

Abstract
Zn2+ toxicity is implicated in pancreatic β-cell death that occurs secondarily to: streptozotocin exposure in vitro; and both autoimmune attack or streptozotocin in vivo models of T1DM. This is demonstrated by reduced β-cell death or diabetic incidence in vitro or in NOD mice after treatment with Zn2+ preferring chelators, pyruvate, nicotinamide, a reduced zinc diet, sirtuin inhibitors, or zinc transporter knockout. These therapeutics are also demonstrated to be efficacious against Zn2+ neurotoxicity.
Aims
To determine if the sirtuin pathway is involved in Zn2+-, streptozotocin-, or cytokine-mediated β-cell death in vitro, and streptozotocin-, or NOD induced T1DM in vivo.
Methods
Sensitivity of MIN6 cells expressing empty vector, sirtuin protein-1 (SIRT1) or its siRNA, to Zn2+, streptozotocin, or cytokines, and effects on NAD+ levels were determined. Covariance of manipulating SIRT1 levels with diabetic incidence was tested in vivo.
Results
1) sirtuin pathway inhibition or SIRT1 knockdown attenuated Zn2+-, STZ-, and cytokine-mediated toxicity and NAD+ loss in β-cells, 2) SIRT1 overexpression potentiated these toxicities, 3) young SIRT1 β-cell transgenic mice have improved glucose tolerance under basal conditions, but upon aging showed increased sensitivity to streptozotocin compared to SIRT1 +/− mice, and 4) SIRT1 +/− mice in an NOD background or exposed to streptozotocin trended toward reduced diabetic incidence and mortality compared to wildtype.
Conclusions
These results have implicated SIRT1-mediated NAD+ loss in Zn2+, STZ, or cytokine toxicities of MIN6, and in NOD or streptozotocin T1DM animal models. Modulation of β-cell Zn2+ and NAD+ levels, and the sirtuin pathway could be novel therapeutic targets for T1DM.
Keywords: Sirtuins, NAD+, MIN6, SIRT1 knockout
Introduction
Type-1 diabetes (T1DM) is an autoimmune disease resulting from specific T-lymphocyte-, ROS-, and cytokine-mediated destruction of the insulin-producing β-cells of the islets of Langerhans resulting in dysregulation of blood glucose [1]. The development of T1DM is reduced by treatment with T-cell and cytokine inhibitors, and ROS scavengers [2]. These oxidative processes are suggested to alter the NAD+/ NADH ratio and inhibit proteins involved in energy metabolism and glycolysis causing the accumulation of triosephosphates [3,4]. Inhibitors of NAD+ catabolism have been demonstrated to attenuate diabetic incidence in models of T1DM [5,6]. NAD+ loss is linked to diabetes. Heterozygous knockout of the rate limiting enzyme in NAD+ synthesis (Nampt), causes reduced insulin secretion [7]. Also, Nampt and NAD+ levels are reduced in T2DM and the aging or high-fat diet models thereof. Restoration of NAD+, by nicotinamide mononucleotide precursor supplementation, attenuates diabetes in aging and high-fat diet mouse models of T2DM [8].
We recently showed that just prior to becoming diabetic, NOD mice demonstrate increased punctate Zn2+ staining in islets which is attenuated by a reduced Zn2+ diet, or zinc transporter 5 (ZnT5) knockout. Triweekly pyruvate or nicotinamide injections, chronic treatment with a reduced zinc diet, or knockout of the zinc transporter 5 (ZnT5) gene delay onset of diabetic incidence and animal mortality by reducing pancreatic zinc and/or maintaining β-cell NAD+ levels and mass [9]. This complements the beneficial effects demonstrated for Zn2+ chelation against acute or multiple low dose streptozotocin exposures [10,11]. Zinc neurotoxicity in vitro or in vivo induces NAD+ loss and glycolytic inhibition resulting in increased triosephosphates and death in a manner exactly equivalent to that seen in β-cells. These results are prevented by genetic or dietary reduction in brain Zn2+, or exogenous pyruvate, nicotinamide, NAD+, or sirtuin inhibition [12–14].
Zn2+ and β-cell death
Zn2+ is present in the pancreas at the highest concentration anywhere in the body, and within the pancreas is concentrated in the secretory granules of β-cells [10]. In the β-cell, Zn2+ allows insulin processing and crystallization in the secretory granules [15,16]. Significant amounts of free Zn2+ are also released from β-cell secretory granules [17,18]. As β-cells contain most of the pancreatic Zn2+, its toxic release, by the immune response or exposure to ROS/streptozotocin, would help explain the specificity of β-cell death in type-1 diabetes [19,20]. Zn2+ released intra- or extra-cellularly by diabetic conditions is postulated to induce detrimental intracellular effects on NAD+ levels through activation of the sirtuin pathway. The NAD+ loss results in glycolytic inhibition which is prevented by restoration of NAD+ levels using nicotinamide, sirtuin inhibition, and pyruvate, but not lactate (Figure 1). In these studies, we examined the deleterious effects that Zn2+-mediated NAD+ loss, had in facilitating β-cell death. We propose that immune-, and ROS-mediated dysfunction of energy metabolic pathways could be potentiated by Zn2+ release, and sirtuin-mediated loss of NAD+. We studied the effects of SIRT1 overexpression or knockdown in MIN6 cultures on NAD+ loss and Zn2+, STZ, or cytokine toxicities, and the effect of SIRT1 expression on the streptozotocin or NOD in vivo models of T1DM.
Figure 1. Model of zinc toxicity and prevention in Type-1 Diabetes.

We hypothesize that ZnT5 and ZnT8 mediate Zn2+ accumulation in secretory granules. The immune cell response injures β-cells causing Zn2+ release from granules, and re-uptake, through Ca2+ channels, in neighboring β-cells. Additionally, the ROS may oxidize metallothionein causing Zn2+ release. The resulting increased [Zn2+]i may cause direct inhibition of mitochondria, and GAPDH, or their indirect inhibition by a reduction in NAD+ levels induced by the NAD+ catabolizing enzyme SIRT1. Pyruvate, nicotinamide, and sirtuin inhibition prevent NAD+ loss and glycolytic inhibition. Black = Toxic, Gray = Therapeutic.
Experimental Procedures
Cell culture and toxicity studies
The insulinoma cell line, MIN6, was used to generate stably transformed cell lines overexpressing SIRT1 (760% of control, MIN6- Sir2OE1), an siRNA to SIRT1 (resulting in 28% of control expression, MIN6-Sir2KD1), and their empty vector control lines [21]. These lines were maintained in modified Dulbecco’s medium (DMEM) + 15% supplemented bovine serum, 1% L-glutamine, 0.1% penicillin/streptomyocin, 200 microg/mL of G418, and 29 micromol/L β-mercaptoethanol. Cell lines were passaged at 50–70% confluent using 0.05% trypsin/ 0.02% EDTA, and Zn2+ loading was achieved by supplementation with 10 micromol/L Zn2+ in the growth medium. High-density cultures (HD) were used for streptozotocin and cytokine toxicities. MIN6 cells were collected, counted, resuspended in DMEM + 15% FBS and plated at HD/low extracellular volume (7.5 x1010 cells/L; in 0.04 milliL) in V-shaped 96 well plates [10]. These HD cultures were plated in 7.5 millimol/L streptozotocin (STZ) or a mixture of cytokines (250 microgram/L IL-1β, 8 microgram/L TNF-α, and 200 microgram/L IFN-γ) with coexposure to the compounds tested. High cytokine levels were required due to the HD cultures and short exposure (6 h). Exposure to 15–40 micromol/L Zn2+ in serum-free MEM, or to 200–300 micromol/L Zn2+ in DMEM + 15% FBS under low density conditions were also utilized. Optimized concentrations of compounds were included during the toxicity exposure as indicated. 2-hydroxy naphthaldehyde (30–60 micromol/L, Naph), or sirtinol are sirtuin inhibitors; and pyruvate, nicotinamide (10–20 millimol/L), and NAD+ (6 millimol/L), restore NAD+ levels [13]. Cell viability was assayed at varying times later by 3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium Bromide (MTT) staining (0.1% final) of individual wells of a tissue culture plate, and the absorbance at 595 nm was then measured (n = 8–20 wells of cells from at least three independent experiments). Staining with propidium iodide (2.5 milligram/L) for normal density Zn2+ toxicity studies followed by measurement of fluorescence was also used. HD cultures were only assayed by MTT because a monolayer culture is required for PI staining [22,23].
Determination of levels of NAD+, and NADH
Measurements of NAD+ and NADH were made on cell lysates prepared immediately after 4 hr Zn2+, STZ or cytokine exposures. Cells were washed three times to remove compounds followed by lysis in NaOH/EDTA. This lysate was split and part of it directly hydrolyzed at 80°C for 20 min, and the other part acidified followed by hydrolysis at 80°C for 20 min. Alkaline hydrolysis destroys NADH, whereas acid hydrolysis destroys NAD+ allowing for their determinations by linked enzymatic cycling reactions. 2 μl of acid extract (~5000 cells) were added directly to 100 μl of NAD+ cycling reagent (100 millimol/L Tris-HCl, pH 8.1, 2 millimol/L β-mercaptoethanol, 2 millimol/L oxaloacetate, 0.3 mol/L ethanol, 0.02% BSA, and yeast alcohol dehydrogenase and 0.5 μg/mL malic dehydrogenase) and incubated at 25°C to obtain 500 cycles of NAD+ amplification. Termination by heating at 100°C for 5 min was followed by addition of 1 mL of malate indicator reagent (50 millimol/L amino-methylpropanol (pH 9.9), 5 millimol/L L-glutamate, 0.2 millimol/L NAD+, 5 μg/mL malic dehydrogenase, and 2 μg/ mL glutamate oxaloacetate transaminase). This reaction was incubated for 10 min at 25°C. The NADH generated from malate was measured fluorimetrically (excitation at 365 nm, emission monitored at 460 nm) [13,24]. For NAD+ additions, a correction was made based on NAD+ addition and washout from control cultures.
SIRT1 gene expression
Total RNA from harvested MIN6 cells (107) exposed to 0 or 40 micromol/L Zn2+ in serum-free MEM for 3 h was extracted as detailed [25]. The RNA concentration and integrity were verified, and reverse transcription was performed using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) on 1 μg of total cellular RNA. Real-time RT-PCR specific primers for SIRT1 and β-actin are as described previously [26]. The reactions were run in duplicate with iQSYBR green Supermix (BioRad) as per manufacturer’s instructions. A melt-curve analysis was performed at the end of each experiment to verify that a single product per primer pair was amplified, and the sizes of the amplified DNA fragments were verified. Samples were compared using the relative CT (the cycle number at the threshold level of log-based fluorescence) method. The percent increase was determined relative to an untreated control culture after normalizing to β-actin expression using 2−ΔΔCT (Livak) method.
Colony maintenance and trials
The NOD inbred mouse strain (Taconic), the β-cell SIRT1 overexpressing mice (BESTO), and the SIRT1 +/− mice were maintained at LSUHSC’s transgenic animal facility. The SIRT1 +/− and BESTO mice were backcrossed onto a C57/Bl6/J background for maintenance. The BESTO mice, line 431–2, showed a 12-fold overexpression of SIRT1 predominantly in β-cells which resulted in transcriptional regulation of target genes [21]. Housing and anesthesia concurred with the institutional Animal Studies Committee guidelines, the PHS Guide for the Care and Use of Laboratory Animals, USDA Regulations, and the AVMA Panel on Euthanasia. SIRT1 heterozygous knockout animals [27] were backcrossed to NOD mice for 10 generations to syngeneity; SIRT1 +/+/NOD animals develop diabetes and mortality at equivalent ages to the parental NOD animals. Upon interbreeding heterozygous animals, SIRT1 −/− animals die peri-natally whether on an SV129 or an NOD background, but survive on a CD1 background [27,28]. This was a double blind trial (both handler and histologist were blind to genotype) of age-matched female NOD mice with either a SIRT1 +/− or a SIRT1 +/+ genotype. Water and food ingestion, and body weight were monitored weekly and did not vary between groups. Fed blood glucose was monitored every Monday afternoon, and fasted blood glucose was also determined periodically (glucose oxidase). Fed and fasted blood glucose gave qualitatively similar results. Animals demonstrating continued akinesia with prodding or inability to eat and drink were euthanized and mortality recorded. MLDS was performed at 4–7 months of age (0.055 g/kg i.p. injection each day for 5 days), and blood glucose was measured on days 0, 1, 4, 7, 14, and 21 (n = 20).
Reagents
Unless otherwise stated, all reagents were from Sigma Chemical Co (St. Louis, MO).
Results
In MIN6 cultures, knockdown of SIRT1 reduced Zn2+ toxicities, and overexpression of SIRT1 potentiated Zn2+ toxicities
As shown in Figure 2, MIN6 cultures over expressing SIRT1 or an overexpression empty vector control were exposed to A) Zn2+ in the absence of serum or B) Zn2+ in the presence of serum. SIRT1 overexpression (760% of control) significantly potentiated Zn2+ toxicity at all levels of exposure in the presence or absence of serum in β-cells, and sirtuin inhibitors (sirtinol or Naph) attenuated this death. As shown in Figure 3, MIN6 cultures over expressing an siRNA to SIRT1 or an siRNA empty vector control were exposed to A) Zn2+ in the absence of serum or B) Zn2+ in the presence of serum. SIRT1 knockdown (28% of control) significantly attenuated Zn2+ toxicity at all levels of exposure in the presence or absence of serum in β-cells and sirtuin inhibitors did not further reduce this toxicity. Real-time PCR for β-cell SIRT1 expression was performed on control MIN6 cultures 3 h after exposure to 40 micromol/L Zn2+, resulting in a 39 ± 4.5% significant increase in expression relative to untreated control at P < 0.05 by student t-test (n = 6).
Figure 2. SIRT1 overexpression in MIN6 cells potentiated Zn2+ toxicity.

Stably transfected MIN6 cell lines were established which overexpress SIRT1 protein (760% of control). These cell lines were tested for their sensitivity to Zn2+ toxicity in the presence or absence of serum. A. SIRT1 overexpressing (SIRT1) and empty vector control (Control) MIN6 cell lines were assayed for their sensitivity to Zn2+ in the absence of serum (MS). Cells were grown to 50% confluence exposed as indicated, and cell death determined by propidium iodide staining (5 microg/mL) 20–22 hrs later compared to the complete death induced by 3 millimol/L Zn2+ (mean ± SEM, n=12–20 from three independent experiments). B. This cell line was also assayed in the presence of serum (requiring higher Zn2+ concentrations) as in A. * signifies difference from Zn2+ exposure in Control cultures; # signifies difference from similarly exposed Control cultures; and Δ signifies difference from Zn2+ exposure in SIRT1 cultures at P < 0.05. S means sirtinol, uM means micromol/L, Zn means zinc, naph means 2-hydroxynaphthaldehyde.
Figure 3. SIRT1 knockdown in MIN6 cells attenuated Zn2+ toxicity.
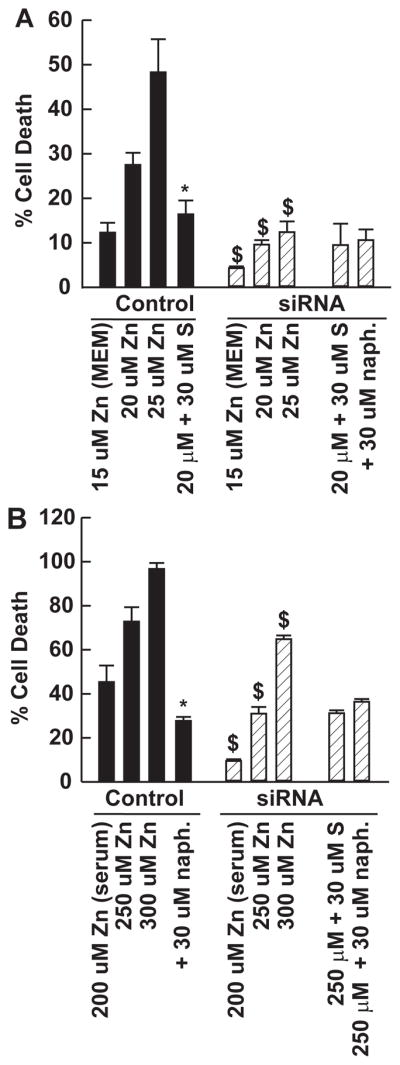
Stably transfected MIN6 cell lines were established which overexpress a small inhibitory RNA to SIRT1 resulting in knockdown of its expression (28% of control). These cell lines were tested for their sensitivity to Zn2+ toxicity in the presence or absence of serum. A. Knockdown of SIRT1 (siRNA) and empty vector siRNA control (Control) MIN6 cell lines were assayed as in Figure 1. Cells were grown to 50% confluence exposed as indicated, and cell death determined by propidium iodide staining (5 microg/mL) 20–22 hrs later compared to the complete death induced by 3 millimol/L Zn2+ (mean ± SEM, n=12–20 from three independent experiments). B. This cell line was also assayed in the presence of serum (requiring higher Zn2+ concentrations) as in A. * signifies difference from Zn2+ exposure alone at P < 0.05. $ signifies difference from similarly exposed Control cultures at P < 0.05. S means sirtinol, uM means micromol/L, Zn means zinc, naph means 2-hydroxynaphthaldehyde.
Knockdown of SIRT1 reduced STZ and cytokine toxicities, and overexpression of SIRT1 potentiated them
MIN6 overexpressing SIRT1, or its siRNA, and control cultures were exposed to 7.5 millimol/L STZ (Figure 4), or mixed cytokines (Figure 5) for 6 hours (high density condition-7.5x1010 cells/L, 0.04 milliL in each 96-well V-bottom pit) in the presence or absence of nicotinamide, NAD+, or Naph. These compounds were chosen because they have been previously demonstrated to restore NAD+ levels, and to attenuate Zn2+ neurotoxicity. Cell viability was determined by MTT staining. Pyruvate, nicotinamide, NAD+, sirtinol, and Naph were applied at optimized concentrations as determined by dose responses (data not shown). These compounds each attenuated STZ and mixed cytokine toxicities, with nicotinamide and NAD+ having the best efficacy. In SIRT1 overexpressing MIN6 cells, 2x naphthaldehyde (60 micromol/L) was required for efficacy.
Figure 4. STZ toxicity in high-density MIN6 cultures was attenuated by SIRT1 knockdown, and potentiated by SIRT1 overexpression.

MIN6 cultures were exposed to 7.5 millimol/L STZ at high density. SIRT1 denotes the SIRT1 overexpressing line, and siRNA denotes the line which overexpresses the SIRT1 siRNA. Cell survival was assayed by 0.1% MTT staining. # signifies difference from STZ exposure alone; $ signifies difference from similarly exposed control cultures at P < 0.05. mM means millimol/L, STZ means streptozotocin.
Figure 5. Cytokine mediated β-cell death was attenuated by SIRT1 knockdown and was potentiated by SIRT1 overexpression.

MIN6 cells were exposed as indicated to cytokines for 6 hrs at high density. Cell survival was assayed by 0.1% MTT staining. In SIRT1 overexpressing MIN6 cells, 2x naphthaldehyde (60 micromol/L) was required for efficacy. # signifies difference from cytokine exposure alone; $ signifies difference from similarly exposed control cultures at P < 0.05.
Zn2+, STZ, and cytokines reduced NAD+ levels which could be restored by SIRT1 knockdown, nicotinamide, and NAD+
MIN6 cultures were exposed to 10 millimol/L streptozotocin, 40 micromol/L Zn2+, or mixed cytokines for 4 hrs (prior to cell death); NAD+ was isolated and measured. Zn2+, streptozotocin, and cytokines induced a significant decrease in NAD+ levels and nicotinamide or NAD+ restored these levels. In addition, SIRT1 knockdown attenuated the loss of NAD+ for Zn2+ and streptozotocin (Figure 6). We previously showed that sirtuin inhibition attenuated the loss of NAD+ for Zn2+ in unmodified MIN6 cells [9].
Figure 6. STZ and Zn2+ reduce NAD+ levels in MIN6 cells which are restored by exogenous NAD+.

MIN6 cells were exposed as indicated for 3 hours. Cells were lysed, NAD+ was extracted and assayed by the enzymatic micro-cycling assay, and the concentration was determined. * signifies a significant difference from control. # indicates a significant difference from 40 micromol/L Zn2+ exposure alone. $ indicates a significant difference from 10 millimol/L STZ exposure alone. & indicates a significant difference from cytokine exposure alone at P <0.05.
Mice overexpressing SIRT1 in β-cells had increased susceptibility to STZ compared to SIRT1 +/−, and SIRT1 +/− bred onto an NOD background had reduced mortality
Two more BESTO mice were susceptible to multiple low dose streptozotocin exposure than their wildtype littermates (Table 1). SIRT1 knockout mice survive on a CD1 background, but are smaller and sickly probably due to developmental problems, and did not survive MLDS exposure [28]. SIRT1 knockout mice on SV129 or NOD backgrounds are embryonic lethal. However, SIRT1 +/− heterozygous mice trended towards reduced susceptibility to MLDS, and when put on an NOD background, trended toward reduced diabetic incidence and mortality with a single mortality time point showing a significant difference. When SIRT1 +/− mice were compared to BESTO mice for susceptibility to MLDS, there was a significant difference (Figure 7, Table 1).
Table 1.
SIRT1 β-cell transgenics trend toward more diabetic incidence after MLDS, and SIRT1 +/− mice trend toward reduced diabetic incidence. Multiple low dose streptozotocin (MLDS) exposure (55 millig/kg i.p. on 5 consecutive days) was administered to mice (5–10 months of age) as indicated in a blinded fashion. Blood glucose was determined on Day 0, 1, 3, 7, 10, and 14 in a blinded fashion. Diabetic incidence was defined as blood glucose > 2.5 g/L. Mortality was recorded in a blinded fashion on the day that animals displayed akinesia after prodding, or inability to reach food.
| Animal Condition | Diabetic Animals by 7 days | Diabetic Animals by 14 days | Surviving Animals by 21 days |
|---|---|---|---|
| Multiple Low Dose STZ in BESTO Tg-Ctrl | 9 out of 20 | 12 out of 20 | 11 out of 20 |
| Multiple Low Dose STZ in BESTO Tg+ | 11 out of 20 | 14 out of 20 | 8 out of 20 |
| Multiple Low Dose STZ in SIRT1 +/+ | 9 out of 20 | 12 out of 20 | 11 out of 20 |
| Multiple Low Dose STZ in SIRT1 +/− | 6 out of 20* | 9 out of 20* | 13 out of 20* |
indicates difference from BESTO Tg+ mice at P < 0.05 by student t-test.
Figure 7. SIRT1 +/− mice on an NOD background trend toward reduced susceptibility to diabetic incidence and mortality compared to SIRT1 +/+/ NOD littermates.

SIRT1 +/− mice were backcrossed into NOD animals for 10 generations, and then intercrossed to generate SIRT1 +/+/NOD, SIRT1 +/−/NOD, but no SIRT1 −/−/NOD littermates. Blood glucose was monitored weekly with a One-Touch Ultra glucose monitor, and animals that demonstrated akinesia with continued prodding by an observer blinded to treatment condition were sacrificed, and mortality recorded. A) The number of animals whose blood glucose (BG) remained below 2.5 g/L for 2 consecutive weeks was plotted as a function of weeks of age. B) The number of surviving animals was plotted as a function of weeks of age. * indicates a significant difference in the age at which 50% of the mice have died compared to SIRT1 +/+/NOD littermate controls at P < 0.05 by a t-test.
Discussion
In these studies we demonstrated that: 1) sirtuin pathway inhibition attenuated Zn2+-, STZ-, and cytokine-mediated toxicity and NAD+ loss in β-cells, 2) SIRT1 overexpression in MIN6 cell lines potentiated NAD+ loss, and Zn2+, STZ, and cytokine toxicities, 3) SIRT1 knockdown using an siRNA expressing MIN6 cell line attenuated NAD+ loss and these toxicities, 4) diabetic incidence and mortality induced in vivo by streptozotocin or NOD, showed covariance with SIRT1 expression levels upon genetic manipulation.
Zn2+ is toxic to insulinoma cells and to isolated islets, and that zinc chelation and pyruvate can prevent toxicity. Zn2+ chelation and pyruvate attenuate hyperglycemia in the acute high dose streptozotocin model though the NAD+-dependent mechanism of action was questioned [10,20]. We have utilized several in vitro and in vivo models of T1DM. These include in vitro exposure of β-cells to zinc, mixed cytokines, or STZ; and in vivo exposure to STZ, or the immune-mediated NOD mouse model. STZ and the NOD mouse cause selective death of insulin-secreting β-cells, inducing reductions in nicotinamide co-factor levels, glucose oxidation, and glucose-induced insulin secretion [29–32]. The NAD+ precursor, nicotinamide, reduces diabetic incidence in both the acute high dose, and the multiple low dose streptozotocin injection paradigms [33,34]. Zn2+ preferring chelators (CaEDTA and clioquinol) reduce diabetic incidence in the acute high dose and multiple low dose streptozotocin injection models as demonstrated by the reduction in Zn2+ staining, β-cell death and diabetic symptoms achieved [10,11,20]. We recently demonstrated that pyruvate attenuates multiple low-dose streptozotocin exposure (MLDS) induced-, and NOD-induced diabetes, and that a zinc reduced diet also attenuates NOD-induced diabetes [9].
Our studies in neurons show that an increase in intracellular Zn2+ causes a loss of NAD+ levels that may be partially mediated by sirtuin or poly-ADP ribosyl polymerase (PARP) activation depending on cell-type [13,35]. The resultant decrease in the NAD+/NADH ratio inhibits the energy metabolic pathway at the susceptible enzymes GAPDH and PDH. Pyruvate, nicotinamide, or exogenous NAD+ restore NAD+ levels, and glycolytic flux, and thereby attenuate death. Pyruvate is converted to lactate regenerating NAD+ at the expense of NADH [12]. Nicotinamide induces increased synthesis of NAD+, or decreases its degradation by NAD+-catabolizing enzymes [35]. Nicotinamide is effective in diabetic models [5,36], with therapeutic effects observed only if it is given to prediabetic patients [37–40]. Its mechanism of action is not well defined, with suggestions that it prevents PARP-activation and NAD+ depletion, thereby reducing apoptosis of β-cells induced by DNA damage [5]. PARP induced NAD+ depletion is also a mechanism for GAPDH inhibition, resulting in triosephosphate accumulation, and β-cell death in diabetes [41]. However, no reduction in diabetes occurs in the NOD/PARP −/− mouse arguing against a causative role for PARP in this animal model of diabetes [42]. The ability of exogenous NAD+ to attenuate Zn2+, STZ, and cytokine toxicities in insulinoma cultures argues that NAD+ levels are involved in the mechanism. The protective effects of sirtinol and SIRT1 knockdown on NAD+ levels and beta-cell death suggested that the sirtuin pathway may be involved.
The sirtuin family (SIRT) and the pancreas
Inhibition of the sirtuin pathway attenuates Zn2+ or STZ toxicities of β-cells in part by preventing NAD+ depletion [9]; as we have also shown for Zn2+ neurotoxicity. The sirtuin pathway is involved in Zn2+-induced neuronal, and β-cell NAD+ depletion and toxicities [9,13]. The sirtuin family of proteins are NAD+-dependent protein deacetylases resulting in NAD+-catabolism, transcriptional silencing, and transcriptional regulation [43]. SIRT1 is ubiquitously expressed; within the pancreas, SIRT1 is expressed strongly in the cytoplasm of α-cells and weakly in both the nucleus and cytoplasm of β-cells [21]. Young β-cell SIRT1 transgenic (BESTO) mice have increased glucose tolerance under basal conditions [21], but lose this effect with age [44]. This suggests that under young physiologic conditions, SIRT1 overexpression may be beneficial, but under pathophysiologic aged T1DM diabetic conditions (MLDS), SIRT1 may be detrimental perhaps due to potentiation of zinc toxicity. Sirtuins appear to mediate part of the NAD+ loss after Zn2+ and STZ exposures of MIN6 cells, as evidenced by the partial restoration of NAD+ levels by sirtuin inhibition for zinc and STZ exposures, but not for cytokine exposures [9]. Sirtuins also act through transcriptional modulation, which may be the predominant mechanism in Zn2+ neurotoxicity, and cytokine toxicity in β-cells. SIRT1 overexpression was shown to attenuate IL-1β and IFN-γ induced toxicity in RIN β-cells [45]. However, this study was not done in the presence of TNF-α, or under high-density conditions where zinc release and toxicity could play a role. Recently, SIRT1 was shown to attenuate pancreatic β-cell expansion [46], and to decrease hepatic insulin responsiveness [47]. However, the SIRT1 and AMP kinase activator, resveratrol, suppresses T-cell immune responses, and attenuates diabetic incidence in NOD mice [48]. These effects of resveratrol may be SIRT1 independent, mediated instead by oxidant scavenger or AMP kinase activation mediated mechanisms [49,50]. In age or high-fat diet models of T2DM, SIRT1 activation partially mediates the beneficial effects of NAD+ restoration in the peripheral tissues affected by T2DM (liver, WAT, skeletal muscle), though effects on the pancreas or beta-cells were not presented [8]. Similar effects in models of T1DM were not presented.
In these studies, we have implicated the sirtuin pathway and SIRT1 in the pancreatic NAD+ loss and beta-cell death induced by the in vitro models of T1DM: Zn2+, STZ, or mixed cytokine exposures. We have also suggested that SIRT1 plays a role in the diabetic incidence and mortality induced by the STZ or NOD in vivo models of T1DM. These results differ from those reported for SIRT1 on T2DM induced affects in peripheral tissues, where SIRT1 activity appears to mediate the effects of reducing or increasing NAD+ levels [8]. The roles of SIRT1 in pancreatic versus peripheral tissue, and in T1DM versus T2DM appear to be varied and complex, and will require further studies to unravel.
Acknowledgments
Supported by NIH grant DK 073446 (CTS). I would like to thank Shin-Ichiro Imai (Washington University) for the BESTO transgenic mice and the MIN6-Sir2OE1, and MIN6-Sir2KD1 cell lines, Dr. Fred Alt (Harvard University) for the SIRT1 knockout mouse line, and Ai-Li Cai and Chunxiao Shi (Washington University) for expert technical assistance with NAD+ measurements, and colony maintenance.
Abbreviations
- BESTO
β-cell SIRT1 Overexpressing mice
- CaEDTA
Ethylene Diamine Tetra-Acetic acid-Calcium salt
- GAPDH
Glyceraldehyde-3-Phosphate Dehydrogenase
- HD
High-Density cultures
- IFN-γ
Interferon Gamma
- IL-1β
Interleukin-1 Beta
- MEM
Minimal Essential Medium
- MLDS
Multiple Low-Dose Streptozotocin
- mM
Millimol/L
- MTT
3-(4,5-dimethylthiazol-2-Yl)-2,5-diphenyltetrazolium Bromide
- Nampt
Nicotinamide Phosphoribosyl Transferase
- Naph
2-hydroxynaphthaldehyde
- N
Nicotinamide
- NAD+
Nicotinamide Adenine Dinucleotide
- NOD
Non-Obese Diabetic
- P
Pyruvate
- PARP
Poly-ADP Ribose Polymerase
- PDH
Pyruvate Dehydrogenase
- ROS
Reactive Oxygen Species
- S
Sirtinol
- siRNA
Small inhibitory RNA
- SIRT1
Sirtuin protein 1
- STZ
Streptozotocin
- TNF-α
Tumor Necrosis Factor-alpha
- T1DM
Ttype-1 Diabetes
- T2DM
Type-2 Diabetes
- uM
Micromol/L
- Zn2+
Zinc
- [Zn2+]
Intracellular Zinc concentration
- ZnT5
Zinc Transporter 5
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Author Contributions
C.T.S. is the guarantor of this manuscript, performed all aspects of this paper, and has no conflicts to report.
References
- 1.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- 2.Mathis D, Vence L, Benoist C. beta-Cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 3.Obrosova I, Faller A, Burgan J, Ostrow E, Williamson JR. Glycolytic pathway, redox state of NAD(P)-couples and energy metabolism in lens in galactose-fed rats: effect of an aldose reductase inhibitor. Curr Eye Res. 1997;16:34–43. doi: 10.1076/ceyr.16.1.34.5113. [DOI] [PubMed] [Google Scholar]
- 4.Trueblood N, Ramasamy R. Aldose reductase inhibition improves altered glucose metabolism of isolated diabetic rat hearts. Am J Physiol. 1998;275:H75–H83. doi: 10.1152/ajpheart.1998.275.1.H75. [DOI] [PubMed] [Google Scholar]
- 5.Kolb H, Burkart V. Nicotinamide in type 1 diabetes. Mechanism of action revisited. Diabetes Care. 1999;22:B16–B20. [PubMed] [Google Scholar]
- 6.Suarez-Pinzon WL, Mabley JG, Power R, Szabo C, Rabinovitch A. Poly (ADP-ribose) polymerase inhibition prevents spontaneous and recurrent autoimmune diabetes in NOD mice by inducing apoptosis of islet-infiltrating leukocytes. Diabetes. 2003;52:1683–1688. doi: 10.2337/diabetes.52.7.1683. [DOI] [PubMed] [Google Scholar]
- 7.Revollo JR, Korner A, Mills KF, Satoh A, Wang T, et al. Nampt/PBEF/ Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheline CT, Shi C, Takata T. Dietary Zinc Reduction, Pyruvate Supplementation, or Zinc Transporter 5 Knockout Attenuate Beta-Cell Death in Non-Obese Diabetic Mice, Islets, and Insulinoma Cells. Journal of Nutrition. 2012 doi: 10.3945/jn.112.167031. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim BJ, Kim YH, Kim S, Kim JW, Koh JY, et al. Zinc as a paracrine effector in pancreatic islet cell death. Diabetes. 2000;49:367–372. doi: 10.2337/diabetes.49.3.367. [DOI] [PubMed] [Google Scholar]
- 11.Priel T, Aricha-Tamir B, Sekler I. Clioquinol attenuates zinc-dependent beta-cell death and the onset of insulitis and hyperglycemia associated with experimental type I diabetes in mice. Eur J Pharmacol. 2007;565:232–239. doi: 10.1016/j.ejphar.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 12.Sheline CT, Behrens MM, Choi DW. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J Neurosci. 2000;20:3139–3146. doi: 10.1523/JNEUROSCI.20-09-03139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai AL, Zipfel GJ, Sheline CT. Zinc neurotoxicity is dependent on intracellular NAD levels and the sirtuin pathway. Eur J Neurosci. 2006;24:2169–2176. doi: 10.1111/j.1460-9568.2006.05110.x. [DOI] [PubMed] [Google Scholar]
- 14.Sheline CT, Cai AL, Zhu J, Shi C. Serum or target deprivation-induced neuronal death causes oxidative neuronal accumulation of Zn2+ and loss of NAD+ Eur J Neurosci. 2010;32:894–904. doi: 10.1111/j.1460-9568.2010.07372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang XF, Arvan P. Intracellular transport of proinsulin in pancreatic beta-cells. Structural maturation probed by disulfide accessibility. J Biol Chem. 1995;270:20417–20423. doi: 10.1074/jbc.270.35.20417. [DOI] [PubMed] [Google Scholar]
- 16.Zalewski PD, Millard SH, Forbes IJ, Kapaniris O, Slavotinek A, et al. Video image analysis of labile zinc in viable pancreatic islet cells using a specific fluorescent probe for zinc. J Histochem Cytochem. 1994;42:877–884. doi: 10.1177/42.7.8014471. [DOI] [PubMed] [Google Scholar]
- 17.Formby B, Schmid-Formby F, Grodsky GM. Relationship between insulin release and 65zinc efflux from rat pancreatic islets maintained in tissue culture. Diabetes. 1984;33:229–234. doi: 10.2337/diab.33.3.229. [DOI] [PubMed] [Google Scholar]
- 18.Qian WJ, Kennedy RT. Spatial organization of Ca(2+) entry and exocytosis in mouse pancreatic beta-cells. Biochem Biophys Res Commun. 2001;286:315–321. doi: 10.1006/bbrc.2001.5379. [DOI] [PubMed] [Google Scholar]
- 19.Tartler U, Kroncke KD, Meyer KL, Suschek CV, Kolb-Bachofen V. Nitric oxide interferes with islet cell zinc homeostasis. Nitric Oxide. 2000;4:609–614. doi: 10.1006/niox.2000.0314. [DOI] [PubMed] [Google Scholar]
- 20.Chang I, Cho N, Koh JY, Lee MS. Pyruvate inhibits zinc-mediated pancreatic islet cell death and diabetes. Diabetologia. 2003;46:1220–1227. doi: 10.1007/s00125-003-1171-z. [DOI] [PubMed] [Google Scholar]
- 21.Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Sheline CT, Choi DW. Neuronal death in cultured murine cortical cells is induced by inhibition of GAPDH and triosephosphate isomerase. Neurobiol Dis. 1998;5:47–54. doi: 10.1006/nbdi.1998.0177. [DOI] [PubMed] [Google Scholar]
- 23.Ying H, Gottron F, Choi D. Assessment of cell viability in primary neuronal cultures. John Wiley & Sons, Inc; New York: 2000. [DOI] [PubMed] [Google Scholar]
- 24.Lin SS, Manchester JK, Gordon JI. Enhanced gluconeogenesis and increased energy storage as hallmarks of aging in Saccharomyces cerevisiae. J Biol Chem. 2001;276:36000–36007. doi: 10.1074/jbc.M103509200. [DOI] [PubMed] [Google Scholar]
- 25.Han JH, Stratowa C, Rutter WJ. Isolation of full-length putative rat lyso-phospholipase cDNA using improved methods for mRNA isolation and cDNA cloning. Biochemistry. 1987;26:1617–1625. doi: 10.1021/bi00380a020. [DOI] [PubMed] [Google Scholar]
- 26.Becerril S, Rodríguez A, Catalan V, Sáinz N, Ramírez B, et al. Deletion of Inducible Nitric-Oxide Synthase in Leptin-Deficient Mice Improves Brown Adipose Tissue Function. PLoS One. 2010;5:e10962. doi: 10.1371/journal.pone.0010962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, et al. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strandell E, Eizirik DL, Korsgren O, Sandler S. Functional characteristics of cultured mouse pancreatic islets following exposure to different streptozotocin concentrations. Mol Cell Endocrinol. 1988;59:83–91. doi: 10.1016/0303-7207(88)90198-0. [DOI] [PubMed] [Google Scholar]
- 30.Eizirik DL, Sandler S, Welsh N, Hellerstrom C. Preferential reduction of insulin production in mouse pancreatic islets maintained in culture after streptozotocin exposure. Endocrinology. 1988;122:1242–1249. doi: 10.1210/endo-122-4-1242. [DOI] [PubMed] [Google Scholar]
- 31.Strandell E, Eizirik DL, Sandler S. Survival and B-cell function of mouse pancreatic islets maintained in culture after concomitant exposure to streptozotocin and nicotinamide. Exp Clin Endocrinol. 1989;93:219–224. doi: 10.1055/s-0029-1210860. [DOI] [PubMed] [Google Scholar]
- 32.Bunik VI. 2-Oxo acid dehydrogenase complexes in redox regulation. Eur J Biochem. 2003;270:1036–1042. doi: 10.1046/j.1432-1033.2003.03470.x. [DOI] [PubMed] [Google Scholar]
- 33.Mendola J, Wright JR, Jr, Lacy PE. Oxygen free-radical scavengers and immune destruction of murine islets in allograft rejection and multiple low-dose streptozocin-induced insulitis. Diabetes. 1989;38:379–385. doi: 10.2337/diab.38.3.379. [DOI] [PubMed] [Google Scholar]
- 34.Hassan N, Janjua MZ. The optimum dose of nicotinamide for protection of pancreatic beta-cells against the cytotoxic effect of streptozotocin in albino rat. J Ayub Med Coll Abbottabad. 2001;13:26–30. [PubMed] [Google Scholar]
- 35.Sheline CT, Wang H, Cai AL, Dawson VL, Choi DW. Involvement of poly ADP ribosyl polymerase-1 in acute but not chronic zinc toxicity. Eur J Neurosci. 2003;18:1402–1409. doi: 10.1046/j.1460-9568.2003.02865.x. [DOI] [PubMed] [Google Scholar]
- 36.O’Brien BA, Harmon BV, Cameron DP, Allan DJ. Nicotinamide prevents the development of diabetes in the cyclophosphamide-induced NOD mouse model by reducing beta-cell apoptosis. J Pathol. 2000;191:86–92. doi: 10.1002/(SICI)1096-9896(200005)191:1<86::AID-PATH573>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 37.Visalli N, Costa C, Natoli A. Hypoprolactinemia in subjects with various pathologies of the reproductive system. Clin Ter. 1986;116:421–423. [PubMed] [Google Scholar]
- 38.Pozzilli P, Browne PD, Kolb H. Meta-analysis of nicotinamide treatment in patients with recent-onset IDDM. The Nicotinamide Trialists. Diabetes Care. 1996;19:1357–1363. doi: 10.2337/diacare.19.12.1357. [DOI] [PubMed] [Google Scholar]
- 39.Behme MT. Nicotinamide and diabetes prevention. Nutr Rev. 1995;53:137–139. doi: 10.1111/j.1753-4887.1995.tb01538.x. [DOI] [PubMed] [Google Scholar]
- 40.Vidal J, Fernandez-Balsells M, Sesmilo G, Aguilera E, Casamitjana R, et al. Effects of nicotinamide and intravenous insulin therapy in newly diagnosed type 1 diabetes. Diabetes Care. 2000;23:360–364. doi: 10.2337/diacare.23.3.360. [DOI] [PubMed] [Google Scholar]
- 41.Wahlberg G, Adamson U, Svensson J. Pyridine nucleotides in glucose metabolism and diabetes: a review. Diabetes Metab Res Rev. 2000;16:33–42. doi: 10.1002/(sici)1520-7560(200001/02)16:1<33::aid-dmrr79>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez C, Menissier De Murcia J, Janiak P, Bidouard JP, Beauvais C, et al. Unexpected sensitivity of nonobese diabetic mice with a disrupted poly(ADP-Ribose) polymerase-1 gene to streptozotocin-induced and spontaneous diabetes. Diabetes. 2002;51:1470–1476. doi: 10.2337/diabetes.51.5.1470. [DOI] [PubMed] [Google Scholar]
- 43.Blander G, Guarente L. The sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 44.Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JH, Song MY, Song EK, Kim EK, Moon WS, et al. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes. 2009;58:344–351. doi: 10.2337/db07-1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bastien-Dionne PO, Valenti L, Kon N, Gu W, Buteau J. Glucagon-like peptide 1 inhibits the sirtuin deacetylase SirT1 to stimulate pancreatic beta-cell mass expansion. Diabetes. 2011;60:3217–3222. doi: 10.2337/db11-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erion DM, Yonemitsu S, Nie Y, Nagai Y, Gillum MP, et al. SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proc Natl Acad Sci U S A. 2009;106:11288–11293. doi: 10.1073/pnas.0812931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee SM, Yang H, Tartar DM, Gao B, Luo X, et al. Prevention and treatment of diabetes with resveratrol in a non-obese mouse model of type 1 diabetes. Diabetologia. 2011;54:1136–1146. doi: 10.1007/s00125-011-2064-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitada M, Kume S, Imaizumi N, Koya D. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes. 2011;60:634–643. doi: 10.2337/db10-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Um JH, Park SJ, Kang H, Yang S, Foretz M, et al. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes. 2010;59:554–563. doi: 10.2337/db09-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]