

Abstract
Airways hyperresponsiveness (AHR) is usually produced within days of first antigen exposure in mouse models of asthma. Furthermore, continual antigen challenge eventually results in the resolution of the AHR phenotype. Human asthma also waxes and wanes with time, suggesting that studying the time course of AHR in the allergic mouse would offer insights into the variation in symptoms seen in asthmatics.
Mice were sensitized with ovalbumin (OVA) on days 0 and 14. As assessed by airway resistance (Rn), lung elastance (H) and tissue damping (G), AHR was measured post an OVA inhalation on day 21 (Short Challenge group), after three days of OVA inhalation on day 25 (Standard Challenge group) and following an OVA inhalation on day 55 in mice previously challenged on days 21–23 (Recall Challenge group). Bronchoalveolar lavage was analyzed for inflammatory cells, cytokines and protein.
AHR in the Short Challenge group was characterized by an increase in Rn and neutrophil accumulation in the lavage. AHR in the Standard Challenge group was characterized by increases in H and G but by only a modest response in Rn, while inflammation was eosinophilic. In the Standard Challenge protocol, mice lacking fibrinogen were no different from control in their AHR response. AHR in the Recall Challenge group was characterized by increases only in G and H and elevated numbers of both neutrophils and eosinophils. Lavage cytokines were only elevated in the Recall Challenge group. Lavage protein was significantly elevated in all groups.
The phenotype in allergically inflamed mice evolves distinctly over time, both in terms of the nature of the inflammation and the location of the AHR response. The study of mouse models of AHR might be better served by focusing on this variation rather than simply on a single time point at which AHR is maximal.
Introduction
One of the key features of asthma is airways hyperresponsiveness (AHR), so this phenotype is a prerequisite in animal models to be used for preclinical asthma research. However, AHR can arise through a variety of different mechanisms, so its mere presence does not guarantee relevance to human asthma. For example, we have shown that AHR in allergic mice is almost entirely reflective of increased closure of small airways in the lung periphery cause by an inflamed and mucus laden epithelium [1–3]. By contrast, we have also shown that direct application of cationic protein to the airways produces AHR as a result of increased smooth muscle contraction, likely due to loss of integrity of the epithelial barrier that normally acts to protect the underlying smooth muscle from agents entering the airway lumen [4,5]. The extent to which either of the above mechanisms of AHR mimics the situation in human asthma remains an open question; indeed, it is possible that both are operative to some degree.
What is more troubling, however, about current mouse models of asthma from the perspective of relevance to the human disease is the acuteness with which AHR is induced. Human asthma is typically a chronic condition that often has a history extending decades back in time, and for which the instigating factor is invariably obscure. This contrasts rather starkly with the production of AHR in a mouse, which is typically manifest via sensitization to and challenge with a foreign antigen in combination with an adjuvant over a few days or weeks [6]. Furthermore, the AHR phenotype in allergically inflamed mice is transient, waxing and waning over the course of a month or so due to the phenomenon of immune tolerization even in the face of continual antigenic challenge [7–10]. These inconvenient facts are often ignored by asthma researchers.
On the other hand, although human asthma is a chronic condition, its inflammatory and symptomatic manifestations generally fluctuate with time due to factors such as variation in seasonal allergens as well as unexplained periods of remission [11]. This led us to suspect that the transient asthma-like phenotype in allergically inflamed mice may be more appropriately viewed as recapitulating an asthma fluctuation rather than as being a model of the entire disease. Such a perspective points to the importance of understanding the temporary dynamics of the allergic AHR phenotype, rather than simply focusing on its most pronounced manifestation at a single point in time. Accordingly, we set out in the present study to examine how the AHR phenotype evolves over time in an ovalbumin sensitized and challenged mouse model of allergic lung disease.
Methods
Animals
Female BALB/cJ mice were purchased from Jackson Laboratories (Bar Harbor, ME). Fibrinogen knock-out mice, littermate heterozygotes and homozygotes (Fgn−/−, Fgn+/− and Fgn+/+) on a C57BL/6J background were bred in house. The mice were housed in an AAALAC and USDA accredited animal facility at the University of Vermont fully equipped for laboratory animal care. The study was approved by the Institutional Animal Care and Use Committee at the University of Vermont.
Assessment of airway hyperresponsiveness (AHR)
The mice were anaesthetized with i.p. sodium pentobarbital (90mg/kg), the trachea cannulated and connected to a flexiVent™ computer controlled small animal ventilator (SCIREQ, QC, Canada), as previously described [12,13,1], and ventilated at 200 breaths/minute. Next the mice were paralyzed with pancuronium bromide i.p. (0.8μg/ kg). The animals were stabilized over about ten minutes of regular ventilation at a positive end-expiratory pressure (PEEP) of 3cm H2O. A standard lung volume history was then established by delivering two total lung capacity maneuvers (TLC) to a pressure limit of 25cm H2O and holding for three seconds. Next, two baseline measurements of respiratory input impedance (Zrs) were obtained. This was followed by an inhalation of aerosolized PBS (control) for 10 s, achieved by an in-line piezo electric nebulizer (Aeroneb, Aerogen, Galway, Ireland). Zrs was then measured every 10 s for 3 min (18 measurements of Zrs in total). This complete sequence of maneuvers and measurements was then repeated for aerosol exposures to three incremental doses of methacholine (3.125, 12.5 and 25 mg/ml). Airways responsiveness was quantified in terms of the average of the 18 measurements obtained at the highest methacholine dose.
Lung mechanics
Zrs over the frequency range 1–20.5 Hz was determined using a two second broadband perturbation in volume applied by the flexiVent. Each determination of Zrs was fit with the constant phase model of impedance [14] given by
| (Eq.1) |
where Rn is a frequency independent Newtonian resistance reflecting that of the conducting airways, I is airway gas inertance, G characterizes tissue resistance, H characterizes tissue stiffness, i is the imaginary unit, and f is frequency in Hz [15,14].
Broncho alveolar lavage analysis
At the end of the AHR protocol the mice were euthanized with a lethal dose of sodium pentobarbital (150mg/kg, i.p.) and the lungs were lavaged with 1 ml of phosphate buffered saline. Total cell counts were obtained, and the lavage was centrifuged and the supernatant used for analysis of cytokines (Bio-Plex®). The cell pellet was then re-suspended and cytospin slides prepared for cell differentials using Hematoxylin - Eosin stain. As it has previously been shown that plasma extravasation can be an important part of the response to inflammatory stimuli such as an antigen challenge [16], we also analyzed the BALF for protein content using standard Bradford analysis. Protein content was calculated using a colorimetric assay (Bio-Rad Laboratories, Hercules, CA), standardized to graded concentrations of bovine serum albumin (BSA).
Statistics
Statistical testing was performed by one-way ANOVA with Bonferroni post-hoc test. A p<0.05 was accepted as a statistically significant difference.
Experimental design
Female BALB/cJ mice, 6 – 8 weeks of age, were sensitized and challenged with chicken ovalbumin (OVA). Briefly, on days 0 and 14, animals were injected (100μl, intraperitoneal - i.p.) with OVA (20μg) emulsified in 2.25mg of aluminum hydroxide/magnesium hydroxide. Control animals had the i.p. injections with OVA + alum but received phosphate buffered saline (PBS) inhalations. Airways responsiveness was determined after subjecting these mice, in separate groups, to 3 different OVA inhalation challenge protocols, as follows (Figure 1):
Figure 1. Outline of the experiments.
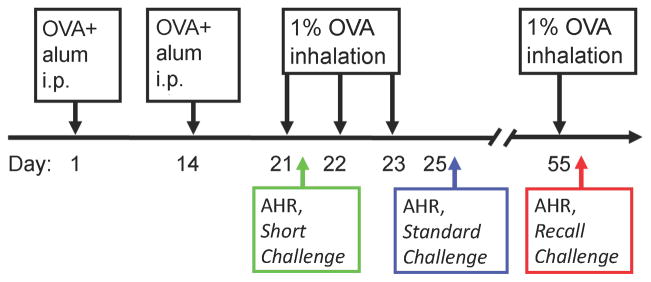
BALB/cJ mice were sensitized with i.p. injections of alum and OVA and then challenged by inhalation of 1% OVA for 30 minutes as indicated. Mice were harvested at three different time points; 16 hrs post the first OVA challenge (Short Challenge), 48 hrs post three repeated OVA challenges (Standard Challenge) and 16 hrs after a single recall OVA challenge at day 55 in mice that had also been OVA challenged on days 21–23(Recall Challenge). Control mice received i.p. OVA+alum but only PBS inhalations. At each harvest respiratory mechanics were assessed and BALF collected.
Short challenge
Mice were exposed to a dilute (1%) OVA aerosol for 30 minutes 1 week after the second i.p. injection and then studied 16 hours later (n = 6 – 8 per group).
Standard challenge
Mice were exposed to a dilute (1%) OVA aerosol for 30 minutes on each of 3 consecutive days beginning each 1 week after the second i.p. injection and then studied 48 hours later (n = 6 – 8 per group).
Recall challenge
Mice were exposed to a dilute (1%) OVA aerosol for 30 minutes on each of 3 consecutive days beginning each 1 week after the second i.p. injection. They were given an additional 30 minute aerosol challenge 32 days later and studied 16 after that (n = 8 per group).
Results
Airways responsiveness in the Short Challenge group was characterized by an increase in Rn (Figure 2), which we interpret as reflecting contraction of the smooth muscle surrounding the conducting airways. Inflammation was dominated mostly by neutrophil accumulation in the BALF (Figure 3) and by IL-4 and IL-5 in the cytokine profile (Figure 4). Despite the presence of neutrophils, no IL-17 was detected in the BALF, in agreement with our previous observation that OVA+ alum sensitization does not elicit a Th17 response [17].
Figure 2. Airways hyperresponsiveness to inhaled methacholine.
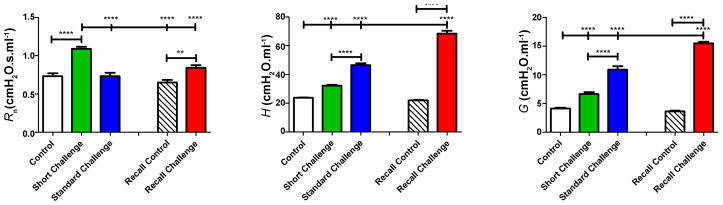
BALB/cJ mice were sensitized and challenged with OVA. AHR was assessed 16 hrs after the first OVA inhalation (green bars, short Challenge), 48 hrs after 3 daily exposures to OVA (blue bars, Standard Challenge) or 16 hrs after a single inhalation of OVA one month after the original challenge (red bars, Recall Challenge). Control mice received i.p. sensitizations but control PBS inhalations (white bars). Following a single exposure to inhaled OVA, methacholine produced a response dominated by an increase in Rn. In contrast, after 3 exposures to OVA the methacholine response was dominated by H and G. After a single recall challenge, the response to methacholine was entirely dominated by increases in G and H. **p<0.01, ****p<0.0001.
Figure 3. Cell counts and differentials from bronchoalveolar lavage.

Neutrophils dominated the BALF after a single OVA challenge (green bars, Short Challenge). Eosinophils dominated the BALF after 3 exposures to OVA (blue bars, Standard Challenge). After a recall challenge, the BALF had a mix of neutrophils and eosinophils (red bars, Recall Challenge). Neither challenge protocol generated significant levels of lymphocytes. **p<0.01, ***p<0.001, ****p<0.0001.
Figure 4. Cytokine titers in bronchoalveolar lavage.

IL-4, IL-5, IL-10, IL-13 and eotaxin were significantly increased after the recall challenge with OVA (red bars, Recall Challenge). IL-5 and Eotaxin were increased after a single OVA challenge (green bars, Short Challenge) whereas eotaxin was the only cytokine increased in the Standard Challenge group (blue bars). *p<0.05, ***p<0.001, ****p<0.0001.
In contrast, responsiveness in the Standard Challenge group was characterized by increases in H and G, while Rn was only modestly elevated relative to control (Figure 2). At this time point, inflammation was dominated by an accumulation of eosinophils (Figure 3) with no cytokine being significantly elevated above its control level (Figure 4).
Finally, responsiveness in the Recall Challenge group was characterized entirely by large increases in G and H relative to control (Figure 2), while the inflammatory picture contained a mix of neutrophils and eosinophils (Figure 3) and elevations in IL-4, IL-5, IL-10, IL-13 and eotaxin, with no detectable IL-17 (Figure 4).
Total protein content in the BALF increased significantly and progressively with the duration of the challenge protocols (Figure 5). We conducted a limited investigation using the Standard Challenge protocol to determine if lack of fibrinogen would modulate the AHR. Figure 6 shows that wild type control C57BL/6J mice have a significantly increased response to aerosolized methacholine relative to control mice, but that Fgn−/− mice are no different from control Fgn+/+ in their response. Fgn+/− mice had an intermediate level of AHR.
Figure 5. Protein in bronchoalveolar lavage.
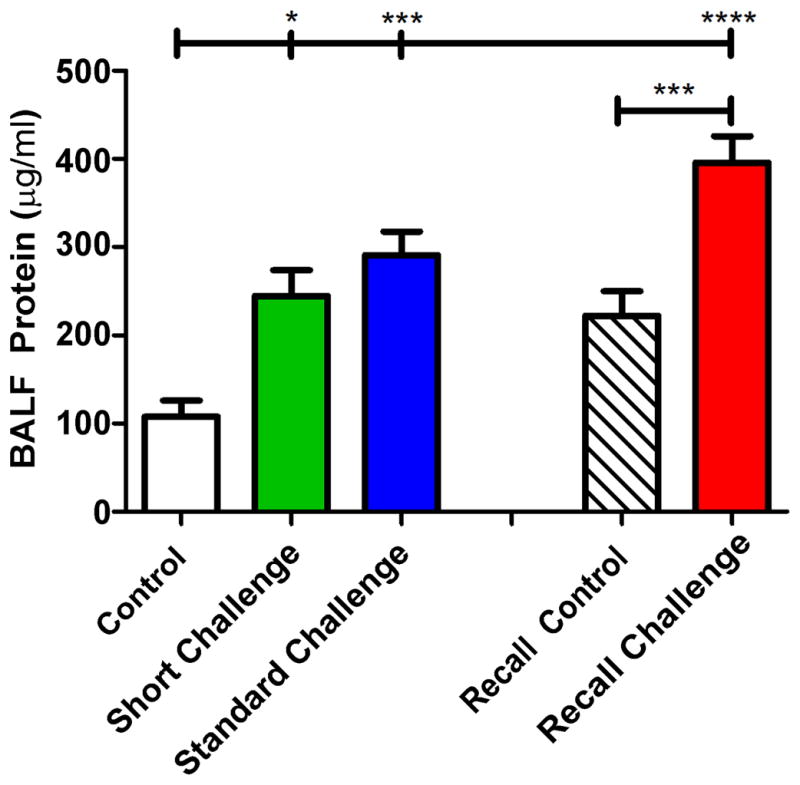
The protein content of the BALF was significantly elevated over control in all groups. The protein level was highest following a recall challenge with OVA (red bar, Recall Challenge). *p<0.05, ***p<0.001, ****p<0.0001.
Figure 6. AHR in fibrinogen knock-out mice.
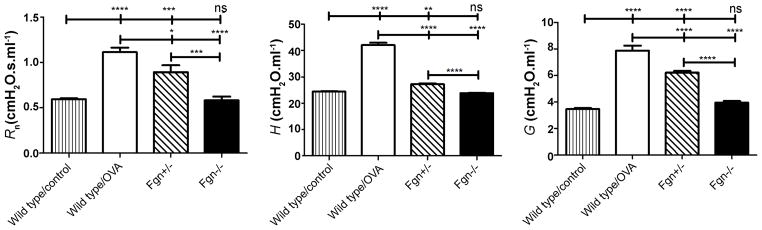
Fibrinogen knock-out mice (Fgn−/−) and heterozygotes (Fgn+/−) on C57BL/6J background were sensitized with two i.p. injections with OVA + alum on days 1 and 14 and then exposed to 3 daily inhalations of nebulized 1% OVA. Non-sensitized and sensitized littermates (Fgn+/+) were used as wild type controls. AHR was assessed 48 hrs after the last exposure to OVA. The control mice had a significantly elevated response to inhaled methacholine affecting all three parameters of respiratory mechanics (Rn, H and G). In contrast, Fgn−/− did not show a response different from wild type control mice. Fgn+/− had an intermediary response. n = 11, *p<0.05, **p<0.01, ****p<0.0001.
Discussion
The inflammatory process in allergically inflamed mice is set up by the antigen sensitization procedure, and gets under way with the first OVA inhalation challenge. It is clear from the present study that this first challenge sets in motion a sequence of events that evolve rapidly over time. Indeed, after only 16 hrs the animals exhibited increased narrowing of the conducting airways to methacholine (Figure 2) and a substantial neutrophilia (Figure 3). A mere 4 days later, this transformed into a methacholine response localized to the lung periphery (Figure 2) and an eosinophilic inflammatory profile (Figure 3). These alterations were accompanied by changes in the inflammatory cytokine profile of the BALF (Figure 4) and progressively increasing BALF protein (Figure 5). However, this evolving picture does not continue indefinitely. We and others have previously observed that more protracted antigen challenges eventually result in tolerization illustrated by a return in the AHR phenotype and airway eosinophilia toward baseline despite continued presence of structural inflammatory changes [7,8]. On the other hand, we found in the present study that if animals are given an extended (30 day) rest from exposure to antigen then they will respond to a subsequent challenge with even more vigor than before (Figure 2–5).
A key question that arises is how the above observations relate to asthma. Specifically, do the changes in phenotype we observed in the present study correspond in any way to events taking place in the course of the human disease? We suspect that they may, given the growing realization that there is more than a single asthma phenotype in humans. Classically, the eosinophil has been implicated as the predominant inflammatory granulocyte driving the pathophysiology of allergic asthma [18,19]. However, the pathogenic role of neutrophils in asthma is now more widely appreciated [20,21]; studies of induced sputum from asthmatics suggest that up to 60% of patients have non-eosinophilic airways inflammation [22], and a direct correlation has been demonstrated between sputum neutrophilia and severity of airflow limitation [23,24]. Furthermore, neutrophilic inflammation has been strongly implicated in asthma exacerbation [25], severe asthma [26–28], corticosteroid-resistant asthma [29,30], and fatal asthma [31]. Indeed, it has been suggested that mice with AHR accompanied by airway neutrophilia model a distinct phenotype of asthma [25,32,33] that mimics severe human asthma better than mice with eosinophilic inflammation [21]. We have shown, for example, that early following a single allergen challenge in OVA + alum sensitized mice the airway epithelium inducibly expresses the neutrophilic chemokine MIP-2 and neutrophil accumulation in the BALF is substantial, whereas eotaxin expression occurs later and precedes the accumulation of eosinophils and airway hyperresponsiveness to methacholine [34]. Furthermore, eliminating neutrophils using a GR1 antibody in an acid-induced AHR murine model eliminated AHR [35], suggesting that neutrophils primarily affect the periphery of the lung. The mice in the Recall Challenge group of the present study also demonstrate that robust AHR can be achieved in the presence of high numbers of neutrophils with few eosinophils. On the other hand, a complete lack of eosinophils prevents AHR in OVA allergic mice [36]. These various results support the view that rather than thinking of one particular cell type as being critical to the generation of AHR, we should rather think of AHR as part of a dynamically evolving phenotype that requires a number of different cell types to be present at various stages throughout its course.
Our conclusions about the nature of the AHR phenotype in this study are predicated on our ability to infer mechanism from the relative changes in the impedance parameters Rn, G and H following methacholine challenge. This is based on a substantial amount of prior work from our laboratory showing that Rn is a good reflection of the flow resistance of the airway tree [37] and that increases in G and H in the same proportion reflect closure of small airways [1,3]. Our inferences are also supported by in silico experimentation with anatomically-based computational models of the mouse lung on the changes in Zrs caused by bronchoconstriction [4,3,38]. Nevertheless, these conclusions remain largely inferential, so it is important to ask how well they hold up to actual findings, as in the present study. Relevant to this question is the finding that BALF protein content increased (Figure 5) commensurately with measures of airway closure (H and G in Figure 2). Increased BALF protein is to be expected since it has been shown that OVA challenge in allergic animals generates significant vascular leakage into the airway lumen [39,40], something that is inhibited by common asthma drugs such as formoterol and glucocorticoids [16,39,40]. In any case, the accumulation of vascular fluid and protein in the airspaces of the lung would be expected to affect surfactant function and so predispose to the closure of small airways, explaining why extensive lung derecruitment seems to accompany experimental AHR in mice [1,3].
We thus decided to take this issue further by investigating whether the airway closure of AHR is due to any particular plasma protein that exudes into the airspace. While plasma extravasation and fibrin accumulation in the airway lining fluid is a well-known culprit of increased surface tension and lung derecruitment in Acute Lung Injury [41], there is also evidence that fibrinogen and fibrin contribute to small airway closure in a BALB/cJ mouse model of asthma using the same Standard Challenge protocol as in this study [42]. In a recent human case study it was found that levels of sputum D-dimer, a common fibrin breakdown product, was elevated in severe asthmatics over that of moderate asthmatics, indicative of fibrin formation and turnover in asthmatic airways [26]. We therefore investigated whether a lack of fibrin offers protection against the rises in G and H that dominates AHR by repeating the Standard Challenge protocol in mice (C57BL/6 background) lacking the ability to produce circulating fibrinogen (Fgn−/−). Figure 6 shows that wild type control Fgn+/+ mice have a significantly increased response to aerosolized methacholine relative to control mice, but that Fgn−/− mice are no different from non-sensitized control in their response. Furthermore, Fgn+/− mice, which have a plasmafibrinogen level of about 70% that of the wild type mice [43], had an intermediate level of airways responsiveness, falling between that of the wild type and Fgn−/− mice, suggesting a dosing effect of fibrinogen on AHR. In addition, the BALF from the Fgn−/− and Fgn+/− mice was dominated by eosinophils (70.9 ± 13.0 % and 64.2 ± 31.2 %, respectively). These results would seem to suggest that fibrin plays a key role in the exacerbated airway closure caused by methacholine challenge in allergically inflamed mice and correlate with our previous findings in BALB/cJ mice [42]. However, the findings are not quite so clear cut because we also assayed the BALF for fibrinogen in the main study mice but did not find any differences between inflamed and control mice (data not shown). Furthermore, in a previous study of acid-induced Acute Lung Injury in which the BALF was conspicuous for high levels of fibrinogen, we found that fibrinogen knockout mice were not protected against lung derecruitment [41]. Hence, any ameliorating effects of fibrin or fibrinogen removal might simply reflect a reduction in the general protein burden in the airspaces, leading to a reduced effect on surfactant function, preservation of reduced surface tension, and less distal airway closure.
In conclusion we have shown that allergically induced AHR in mice varies in both nature and severity over time, accompanied by concomitant variations in cellular inflammation and cytokine profile. This reinforces the notion that AHR is an evolving phenotype, and that the study of mouse models of AHR might be better served by focusing on this variation rather than simply on a single time point at which AHR is maximal. This might make such mouse models more relevant to human asthma. Indeed our novel finding showing that a recall challenge generates a unique phenotype might be more comparable to clinical asthma with established immunological memory, presence of neutrophils and peripheral airway closure.
Acknowledgments
Breeding pairs of Fgn+/− mice were generously supplied by Dr. Jay Degen (Children’s Hospital Medical Center, Cincinnati, OH), through the Trudeau Institute Breeding Facility in Saranac Lake, NY [44]. The study was supported by NHLBI R01 HL089177, NCRR-COBRE P20RR15557. Dr. Riesenfeld was supported by a training grant NIH T32-HL076122.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Lundblad LKA, Thompson-Figueroa J, Allen GB, Rinaldi L, Norton RJ, et al. Airway hyperresponsiveness in allergically inflamed mice: the role of airway closure. Am J Respir Crit Care Med. 2007;175:768–774. doi: 10.1164/rccm.200610-1410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riesenfeld EP, Sullivan MJ, Thompson-Figueroa JA, Haverkamp HC, Lundblad LK, et al. Inhaled salmeterol and/or fluticasone alters structure/function in a murine model of allergic airways disease. Respir Res. 2010;11:22. doi: 10.1186/1465-9921-11-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagers S, Lundblad LKA, Ekman M, Irvin CG, Bates JHT. The allergic mouse model of asthma: normal smooth muscle in an abnormal lung? J Appl Physiol. 2004;96:2019–2027. doi: 10.1152/japplphysiol.00924.2003. [DOI] [PubMed] [Google Scholar]
- 4.Bates JH, Wagers SS, Norton RJ, Rinaldi LM, Irvin CG. Exaggerated airway narrowing in mice treated with intratracheal cationic protein. J Appl Physiol. 2006;100:500–506. doi: 10.1152/japplphysiol.01013.2005. [DOI] [PubMed] [Google Scholar]
- 5.Homma T, Bates JHT, Irvin CG. Airway hyperresponsiveness induced by cationic proteins in vivo: site of action. Am J Physiol Lung Cell Mol Physiol. 2005;289:L413–418. doi: 10.1152/ajplung.00059.2005. [DOI] [PubMed] [Google Scholar]
- 6.Cieslewicz G, Tomkinson A, Adler A, Duez C, Schwarze J, et al. The late, but not early, asthmatic response is dependent on IL-5 and correlates with eosinophil infiltration. J Clin Invest. 1999;104:301–308. doi: 10.1172/JCI7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lundblad KAL, Gustafsson B, Johansson U, Ottosson P, Persson PT, et al. Airways hyperreactivity does not correlate with morphometry in allergic mice. Am J Respir Crit Care Med. 1999;159:A408. [Google Scholar]
- 8.Schramm CM, Puddington L, Wu C, Guernsey L, Gharaee-Kermani M, et al. Chronic inhaled ovalbumin exposure induces antigen-dependent but not antigen-specific inhalational tolerance in a murine model of allergic airway disease. Am J Pathol. 2004;164:295–304. doi: 10.1016/S0002-9440(10)63119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vissers JL, van Esch BC, Hofman GA, Kapsenberg ML, Weller FR, et al. Allergen immunotherapy induces a suppressive memory response mediated by IL-10 in a mouse asthma model. J Allergy Clin Immunol. 2004;113:1204–1210. doi: 10.1016/j.jaci.2004.02.041. [DOI] [PubMed] [Google Scholar]
- 10.Yiamouyiannis CA, Schramm CM, Puddington L, Stengel P, Baradaran-Hosseini E, et al. Shifts in lung lymphocyte profiles correlate with the sequential development of acute allergic and chronic tolerant stages in a murine asthma model. Am J Pathol. 1999;154:1911–1921. doi: 10.1016/S0002-9440(10)65449-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lemanske RF, Busse WW. Asthma: clinical expression and molecular mechanisms. J Allergy Clin Immunol. 2010;125:S95–102. doi: 10.1016/j.jaci.2009.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lundblad LK, Rinaldi LM, Poynter ME, Riesenfeld EP, Wu M, et al. Detrimental effects of albuterol on airway responsiveness requires airway inflammation and is independent of beta-receptor affinity in murine models of asthma. Respir Res. 2011;12:27. doi: 10.1186/1465-9921-12-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lundblad LKA, Irvin CG, Adler A, Bates JH. A reevaluation of the validity of unrestrained plethysmography in mice. J Appl Physiol. 2002;93:1198–1207. doi: 10.1152/japplphysiol.00080.2002. [DOI] [PubMed] [Google Scholar]
- 14.Schuessler T, Bates J. A computer-controlled research ventilator for small animals: design and evaluation. IEEE Trans Biomed Eng. 1995;42:860–866. doi: 10.1109/10.412653. [DOI] [PubMed] [Google Scholar]
- 15.Hantos Z, Daroczy B, Suki B, Nagy S, Fredberg JJ. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol. 1992;72:168–178. doi: 10.1152/jappl.1992.72.1.168. [DOI] [PubMed] [Google Scholar]
- 16.Erjefalt, Andersson, Gustafsson, Korsgren, Sonmark, et al. Allergen challenge-induced extravasation of plasma in mouse airways. Clin Exp Allergy. 1998;28:1013–1020. doi: 10.1046/j.1365-2222.1998.00372.x. [DOI] [PubMed] [Google Scholar]
- 17.Ather JL, Ckless K, Martin R, Foley KL, Suratt BT, et al. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol. 2011;187:64–73. doi: 10.4049/jimmunol.1100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bousquet J, Chanez P, Lacoste JY, Barneon G, Ghavanian N, et al. Eosinophilic inflammation in asthma. N Engl J Med. 1990;323:1033–1039. doi: 10.1056/NEJM199010113231505. [DOI] [PubMed] [Google Scholar]
- 19.Wardlaw AJ, Brightling C, Green R, Woltmann G, Pavord I. Eosinophils in asthma and other allergic diseases. Br Med Bull. 2000;56:985–1003. doi: 10.1258/0007142001903490. [DOI] [PubMed] [Google Scholar]
- 20.Kamath AV, Pavord ID, Ruparelia PR, Chilvers ER. Is the neutrophil the key effector cell in severe asthma? Thorax. 2005;60:529–530. doi: 10.1136/thx.2005.043182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woodruff PG, Fahy JV. A role for neutrophils in asthma? Am J Med. 2002;112:498–500. doi: 10.1016/s0002-9343(02)01105-1. [DOI] [PubMed] [Google Scholar]
- 22.Gibson PG, Simpson JL, Saltos N. Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin-8. Chest. 2001;119:1329–1336. doi: 10.1378/chest.119.5.1329. [DOI] [PubMed] [Google Scholar]
- 23.Little SA, MacLeod KJ, Chalmers GW, Love JG, McSharry C, et al. Association of forced expiratory volume with disease duration and sputum neutrophils in chronic asthma. Am J Med. 2002;112:446–452. doi: 10.1016/s0002-9343(02)01047-1. [DOI] [PubMed] [Google Scholar]
- 24.Woodruff PG, Khashayar R, Lazarus SC, Janson S, Avila P, et al. Relationship between airway inflammation, hyperresponsiveness, and obstruction in asthma. J Allergy Clin Immunol. 2001;108:753–758. doi: 10.1067/mai.2001.119411. [DOI] [PubMed] [Google Scholar]
- 25.Fahy JV, Kim KW, Liu J, Boushey HA. Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol. 1995;95:843–852. doi: 10.1016/s0091-6749(95)70128-1. [DOI] [PubMed] [Google Scholar]
- 26.Brims FJ, Chauhan AJ, Higgins B, Shute JK. Up-regulation of the extrinsic coagulation pathway in acute asthma--a case study. J Asthma. 2010;47:695–698. doi: 10.3109/02770901003682802. [DOI] [PubMed] [Google Scholar]
- 27.Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF, et al. Neutrophilic inflammation in severe persistent asthma. Am J Respir Crit Care Med. 1999;160:1532–1539. doi: 10.1164/ajrccm.160.5.9806170. [DOI] [PubMed] [Google Scholar]
- 28.Wenzel SE, Szefler SJ, Leung DY, Sloan SI, Rex MD, et al. Bronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoids. Am J Respir Crit Care Med. 1997;156:737–743. doi: 10.1164/ajrccm.156.3.9610046. [DOI] [PubMed] [Google Scholar]
- 29.Green RH, Brightling CE, Woltmann G, Parker D, Wardlaw AJ, et al. Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax. 2002;57:875–879. doi: 10.1136/thorax.57.10.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavord ID, Brightling CE, Woltmann G, Wardlaw AJ. Non-eosinophilic corticosteroid unresponsive asthma. Lancet. 1999;353:2213–2214. doi: 10.1016/S0140-6736(99)01813-9. [DOI] [PubMed] [Google Scholar]
- 31.Sur S, Crotty TB, Kephart GM, Hyma BA, Colby TV, et al. Sudden-onset fatal asthma. A distinct entity with few eosinophils and relatively more neutrophils in the airway submucosa? Am Rev Respir Dis. 1993;148:713–719. doi: 10.1164/ajrccm/148.3.713. [DOI] [PubMed] [Google Scholar]
- 32.Douwes J, Gibson P, Pekkanen J, Pearce N. Non-eosinophilic asthma: importance and possible mechanisms. Thorax. 2002;57:643–648. doi: 10.1136/thorax.57.7.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner MO, Hussack P, Sears MR, Dolovich J, Hargreave FE. Exacerbations of asthma without sputum eosinophilia. Thorax. 1995;50:1057–1061. doi: 10.1136/thx.50.10.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poynter ME, Irvin CG, Janssen-Heininger YM. Rapid activation of nuclear factor-kappaB in airway epithelium in a murine model of allergic airway inflammation. Am J Pathol. 2002;160:1325–1334. doi: 10.1016/s0002-9440(10)62559-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen GB, Cloutier ME, Larrabee YC, Suratt BT, Bates JHT. Airways Hyperresponsiveness in Mice Following Acid Aspiration Is Neutrophil Dependent. Am J Respir Crit Care Med. 2008;177:A62. [Google Scholar]
- 36.Lee JJ, Dimina D, Macias MP, Ochkur SI, McGarry MP, et al. Defining a link with asthma in mice congenitally deficient in eosinophils. Science. 2004;305:1773–1776. doi: 10.1126/science.1099472. [DOI] [PubMed] [Google Scholar]
- 37.Tomioka S, Bates JH, Irvin CG. Airway and tissue mechanics in a murine model of asthma: alveolar capsule vs. forced oscillations. J Appl Physiol. 2002;93:263–270. doi: 10.1152/japplphysiol.01129.2001. [DOI] [PubMed] [Google Scholar]
- 38.Wagers SS, Haverkamp HC, Bates JH, Norton RJ, Thompson-Figueroa JA, et al. Intrinsic and antigen-induced airway hyperresponsiveness are the result of diverse physiological mechanisms. J Appl Physiol. 2007;102:221–230. doi: 10.1152/japplphysiol.01385.2005. [DOI] [PubMed] [Google Scholar]
- 39.Erjefalt I, Luts A, Persson CG. Appearance of airway absorption and exudation tracers in guinea pig tracheobronchial lymph nodes. J Appl Physiol. 1993;74:817–824. doi: 10.1152/jappl.1993.74.2.817. [DOI] [PubMed] [Google Scholar]
- 40.Erjefalt I, Persson CG. Allergen, bradykinin, and capsaicin increase outward but not inward macromolecular permeability of guinea-pig tracheobronchial mucosa. Clin Exp Allergy. 1991;21:217–224. doi: 10.1111/j.1365-2222.1991.tb00833.x. [DOI] [PubMed] [Google Scholar]
- 41.Allen GB, Cloutier ME, Larrabee YC, Tetenev K, Smiley ST, et al. Neither fibrin nor plasminogen activator inhibitor-1 deficiency protects lung function in a mouse model of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;296:L277–285. doi: 10.1152/ajplung.90475.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wagers SS, Norton RJ, Rinaldi LM, Bates JHT, Sobel BE, et al. Extravascular fibrin, plasminogen activator, plasminogen activator inhibitors, and airway hyperresponsiveness. J Clin Invest. 2004;114:104–111. doi: 10.1172/JCI19569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hattori N, Degen JL, Sisson TH, Liu H, Moore BB, et al. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J Clin Invest. 2000;106:1341–1350. doi: 10.1172/JCI10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suh TT, Holmback K, Jensen NJ, Daugherty CC, Small K, et al. Resolution of spontaneous bleeding events but failure of pregnancy in fibrinogen-deficient mice. Genes Dev. 1995;9:2020–2033. doi: 10.1101/gad.9.16.2020. [DOI] [PubMed] [Google Scholar]