertson JH, Gardner G, Cocke EW. Glomus jugulare tumors. Clin Neurosurg. 1994;41:39–61. [PubMed] [Google Scholar]
- 9.Lo WW, Solti-Bohman LG. Tumors of the temporal bone and the cerebellopontine angle. In: Som PM, Curtin HD, editors. Head and neck imaging, 3rd ed. St Louis, MO: Mosby-Year Book; 1996. pp. 1449–534. [Google Scholar]
- 10.Bertherat J, Gimenez-Roqueplo AP. New insights in the genetics of adrenocortical tumors, pheochromocytomas and paragangliomas. Horm Metab Res. 2005;37:384–390. doi: 10.1055/s-2005-870156. [DOI] [PubMed] [Google Scholar]
- 11.Bhatt S, Vanderlinde S, Farag R, Dogra VS. Pararectal paraganglioma. Br J Radiol. 2007;80:e253–e256. doi: 10.1259/bjr/21661275. [DOI] [PubMed] [Google Scholar]
- 12.Olsen WL, Dillon WP, Kelly WM, Norman D, Brant-Zawadzki M, Newton TH. MR imaging of paragangliomas. AJR Am J Roentgenol. 1987;148:201–204. doi: 10.2214/ajr.148.1.201. [DOI] [PubMed] [Google Scholar]
- 13.van Gils AP, Falke TH, van Erkel AR, Arndt JW, Sandler MP, van der Mey AG, Hoogma RP. MR imaging and MIBG scintigraphy of pheochromocytomas and extraadrenal functioning paragangliomas. Radiographics. 1991;11:37–57. doi: 10.1148/radiographics.11.1.1671719. [DOI] [PubMed] [Google Scholar]
- 14.Brink I, Schaefer O, Walz M, Neumann HP. Fluorine-18 DOPA PET imaging of paraganglioma syndrome. Clin Nucl Med. 2006;31:39–41. doi: 10.1097/01.rlu.0000191577.39458.a0. [DOI] [PubMed] [Google Scholar]
- 15.Lack EE. Tumors of the adrenal gland and extra-adrenal paraganglia. In: Atlas of tumor Pathology, 3rd series, Fascicle19 , editors. Washington DC: Armed Forceds Institute of Pathology; 1974. [Google Scholar]
- 16.Linnoila RI, Keeiser HR, Steinberg SM, Lack EE. Histopathology of benign versus malignant sympathoadrenal paragangliomas: Clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol. 1990;21:1168–1180. doi: 10.1016/0046-8177(90)90155-x. [DOI] [PubMed] [Google Scholar]
- 17.Zheng YY, Chen G, Zhou XG, Jin Y, Xie JL, Zhang SH, Zhang YN. [Retrospective analysis of 4 cases of the so-called blastic NK-cell lymphoma, with reference to the 2008 WHO classification of tumours of haematopoietic and lymphoid tissues] Zhonghua Binglixue Zazhi. 2010;39:600–605. [PubMed] [Google Scholar]
- 18.Arean VM, Ramirez DE, Arellano GA. Intra-abdominal non-chromaffin paraganglioma. Ann Surg. 1956;144:133–137. doi: 10.1097/00000658-195607000-00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carmichael JD, Daniel WA, Lamon EW. Mesenteric chemodectoma. Report of a case. Arch Surg. 1970;101:630–631. doi: 10.1001/archsurg.1970.01340290086021. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka S, Ooshita H, Kaji H. Extraadrenal paraganglioma of the mesenterium. Rinsyo geka. 1991;46:503–506. [Google Scholar]
- 21.Ishikura H, Miura K, Morita J. A case of mesenteric paraganglioma. Syokakigeka. 1996;19:651–655. [Google Scholar]
- 22.Onoue S, Katoh T, Chigura H, Matsuo K, Suzuki M, Shibata Y. A case of malignant paraganglioma arising in the mesentery. J Jpn Surg Assoc. 1999;60:3297–3300. [Google Scholar]
- 23.Muzaffar S, Fatima S, Siddiqui MS, Kayani N, Pervez S, Raja AJ. Mesenteric paraganglioma. Can J Surg. 2002;45:459–460. [PMC free article] [PubMed] [Google Scholar]
- 24.Ponsky LE, Gill IS. Laparoscopic excision of suspected extra-adrenal pheochromocytoma located in the mesenteric root. J Endourol. 2002;16:303–305. doi: 10.1089/089277902760102794. [DOI] [PubMed] [Google Scholar]
- 25.Kudoh A, Tokuhisa Y, Morita K, Hiraki S, Fukuda S, Eguchi N, Iwata T. Mesenteric paraganglioma: report of a case. Surg Today. 2005;35:594–597. doi: 10.1007/s00595-004-2966-3. [DOI] [PubMed] [Google Scholar]
- 26.Nobeyama I, Sano T, Yasuda K, Kikuchi C, Sone K, Kudo J, Oikawa M, Tamahashi N. [Case report of a paraganglioma of the mesenterium] Nihon Shokakibyo Gakkai Zasshi. 2004;101:998–1003. [PubMed] [Google Scholar]
- 27.Matsumoto K, Hirata K, Kanemitsu S, Kawakami S, Aoki T, Nagata N, ITO H. A case of mesenteric paraganglioma. Nihon Shokaki Geka Gakkai Zasshi. 2006;39:84–89. [Google Scholar]