

Abstract
Current treatment of hemophilia A by intravenous infusion of factor VIII (fVIII) concentrates is very costly and has a potential adverse effect of developing inhibitors. Gene therapy, on the other hand, can potentially overcome these limitations associated with fVIII replacement therapy. Although hemophilia B gene therapy has achieved promising outcomes in human clinical trials, hemophilia A gene therapy lags far behind. Compared to factor IX, fVIII is a large protein which is difficult to express at sustaining therapeutic levels when delivered by either viral or non-viral vectors. To improve fVIII gene delivery, numerous strategies have been exploited to engineer the fVIII molecule and overcome the hurdles preventing long term and high level expression. Here we reviewed these strategies, and discussed their pros and cons in human gene therapy of hemophilia A.
Introduction
Hemophilia A is an X-linked, recessive bleeding disorder that affects approximately 1 in 5000 males. This disease is caused by hereditary defects in factor VIII (fVIII) [1]. Clinically, it is characterized by frequent and spontaneous joint hemorrhages, easy bruising and prolonged bleeding. The consequences of bleeding into critical closed spaces, such as the intracranial or the retroperitoneal spaces, are severe and potentially life-threatening. The severity of clinical symptoms is directly related to the coagulation activity of FVIII (<1%, severe; 2-5%, moderate; and 5-30%, mild). Approximately 50% of all cases are classified as severe [2].
Gene therapy has the potential to overcome some of the shortcomings associated with conventional protein replacement therapy for hemophilia A. Not only can it eliminate the risk of infectious diseases, but the appropriate vector can maintain long term gene expression and eliminate the need for repeated treatments. In addition, gene therapy may be more likely to avoid the formation of significant titers of neutralizing antibodies. Evidence shows that daily infusions with high doses of clotting factor concentrate can eliminate neutralizing antibodies among 70-90% of patients. The continuous production of clotting factors from a rAAV-delivered transgene may similarly limit the formation of these neutralizing antibodies [3-6].
The fVIII protein is a particularly challenging target for gene therapy. The coding region of full length fVIII is approximately 7.0 kb. The translated FVIII protein is a 2,351 aa single-chain glycoprotein of 280 kDa, which is predominately synthesized and secreted by the hepatocytes or sinusoidal endothelial cells in the liver [1]. This protein has six domains following its signal peptide (A1-A2-B-A3-C1-C2), in which the B domain has no effect on FVIII activity in in vitro or in vivo assays [7,8]. The human B-domain deleted fVIII DNA is 4371 bp in length. The large cDNA size, short half-life and strong immunogenecity make it difficult to achieve sustained therapeutic levels of gene expression in vivo. Even though the results from hemophilia B gene therapy clinical trials are encouraging [9,10], the prospect of curing hemophilia A using a gene therapy approach does not appear to be imminent.
A variety of vectors have been explored for delivering the fVIII gene for treating hemophilia, which include both non-viral and viral vectors. Viral vectors that have been explored include adenovirus, retrovirus, lentiviral vectors and adeno-associated virus (AAV) vectors [11-16]. The results from these pioneering studies suggest that the sustained expression of therapeutic levels of fVIII in a hemophilic animal model necessitates improvements not only on delivering vectors but also the fVIII gene itself. In this review, a variety of bioengineering approaches exploited to enhance the fVIII transgene and transgene products will be discussed. These approaches hold significant potential to maximize the expression of the therapeutic products.
Engineering fVIII with enhanced expression
Synonymous mutations (mutations that alter the coding DNA and RNA sequence without affecting the amino acid sequence of the underlying protein), i.e codon optimization, is a powerful approach for enhancing gene expression in mammalian systems. The benefits of codon optimization have been previously reviewed [17]. By optimizing the codons for a cDNA encoding the therapeutic protein, a higher level of expression may be achieved from two different mechanisms. First, it may increase transcriptional efficacy. GC content, CpG dinucleotides content, cryptic splicing sites, negative CpG islands, Shine-Dalgarno sequence codon-context, TATA boxes and terminal signals can all be optimized to increase transcriptional efficacy. Second, it may enhance the translation efficiency. Codon usage bias, GC content, mRNA secondary structure, premature polyA sites, RNA instability motif (ARE), stable free energy of mRNA, internal chi sites and ribosomal binding sites are known factors that can affect translation efficiency. Therefore, codon optimization which reduces the complications associated with these issues will increase therapeutic gene expression.
Codon optimization for fVIII has been attempted by two different groups [18,19]. Radcliffe et al reported that codon optimization of the fVIII nucleic acid sequence increases lentiviral vector titre and transgene expression [18]. Codon-optimizing of the BDD fVIII gene increased the vector titer modestly by 2-3 fold. In contrast, codon optimization of the full-length fVIII gene improved vector titre by approximately one order of magnitude. There were also substantial increases in transgene expression per integrated vector copy. In a separate study, it was shown that a different codon-optimized construct led to a 29-to 44-fold increase in expression of fVIII in neonatal hemophilia mice with lentiviral vectors [19]. The protein level can reach more than 200% of the normal human fVIII levels in animal sera. There may be a difference with the algorithm used for codon optimization in these two studies. Based on the enhancing mechanisms associated with codon optimization, it should be useful universally for any gene therapy vector.
Engineering fVIII with higher specific activity
Due to the limitations of transfer vectors it is desirable to achieve a higher coagulation activity out of the limited therapeutic protein that can be expressed. Therefore, engineered fVIII with higher specific activity may lower the vector doses needed in a clinical setting. Fay's group reported that replacement of individual charged residues (D519, E665 or E1984) with either Ala or Val increased procofactor stability at elevated temperature and cofactor stability over an extended time course [20]. Variants with mutations at D519 and either E665 or E1984 exhibited significantly better stability over single mutants [21]. Other fVIII muteins that prevent the dissociation of the A2 subunit in the activated fVIII (fVIIIa) heterotrimer have been shown to possess enhanced cofactor stability. Mutein fVIII IR8 was generated by deleting residues from aa 795–1689 and including Arg-336-Ile, Arg-562-Lys and Arg-740-Ala mutations [22]. Since the A2 domain is covalently attached to the light chain, these mutations resulted in an inactivation-resistant coagulation fVIIIa. IR8 exhibited approximately 5-fold higher specific activity than wild-type fVIII. The deletion in the ar3 region of fVIII leads to a 10-fold weaker affinity for VWF, which may affect its serum half-life. However, the efficacy of IR8 was inconsistent in vivo when delivered using platelets as a delivery vehicle in the bleeding models [23]. Therefore, its usefulness in gene therapy needs to be further evaluated.
Activated fVIII (fVIIIa) typically has a very short half-life, partly due to spontaneous dissociation of the A2 domain from the fVIIIa heterotrimer [24]. Disulfide bridges covalently linking the A2 subunit within fVIIIa have also been explored to enhance fVIII specific activity. As reported by Gale et al, changes in C664 in A2 and C1826 in A3 could allow a disulfide bond to be established between these two amino acids [25]. Similarly, an engineered disulfide bond can be formed between C662–C1828 in fVIIIa [26]. C662–C1828 fVIIIa had normal activity in FX activation while C664–C1826 fVIIIa had reduced activity. They also exhibited approximately a 50%∼100% higher specific activity than wild type (wt) fVIII. Both disulfide bond-stabilized variants show improved affinity for von Willebrand factor (VWF), which may suggest a longer plasma half-life. These variants had approximately a 5-fold increase in half-life relative to wt fVIIIa during clot formation when reconstituted in whole blood [27].
It has been previously demonstrated that fVIII from species other than human can exhibit a higher specific activity than their human counterpart. The recombinant B-domain–deleted canine fVIII showed a 3-fold increased specific activity over that of human BDD-fVIII [28,29]. A hybrid of canine and human fVIII that retains the higher specific activity of canine fVIII may be useful for human gene therapy.
Most of the above modifications altered the codons of the fVIII molecules in order to achieve the improved specific activities. However, one main concern is whether or not such changes increase inhibitor formation when these modified fVIII proteins are expressed in human patients. Whether or not the alterations give rise to better candidates for gene therapy remains to be confirmed.
Engineering fVIII with enhanced secretion
After fVIII synthesis, the fVIII protein is transported to the lumen of the endoplasmic reticulum (ER). In the ER, the fVIII protein is associated with several protein chaperones including immunoglobin binding protein (BiP), calnexin and calreticulin [30,31]. The release of fVIII from BiP is an ATP dependent process, which is one of the limiting factors for efficient fVIII secretion. Both calnexin and calreticulin enhance fVIII secretion and degradation. LMAN1(ERGIC-53) is a chaperone protein in the ER-Golgi intermediate compartment that is required for efficient fVIII and V secretion [32]. Post-translational modifications of fVIII take place in the ER and Golgi apparatus, including N-, O-linked glycosylation and sulfation of tyrosine residues of the heavy chain and light chain. Those modifications are important for proper folding that leads to full procoagulant activity and its interaction with von Willebrand factor [33,34]. The mis-folded fVIII is degraded in the ER and the correctly processed fVIII molecules enter the Golgi apparatus [35]. Additional studies have demonstrated that fVIII secretion is affected by the oligosaccharide content in the B domain. Pipe's group showed that addition of the first 226 amino acid residues with six potential asparagine-linked glycosylation sites (N6) increases the secretion of fVIII by 10-fold while maintaining the same mRNA level as B-domain deleted fVIII [30,35]. Analysis of the differences between human and porcine fVIII suggested amino acids in the A1 and apA3 domain are key determinants of fVIII efficient secretion [36]. Factor VIII with F309S mutation and N6 have been reported to increase the antigen secretion significantly [37]. However, it was found that there was no significant improvement when delivered by lentiviral vectors in a different setting [19,38]. In another study, it also did not show an effect on fVIII heavy chain secretion [39].
Recombinant porcine fVIII secretes 10-100 fold better than human fVIII [40]. Hybrid human/porcine fVIII was shown to express up to 100-fold greater than human fVIII [40,41]. Although the specific activity of plasma-derived porcine fVIII is generally low at ∼100U/mg, the specific activity differences between highly purified recombinant porcine fVIII (∼12,400U/mg) and human fVIII (4000-10000U/mg) are insignificant [40,42]. There is no loss of specific activity with the chimeric ET-801i, which is 88% identical to human BDD fVIII at the protein level. Human cells transduced with lentiviral vectors encoding ET-801i demonstrated an expression level that was 16∼160 fold higher than that of human fVIII [43]. The chimeric porcine and human fVIII may be useful in correcting bleeding phenotype in a special group of patients.
Secretory signal peptides (SP) play a critical role in mediating eukaryotic protein secretion. The hidden Markov model (HMM) provides a protocol to describe and predict relative strengths of secretory signals [44]. Based on this model, the SP-HMM bit score of fVIII is low at 12.5 when compared to the commonly known apolipoprotein C which has an HMM score of 20.9. We explored the possibility of using a high HMM score signal peptide to increase fVIII secretion. In the results summarized in figure 1, even synthetic signal peptides with HMM score as high as 34 and 38 did not increase fVIII secretion over the endogenous fVIII signal peptide. Signal peptides from apolipoprotein C-III (HMM score 20.9), alpha-1-antichymotrypsin (HMM score 19.1) also performed poorly. The signal peptide from albumin (HMM score 18.7) failed to mediate fVIII secretion at all (data not shown). Based on this information, we concluded that the endogenous fVIII signal peptide is actually the most potent one at directing fVIII secretion.
Figure 1.

Effects of heterogeneous secretory signal peptides on fVIII expression. The original signal peptide of fVIII (BDD) was replaced with the various signal peptides listed in the table. The HMM score of each signal peptide is shown in column 3. All factor constructs were under the control of b-actin promoter with a CMV enhancer. At 24 hours post transfection into 293 cells, the secreted fVIII was collected and the coagulation activity was determined by aPTT assay.
Engineering fVIII with prolonged plasma circulation half-life
Intracellular proteolytic processing within the B domain generates one light chain, approximately 80 kDa composed of A3-C1-C2 domains, and one heavy chain containing A1-A2-B domains ranging in size from 90-200 kDa. The heavy and light chains are associated as a heterodimer through a divalent metal-ion-dependent linkage between the A1 and A3 domains. Factor VIII circulates in the human blood with a very short plasma half-life without vWF co-factor. Once fVIII is noncovalently associated with vWF to form a complex, the half-life of fVIII in plasma can be extended from 2 to 12 hours [33,34].
Factor VIII with a longer half-life will be highly desirable for maintaining therapeutic levels. Pegylation is a common strategy for increasing in vivo half-life of a protein [45]. However, it doesn't have an impact on a gene delivery strategy because of the requirement for a chemical conjugation reaction. An alternative approach is to take advantage of the exceptionally long circulation half-life of serum albumin or IgG (approximately 20 days). These carrier proteins mediate a pH-dependent interaction with the neonatal Fc receptor (FcRn) rescuing them from intracellular degradation. The fusion proteins of coagulation factors such as factor VII-Fc or factor IX-albumin have been previously shown to increase the in vivo half-life of therapeutic proteins without affecting the coagulation factors [46-48]. Factor VIII-Fc fusion proteins have demonstrated an increase in plasma half-life by two fold. Due to an increase in the size of the gene upon fusion with the carrier protein, the fVIII fusion proteins are excellent candidates for use in non-viral gene delivery or vectors which are not constrained by size limitation.
The low density lipoprotein receptor-related protein (LPR) is a liver multi-ligand endocytic receptor which is known to play a role in fVIII catabolism [49]. Cell surface heparan sulfate proteoglycans (HSPGs) have also been shown to facilitate this process [50]. The A2 domain residues 484-509 were identified to mediate fVIII-LRP binding. The binding site for heparan sulfate proteoglycans (HSPGs) within the A2 domain of fVIII is residues 558-565. In conditional LRP-deficient mice, inactivation of the LRP gene led to a two-fold increase in plasma fVIII levels and a two-fold prolongation of half-life of injected fVIII [51]. Reducing fVIII's affinity for LRP and/or HSPGs therefore is considered a mechanism to extend its plasma circulation half-life [52]. Factor VIII with mutations in charged residues clustered within the 484-509 region may be a potential target for human gene therapy.
Engineering fVIII to evade inhibitors
Factor VIII inhibitors can occur in hemophilia patients receiving fVIII concentrate (alloantibodies) or in patients with a normal fVIII gene in certain underlying conditions such as autoimmune disorder, lymphoproliferative disorder, malignancy, pregnancy, and certain drugs (autoantibodies). Approximately 30% of patients with severe hemophilia A develop inhibitors against fVIII. The inhibitors are usually polyclonal IgG with specifcity against various epitopes on fVIII, especially the A2, A3 and C2 domain [53,54]. Because the inhibitors are constantly present in excess over fVIII in the plasma, they partially or completely neutralize fVIII molecules, resulting in severe bleeding episodes. The fVIII inhibitors are quantified by Bethesda assay. One Bethesda unit (BU) is defined as the quantity of inhibitor that neutralizes 50% of the fVIII in normal plasma in 2 hours at 37°C. In patients with low (<5 BU) fVIII inhibitor titers, DDAVP and human fVIII are effective. In patients with high titer, raising the fVIII level is no longer effective and the administration of porcine fVIII, activated prothrombin complex concentrates (APCC), or recombinant human activated factor VII (rFVIIa) is necessary [42,55]. Thus, engineering fVIII that can avoid existing inhibitors represents another therapeutic option in addition to tolerance induction [56]. Lollar et al generated a chimeric porcine and human fVIII in which porcine codons replaced their countparts in the A2 and C2 domains of human fVIII [57]. The resulting chimeric molecules significantly decrease fVIII antigenicity when tested in vivo. Whether these chimeric molecules can be used for long term fVIII expression warrants further research.
Another approach to circumvent the problems associated with fVIII inhibitors is to bypass the use of fVIII altogether. This is exemplifed by delivering factor VIIa using AAV vectors [58,59], which would be exceptionally useful for treatment of patients with pre-existing fVIII inhibitors. However, when factor VIIa was expressed at 2 μg/ml or higher in mice, it was associated with thrombosis and early mortality. Therefore, it is necessary to regulate the expression level of factor VIIa to avoid toxicity.
FXa variants have also been utilized to bypass fVIII for hemophilia A treatment [60]. Camire's group showed factor Xa mutants (FXaI16L and FXaV17A) behaved like a zymogen and could activate systemic coagulation in a controlled fashion [60]. They exhibited 60 fold longer half-lives over wild-type factor Xa in hemophilic plasma and could promote robust thrombin generation that bypasses the intrinsic pathway. In vivo, FXaI16L protein appeared to be more efficacious than FVIIa in controlling clot formation [61]. These variants may also be explored as a target for gene therapy of hemophilia A.
Engineering fVIII for delivery by AAV vectors
The choice of vector is perhaps the most critical issue for gene therapy of human hemophilia. Adenovirus generally remains as an episome and the transgene expression is transient. Retroviruses require target cells that are cycling or undergoing cell division at the time of delivery in order to achieve transduction [13]. Complex and sophisticated delivery routes or pretreatment of the animals with agents or surgical procedures designed to promote hepatocyte proliferation are generally essential. In contrast, the AAV vector has been shown to be capable of delivering long term gene expression in post-mitotic cells. Although rAAV has advantages over adenoviral or retrovirus vectors, it faces a tremendous challenge in delivering fVIII gene. The size constraint of AAV for fVIII delivery is illustrated in Figure 2.
Figure 2.
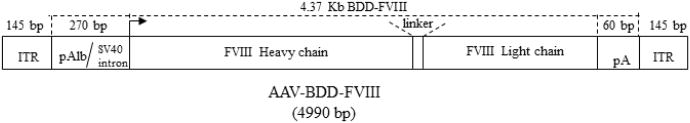
Illustration of the size constraint for expressing fVIII in a single AAV vector. Major elements essential for fVIII expression have been identified. Despite the use of the mini-albumin promoter and small poly A sequence, the rAAV genome is already close to 5kb, more than the optimal ∼4.7-4.8kb AAV packaging capacity.
Although human B-domain deleted fVIII (4371bp) is fully functional and substantially smaller than full length fVIII (7053bp), the AAV vector can only accommodate 4.7kb including the 290bp ITR. Using short regulatory elements like a mini-promoter generally leads to unsatisfactory levels of expression. Despite the size constraints, efforts have been put forward to package B domain deleted fVIII DNA into a single AAV vector conforming with the 4.7 kb size limitation [62-65]. The expression of fVIII by this strategy could yield as much as 27% of normal level using a herpes TK promoter plus hepatitis B enhancer. With AAV8 vector, complete correction of hemophilia A phenotype was also reported [66]. Small elements such as 297 b human α1 antitrypsin promoter, 163 b the hepatic control region of apolipoprotein E gene, and the human α1 antitrypsin promoter (193 b) have also been explored and were able to obtain a moderate level of canine BDD fVIII expression in mice [67].
Ignoring the size limitation, it has been demonstrated that AAV containing an extra-large size expression cassette and strong promoters can fully correct hemophilia A phenotype in a mouse model [68]. This approach has several drawbacks. 1) The vector has a yield which is at least 10 fold lower than the regular vectors; 2) Partially packaged vectors require a high dose in order to restore the expression cassette through complementation; 3) It results in heterogeneity of vector genomes. Single molecule sequencing and other traditional molecular biology methods confrmed that there are no intact genomes packaged when the vector cassette exceeds the size limit [69-72]. This strategy faces significant challenges that must be overcome before moving into clinical trials.
To solve the size constraint for delivering fVIII using an AAV vector, many approaches have been explored using two AAV vectors simultaneously [73-77]. Despite their variance, these approaches can be divided into two categories: 1) Utilizing the properties of AAV DNA concatemerization in vivo [73-75] 2) fVIII polypeptide re-assembly/association [39,76-78].
To take advantages of AAV concatemization in vivo, the dual vector systems often include an intron to split the extra-large expression cassette. The flexibility of an intron allows the unpredictable and un-precise nucleic acid sequences formed at the juncture of concatemerization to be removed at the RNA splicing step. As illustrated in Figure 3, this approach would give rise to a wild type like molecule and has been demonstrated in other disease models [73-75]. In spite of its elegance in design, the drawback is that only approximately 10% of all concatemerizations can lead to DNA that is capable of therapeutic gene expression. Chao et al used this approach and split the fVIII gene at exon 12 [79]. In vivo testing demonstrated approximately 2% of the normal fVIII level for four months in an immunodeficient hemophilia A mouse model. A second approach using dual vectors is to add an enhancer vector and eliminate the requirement for an intron. Upon concatemerization, the enhancer would function in either configuration, before or after the basic vectors. So far this approach has not demonstrated satisfactory results even in small animal studies. A study by Duan's group suggests that the selection of the intron and splice sites have an immense impact on the final outcome [80].
Figure 3.

Illustration of the intron mediated, dual AAV system for expressing fVIII utilizing the concatemerization of AAV vectors. Major steps are shown in the figure. Note there is no need for an intron if the first vector contains enhancer only.
The first strategy for delivering fVIII using two AAV vectors that do not rely on AAV concatemerization was to express the heavy chain and light chain separately [76,81,82]. The re-association of both heavy chain and light chain then restores fVIII coagulation activity. In C57Bl/6 mice, up to 4000ng/ml of antigen expression could be achieved at a dose 3×1011 vg/mouse. In hemophilia A mice the bleeding phenotype was corrected. Similar results were also obtained in hemophilia A dog using AAV8 and AAV9 [83]. The foremost problem associated with this strategy is that there is an imbalance of the secreted heavy chain and light chain. When the vectors having the same promoter were injected at a ratio of 1:1, the expression of LC can be 100 fold higher than that of HC. This chain imbalance also affects the specific activity of the secreted fVIII protein.
Although fVIII light chain has been shown to facilitate fVIII heavy chain secretion, the addition of various domains from the light chain failed to increase fVIII heavy chain secretion [78]. The only exception is a special heavy chain molecule named HCHL, which includes the addition acidic region 3 (ar3) [78]. When compared to the normal HC, HCHL exhibited 3-5 fold increase in secretion. While it is possible to achieve a balance of HC and LC at the antigen level using HCHL, it led to the suboptimal utilization of all HC molecules expressed. This suggested that a certain level of excessive light chain is necessary to allow all HCs expressed to form functional fVIII complex.
The dual vector system carrying HC and LC requires fVIII to be split into heavy chain and light chain. A second approach based on intein mediated “protein splicing” allows fVIII to be split in many different locations. Inteins are essentially “protein introns” which can be precisely excised from a precursor protein and rejoins the surrounding sequences in N- and C-terminus with a peptide bond [84]. Protein splicing is directly analogous to RNA splicing of introns. Unlike posttranscriptional RNA splicing, intein mediated protein splicing takes place posttranslationally. The N- and C- terminus of the precursor protein are named N-extein and C-extein respectively. A mature peptide is formed when the exteins are ligated together after the intein removal. An intein based strategy for expressing fVIII is illustrated in Figure 4. The primary difference between this approach and an intron mediated split fVIII is the substrate. The intron strategy requires the concatermerization of input vectors, which is one of the limiting factors. The intein strategy uses substrates resulting from the transcription/translation of input vectors. Thus, there is a substantial amplification of spliceable precursors over the initial input of viral genomes. However, the limitation is that one splicing reaction only leads to one product. Intron mediated concatemerization actually give rises to a template that can be transcribed over and over.
Figure 4.

Illustration of the intein mediated dual AAV system for expressing fVIII. Note that splicing reaction occurs in the cytoplasm after the polypeptides for fVIII have been translated.
Factor VIII has been successfully manipulated with the mini Ssp DnaB intein, which has a total of 154 amino acids and can be split into a 106 amino acid N-terminal, and a 48 aa C-terminal fragments. The split DnaB intein can still come together through non-covalent interaction to undergo protein splicing [85,86]. By using this approach, the light chain was identified to facilitate fVIII heavy chain secretion [39]. A drawback for this strategy in gene delivery is that it will not solve the chain imbalance issue. In addition, the un-spliced intein will lead to the secretion of a fVIII related neo-antigen, which is likely to complicate immune response in the host.
Summary and Future Prospects
Currently, protein replacement therapy offers hemophilia A patients an effective treatment option, which raises the safety profle requirement for hemophilia A gene therapy. Any fVIII bio-engineering that increases the probability of inhibitor formation is unlikely to be adopted for use as a therapy. Among all fVIII bioengineering approaches discussed, manipulations at the DNA or RNA level, such as codon optimization, appear to be useful and can be universally adopted for all gene delivery strategies. Manipulations at the protein level, i.e., altering the native human fVIII amino acids, have an inherent risk/limitation to be applied to the general hemophilia patient population. Amino acids alterations in fVIII require extensive testing prior to human clinical trials to avoid unexpected inhibitor formation. Ideally, factor VIII engineering should result in a product that meets the following two recommendations. First, a minimal number of non-native amino acids should be used; and second, there should be no significant loss of fVIII functionality such as specific activity or plasma half-life. Development of efficient mini-promoters or micro-fVIII genes smaller than BDD-fVIII is the future direction for using a single AAV vector delivery strategy. The addition of acidic region 3 into fVIII heavy chain that enhances secretion is currently the best option for the dual AAV strategy. In addition, fVIII bypassing technology offers an alternative option that avoids many limitation of bio-engineering fVIII molecules altogether [59-61,87].
Acknowledgments
WX is supported by National Institutes of Health grants HL080789 and HL084381. SAR is supported by a supplement to HL084381. ARM is supported by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award T32-HL-007777 from the National Heart Lung and Blood Institute.
Footnotes
This article was originally published in a journal published by OMICS Publishing Group, and the attached copy is provided by OMICS Publishing Group for the author's benefit and for the benefit of the author's institution, for commercial/research/educational use including without limitation use in instruction at your institution, sending it to specific colleagues that you know, and providing a copy to your institution's administrator.
All other uses, reproduction and distribution, including without limitation commercial reprints, selling or licensing copies or access, or posting on open internet sites, your personal or institution's website or repository, are requested to cite properly.
References
- 1.Mann KG. Biochemistry and physiology of blood coagulation. Thromb Haemost. 1999;82:165–74. [PubMed] [Google Scholar]
- 2.Soucie JM, Evatt B, Jackson D. Occurrence of hemophilia in the United States. The Hemophilia Surveillance System Project Investigators. Am J Hematol. 1998;59:288–294. doi: 10.1002/(sici)1096-8652(199812)59:4<288::aid-ajh4>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 3.Chao H, Walsh CE. Induction of tolerance to human factor VIII in mice. Blood. 2001;97:3311–3312. doi: 10.1182/blood.v97.10.3311. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman BE, Martino AT, Sack BK, Cao O, Liao G, et al. Nonredundant Roles of IL-10 and TGF-beta in Suppression of Immune Responses to Hepatic AAV-Factor IX Gene Transfer. Mol Ther. 2011;19:1263–1272. doi: 10.1038/mt.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LoDuca PA, Hoffman BE, Herzog RW. Hepatic gene transfer as a means of tolerance induction to transgene products. Curr Gene Ther. 2009;9:104–114. doi: 10.2174/156652309787909490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finn JD, Ozelo MC, Sabatino DE, Franck HW, Merricks EP, et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. 2010;116:5842–5848. doi: 10.1182/blood-2010-06-288001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toole JJ, Pittman DD, Orr EC, Murtha P, Wasley LC, et al. A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci U S A. 1986;83:5939–5942. doi: 10.1073/pnas.83.16.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorner AJ, Wasley LC, Kaufman RJ. Increased synthesis of secreted proteins induces expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells. J Biol Chem. 1989;264:20602–20607. [PubMed] [Google Scholar]
- 9.Kay MA, Manno CS, Ragni MV, Larson PJ, Couto LB, et al. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet. 2000;24:257–261. doi: 10.1038/73464. [DOI] [PubMed] [Google Scholar]
- 10.Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- 11.Andrews JL, Kadan MJ, Gorziglia MI, Kaleko M, Connelly S. Generation and characterization of E1/E2a/E3/E4-defcient adenoviral vectors encoding human factor VIII. Mol Ther. 2001;3:329–336. doi: 10.1006/mthe.2001.0264. [DOI] [PubMed] [Google Scholar]
- 12.Lipshutz GS, Sarkar R, Flebbe-Rehwaldt L, Kazazian H, Gaensler KM. Short-term correction of factor VIII deficiency in a murine model of hemophilia A after delivery of adenovirus murine factor VIII in utero. Proc Natl Acad Sci U S A. 1999;96:13324–13329. doi: 10.1073/pnas.96.23.13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Powell JS, Ragni MV, White GC, Lusher JM, Hillman-Wiseman C, et al. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood. 2003;102:2038–2045. doi: 10.1182/blood-2003-01-0167. [DOI] [PubMed] [Google Scholar]
- 14.Israel DI, Kaufman RJ. Retroviral-mediated transfer and amplification of a functional human factor VIII gene. Blood. 1990;75:1074–1080. [PubMed] [Google Scholar]
- 15.Brown BD, Shi CX, Rawle FE, Tinlin S, McKinven A, et al. Factors influencing therapeutic efficacy and the host immune response to helper-dependent adenoviral gene therapy in hemophilia A mice. J Thromb Haemost. 2004;2:111–118. doi: 10.1111/j.1538-7836.2004.00552.x. [DOI] [PubMed] [Google Scholar]
- 16.Hoeben RC, Einerhand MP, Briet E, van Ormondt H, Valerio D, et al. Toward gene therapy in haemophilia A: retrovirus-mediated transfer of a factor VIII gene into murine haematopoietic progenitor cells. Thromb Haemost. 1992;67:341–345. [PubMed] [Google Scholar]
- 17.Fath S, Bauer AP, Liss M, Spriestersbach A, Maertens B, et al. Multiparameter RNA and codon optimization: a standardized tool to assess and enhance autologous mammalian gene expression. PloS one. 2011;6:e17596. doi: 10.1371/journal.pone.0017596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radcliffe PA, Sion CJ, Wilkes FJ, Custard EJ, Beard GL, et al. Analysis of factor VIII mediated suppression of lentiviral vector titres. Gene ther. 2008;15:289–297. doi: 10.1038/sj.gt.3303080. [DOI] [PubMed] [Google Scholar]
- 19.Ward NJ, Buckley SM, Waddington SN, Vandendriessche T, Chuah MK, et al. Codon optimization of human factor VIII cDNAs leads to high-level expression. Blood. 2011;117:798–807. doi: 10.1182/blood-2010-05-282707. [DOI] [PubMed] [Google Scholar]
- 20.Wakabayashi H, Varfaj F, Deangelis J, Fay PJ. Generation of enhanced stability factor VIII variants by replacement of charged residues at the A2 domain interface. Blood. 2008;112:2761–2769. doi: 10.1182/blood-2008-02-142158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wakabayashi H, Griffths AE, Fay PJ. Combining mutations of charged residues at the A2 domain interface enhances factor VIII stability over single point mutations. J Thromb Haemost. 2009;7:438–444. doi: 10.1111/j.1538-7836.2008.03256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pipe SW, Kaufman RJ. Characterization of a genetically engineered inactivation-resistant coagulation factor VIIIa. Proc Natl Acad Sci U S A. 1997;94:11851–11856. doi: 10.1073/pnas.94.22.11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greene TK, Wang C, Hirsch JD, Zhai L, Gewirtz J, et al. In vivo efficacy of platelet-delivered, high specifc activity factor VIII variants. Blood. 2010;116:6114–6122. doi: 10.1182/blood-2010-06-293308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson AR. Structure and function of the factor VIII gene and protein. Semin Thromb Hemost. 2003;29:11–22. doi: 10.1055/s-2003-37935. [DOI] [PubMed] [Google Scholar]
- 25.Gale AJ, Pellequer JL. An engineered interdomain disulfide bond stabilizes human blood coagulation factor VIIIa. J Thromb Haemost. 2003;1:1966–1971. doi: 10.1046/j.1538-7836.2003.00348.x. [DOI] [PubMed] [Google Scholar]
- 26.Gale AJ, Radtke KP, Cunningham MA, Chamberlain D, Pellequer JL, et al. Intrinsic stability and functional properties of disulfide bond-stabilized coagulation factor VIIIa variants. J Thromb Haemost. 2006;4:1315–1322. doi: 10.1111/j.1538-7836.2006.01951.x. [DOI] [PubMed] [Google Scholar]
- 27.Radtke KP, Griffn JH, Riceberg J, Gale AJ. Disulfide bond-stabilized factor VIII has prolonged factor VIIIa activity and improved potency in whole blood clotting assays. J Thromb Haemost. 2007;5:102–108. doi: 10.1111/j.1538-7836.2006.02283.x. [DOI] [PubMed] [Google Scholar]
- 28.Sabatino DE, Freguia CF, Toso R, Santos A, Merricks EP, et al. Recombinant canine B-domain-deleted FVIII exhibits high specific activity and is safe in the canine hemophilia A model. Blood. 2009;114:4562–4565. doi: 10.1182/blood-2009-05-220327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fay PJ, Chavin SI, Schroeder D, Young FE, Marder VJ. Purification and characterization of a highly purified human factor VIII consisting of a single type of polypeptide chain. Proc Natl Acad Sci U S A. 1982;79:7200–7204. doi: 10.1073/pnas.79.23.7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miao HZ, Sirachainan N, Palmer L, Kucab P, Cunningham MA, et al. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103:3412–3419. doi: 10.1182/blood-2003-10-3591. [DOI] [PubMed] [Google Scholar]
- 31.Kaufman RJ, Pipe SW, Tagliavacca L, Swaroop M, Moussalli M. Biosynthesis, assembly and secretion of coagulation factor VIII. Blood Coagul Fibrinolysis. 1997;2:S3–S14. [PubMed] [Google Scholar]
- 32.Cunningham MA, Pipe SW, Zhang B, Hauri HP, Ginsburg D, et al. LMAN1 is a molecular chaperone for the secretion of coagulation factor VIII. J Thromb Haemost. 2003;1:2360–2367. doi: 10.1046/j.1538-7836.2003.00415.x. [DOI] [PubMed] [Google Scholar]
- 33.Weiss HJ, Sussman II, Hoyer LW. Stabilization of factor VIII in plasma by the von Willebrand factor. Studies on posttransfusion and dissociated factor VIII and in patients with von Willebrand's disease. J Clin Invest. 1977;60:390–404. doi: 10.1172/JCI108788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mannucci PM, Tenconi PM, Castaman G, Rodeghiero F. Comparison of four virus-inactivated plasma concentrates for treatment of severe von Willebrand disease: a cross-over randomized trial. Blood. 1992;79:3130–3137. [PubMed] [Google Scholar]
- 35.Pipe SW. Coagulation factors with improved properties for hemophilia gene therapy. Semin Thromb Hemost. 2004;30:227–237. doi: 10.1055/s-2004-825636. [DOI] [PubMed] [Google Scholar]
- 36.Doering CB, Healey JF, Parker ET, Barrow RT, Lollar P. Identification of porcine coagulation factor VIII domains responsible for high level expression via enhanced secretion. J Biol Chem. 2004;279:6546–6552. doi: 10.1074/jbc.M312451200. [DOI] [PubMed] [Google Scholar]
- 37.Pipe SW, Miao H, Butler SP, Calcaterra J, Velander WH. Functional factor VIII made with von Willebrand factor at high levels in transgenic milk. J Thromb Haemost. 2011;9:2235–2242. doi: 10.1111/j.1538-7836.2011.04505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dooriss KL, Denning G, Gangadharan B, Javazon EH, McCarty DA, et al. Comparison of factor VIII transgenes bioengineered for improved expression in gene therapy of hemophilia A. Hum Gene Ther. 2009;20:465–478. doi: 10.1089/hum.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, Zhu F, Li J, Lu H, Jiang H, et al. The enhancing effects of the light chain on heavy chain secretion in split delivery of factor VIII gene. Mol Ther. 2007;15:1856–1862. doi: 10.1038/sj.mt.6300268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doering CB, Healey JF, Parker ET, Barrow RT, Lollar P. High level expression of recombinant porcine coagulation factor VIII. J Biol Chem. 2002;277:38345–38349. doi: 10.1074/jbc.M206959200. [DOI] [PubMed] [Google Scholar]
- 41.Doering CB, Denning G, Dooriss K, Gangadharan B, Johnston JM, et al. Directed engineering of a high-expression chimeric transgene as a strategy for gene therapy of hemophilia A. Mol Ther. 2009;17:1145–1154. doi: 10.1038/mt.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hay CR. Porcine factor VIII: current status and future developments. Haemophilia. 2002;1:24–27. doi: 10.1046/j.1365-2516.2002.00125.x. [DOI] [PubMed] [Google Scholar]
- 43.Spencer HT, Denning G, Gautney RE, Dropulic B, Roy AJ, et al. Lentiviral vector platform for production of bioengineered recombinant coagulation factor VIII. Mol Ther. 2011;19:302–309. doi: 10.1038/mt.2010.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barash S, Wang W, Shi Y. Human secretory signal peptide description by hidden Markov model and generation of a strong artificial signal peptide for secreted protein expression. Biochem Biophys Res Commun. 2002;294:835–842. doi: 10.1016/S0006-291X(02)00566-1. [DOI] [PubMed] [Google Scholar]
- 45.Mei B, Pan C, Jiang H, Tjandra H, Strauss J, et al. Rational design of a fully active, long-acting PEGylated factor VIII for hemophilia A treatment. Blood. 2010;116:270–279. doi: 10.1182/blood-2009-11-254755. [DOI] [PubMed] [Google Scholar]
- 46.Schulte S. Use of albumin fusion technology to prolong the half-life of recombinant factor VIIa. Thromb Res. 2008;4:S14–S19. doi: 10.1016/S0049-3848(08)70029-X. [DOI] [PubMed] [Google Scholar]
- 47.Valentino LA. Recombinant FIXFc: a novel therapy for the royal disease? Expert Opin Biol Ther. 2011;11:1361–1368. doi: 10.1517/14712598.2011.603304. [DOI] [PubMed] [Google Scholar]
- 48.Peters RT, Low SC, Kamphaus GD, Dumont JA, Amari JV, et al. Prolonged activity of factor IX as a monomeric Fc fusion protein. Blood. 2010;115:2057–2064. doi: 10.1182/blood-2009-08-239665. [DOI] [PubMed] [Google Scholar]
- 49.Saenko EL, Yakhyaev AV, Mikhailenko I, Strickland DK, Sarafanov AG. Role of the low density lipoprotein-related protein receptor in mediation of factor VIII catabolism. J Biol Chem. 1999;274:37685–37692. doi: 10.1074/jbc.274.53.37685. [DOI] [PubMed] [Google Scholar]
- 50.Sarafanov AG, Ananyeva NM, Shima M, Saenko EL. Cell surface heparan sulfate proteoglycans participate in factor VIII catabolism mediated by low density lipoprotein receptor-related protein. J Biol Chem. 2001;276:11970–11979. doi: 10.1074/jbc.M008046200. [DOI] [PubMed] [Google Scholar]
- 51.Bovenschen N, Herz J, Grimbergen JM, Lenting PJ, Havekes LM, et al. Elevated plasma factor VIII in a mouse model of low-density lipoproteinreceptor-related protein deficiency. Blood. 101:3933–3939. doi: 10.1182/blood-2002-07-2081. [DOI] [PubMed] [Google Scholar]
- 52.Ananyeva NM, Kouiavskaia DV, Shima M, Saenko EL. Catabolism of the coagulation factor VIII: can we prolong lifetime of f VIII in circulation? Trends Cardiovasc Med. 2001;11:251–257. doi: 10.1016/s1050-1738(01)00124-4. [DOI] [PubMed] [Google Scholar]
- 53.Lollar P. Mapping factor VIII inhibitor epitopes using hybrid human/porcine factor VIII molecules. Haematologica. 2000;85:26–28. [PubMed] [Google Scholar]
- 54.Lollar P. Molecular characterization of the immune response to factor VIII. Vox sang. 2002;1:403–408. doi: 10.1111/j.1423-0410.2002.tb05342.x. [DOI] [PubMed] [Google Scholar]
- 55.DiMichele DM, Gorman PO, Kasper CK, Mannucci PM, Santagostino E, et al. Continuous infusion of porcine factor VIII: stability, microbiological safety and clinical experience. Haemophilia. 2002;1:9–12. doi: 10.1046/j.1365-2516.2002.00129.x. [DOI] [PubMed] [Google Scholar]
- 56.Hausl C, Ahmad RU, Sasgary M, Doering CB, Lollar P, et al. High-dose factor VIII inhibits factor VIII-specific memory B cells in hemophilia A with factor VIII inhibitors. Blood. 2005;106:3415–3422. doi: 10.1182/blood-2005-03-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barrow RT, Healey JF, Gailani D, Scandella D, Lollar P. Reduction of the antigenicity of factor VIII toward complex inhibitory antibody plasmas using multiply-substituted hybrid human/porcine factor VIII molecules. Blood. 2000;95:564–568. [PubMed] [Google Scholar]
- 58.Aljamali MN, Margaritis P, Schlachterman A, Tai SJ, Roy E, et al. Long-term expression of murine activated factor VII is safe, but elevated levels cause premature mortality. J Clin Invest. 2008;118:1825–1834. doi: 10.1172/JCI32878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Margaritis P, Arruda VR, Aljamali M, Camire RM, Schlachterman A, et al. Novel therapeutic approach for hemophilia using gene delivery of anengineered secreted activated. Factor VII J Clin Invest. 113:1025–1031. doi: 10.1172/JCI20106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bunce MW, Toso R, Camire RM. Zymogen-like factor Xa variants restore thrombin generation and effectively bypass the intrinsic pathway in vitro. Blood. 2011;117:290–298. doi: 10.1182/blood-2010-08-300756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ivanciu L, Toso R, Margaritis P, Pavani G, Kim H, et al. A zymogen-like factor Xa variant corrects the coagulation defect in hemophilia. Nat Biotechnol. 2011;29:1028–1033. doi: 10.1038/nbt.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chao H, Mao L, Bruce AT, Walsh CE. Sustained expression of human factor VIII in mice using a parvovirus-based vector. Blood. 2000;95:1594–1399. [PubMed] [Google Scholar]
- 63.Gnatenko DV, Saenko EL, Jesty J, Cao LX, Hearing P, et al. Human factor VIII can be packaged and functionally expressed in an adeno-associated virus background: applicability to haemophilia A gene therapy. Br J Haematol. 1999;104:27–36. doi: 10.1046/j.1365-2141.1999.01137.x. [DOI] [PubMed] [Google Scholar]
- 64.Sarkar R, Xiao W, Kazazian HH., Jr A single adeno-associated virus (AAV)-murine factor VIII vector partially corrects the hemophilia A phenotype. J Thromb Haemost. 2003;1:220–226. doi: 10.1046/j.1538-7836.2003.00096.x. [DOI] [PubMed] [Google Scholar]
- 65.Scallan CD, Lillicrap D, Jiang H, Qian X, Patarroyo-White SL, et al. Sustained phenotypic correction of canine hemophilia A using an adeno-associated viral vector. Blood. 2003;102:2031–2037. doi: 10.1182/blood-2003-01-0292. [DOI] [PubMed] [Google Scholar]
- 66.Sarkar R, Tetreault R, Gao G, Wang L, Bell P, et al. Total correction of hemophilia A mice with canine FVIII using an AAV 8 serotype. Blood. 2004;103:1253–1260. doi: 10.1182/blood-2003-08-2954. [DOI] [PubMed] [Google Scholar]
- 67.Ishiwata A, Mimuro J, Mizukami H, Kashiwakura Y, Takano K, et al. Liver-restricted expression of the canine factor VIII gene facilitates prevention of inhibitor formation in factor VIII-defcient mice. J Gene Med. 2009;11:1020–1029. doi: 10.1002/jgm.1391. [DOI] [PubMed] [Google Scholar]
- 68.Lu H, Chen L, Wang J, Huack B, Sarkar R, et al. Complete correction of hemophilia A with adeno-associated viral vectors containing a full-size expression cassette. Hum Gene Ther. 2008;19:648–654. doi: 10.1089/hum.2007.0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kapranov P, Chen L, Dederich D, Dong B, He J, et al. Native Molecular State of Adeno-Associated Viral Vectors Revealed by Single-Molecule Sequencing. Hum Gene Ther. 2011 doi: 10.1089/hum.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dong B, Nakai H, Xiao W. Characterization of genome integrity for oversized recombinant AAV vector. Mol Ther. 2010;18:87–92. doi: 10.1038/mt.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lai Y, Yue Y, Duan D. Evidence for the failure of adeno-associated virus serotype 5 to package a viral genome > or = 8.2 kb. Mol Ther. 2010;18:75–79. doi: 10.1038/mt.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18:80–86. doi: 10.1038/mt.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yan Z, Zhang Y, Duan D, Engelhardt JF. Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc Natl Acad Sci U S A. 2000;97:6716–6721. doi: 10.1073/pnas.97.12.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun L, Li J, Xiao X. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat Med. 2000;6:599–602. doi: 10.1038/75087. [DOI] [PubMed] [Google Scholar]
- 75.Nakai H, Storm TA, Kay MA. Increasing the size of rAAV-mediated expression cassettes in vivo by intermolecular joining of two complementary vectors. Nat Biotechnol. 2000;18:527–532. doi: 10.1038/75390. [DOI] [PubMed] [Google Scholar]
- 76.Burton M, Nakai H, Colosi P, Cunningham J, Mitchell R, et al. Coexpression of factor VIII heavy and light chain adeno-associated viral vectors produces biologically active protein. Proc Natl Acad Sci U S A. 1999;96:12725–12730. doi: 10.1073/pnas.96.22.12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scallan CD, Liu T, Parker AE, Patarroyo-White SL, Chen H, et al. Phenotypic correction of a mouse model of hemophilia A using AAV2 vectors encoding the heavy and light chains of FVIII. Blood. 2003;102:3919–3926. doi: 10.1182/blood-2003-01-0222. [DOI] [PubMed] [Google Scholar]
- 78.Chen L, Lu H, Wang J, Sarkar R, Yang X, et al. Enhanced factor VIII heavy chain for gene therapy of hemophilia A. Mol Ther. 2009;17:417–424. doi: 10.1038/mt.2008.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chao H, Sun L, Bruce A, Xiao X, Walsh CE. Expression of human factor VIII by splicing between dimerized AAV vectors. Mol Ther. 2002;5:716–722. doi: 10.1006/mthe.2002.0607. [DOI] [PubMed] [Google Scholar]
- 80.Lai Y, Yue Y, Liu M, Ghosh A, Engelhardt JF, et al. Effcient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol. 2005;23:1435–1439. doi: 10.1038/nbt1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mah C, Sarkar R, Zolotukhin I, Schleissing M, Xiao X, et al. Dual vectors expressing murine factor VIII result in sustained correction of hemophilia A mice. Hum Gene Ther. 2003;14:143–152. doi: 10.1089/104303403321070838. [DOI] [PubMed] [Google Scholar]
- 82.Scallan CD, Liu T, Parker AE, Patarroyo-White SL, Chen H, et al. Phenotypic correction of a mouse model of hemophilia A using AAV2 vectors encoding the heavy and light chains of FVIII. Blood. 2003;102:3919–3926. doi: 10.1182/blood-2003-01-0222. [DOI] [PubMed] [Google Scholar]
- 83.Sabatino DE, Lange AM, Altynova ES, Sarkar R, Zhou S, et al. Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther. 2011;19:442–449. doi: 10.1038/mt.2010.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Perler FB. InBase: the Intein Database. Nucleic Acids Res. 2002;30:383–384. doi: 10.1093/nar/30.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu XQ, Hu Z. A DnaB intein in Rhodothermus marinus: indication of recent intein homing across remotely related organisms. Proc Natl Acad Sci U S A. 1997;94:7851–7856. doi: 10.1073/pnas.94.15.7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu H, Xu MQ, Liu XQ. Protein trans-splicing and functional mini-inteins of a cyanobacterial dnaB intein. Biochim Biophys Acta. 1998;1387:422–432. doi: 10.1016/s0167-4838(98)00157-5. [DOI] [PubMed] [Google Scholar]
- 87.Milanov P, Ivanciu L, Abriss D, Quade-Lyssy P, Miesbach W, et al. Engineered factor IX variants bypass FVIII and correct hemophilia A phenotype in mice. Blood. 2011 doi: 10.1182/blood-2011-05-353672. [DOI] [PubMed] [Google Scholar]