

Abstract
Drug discovery in the field of oncology has been advanced mainly through the targeting of receptor tyrosine kinases. Both antibodies and small molecule inhibitors have been found to have successful applications in blocking the proliferative functions of these cell surface receptors. Based on these early successes, additional kinases within the cytoplasm have been found to promote cancer and, as such, have been recognized as feasible targets for additional modes of therapies. Unlike these oncogene targets, most tumor suppressors are irreversibly altered during cancer progression and therefore are not feasible targets for therapy. However, a subset of these genes is reversibly epigenetically suppressed. One such gene is BRM, and when it is re-expressed in cancer cells, this gene halts their growth. Moreover, as the key catalytic subunit of the SWI/SNF complex, BRM is centrally important to a host of anticancer pathways and cellular mechanisms, and its status may serve as a biomarker. Restoring its expression will both reconnect a number of growth-controlling pathways and affect cellular adhesion, DNA repair, and immune functions. For these reasons, restoring BRM expression is not only feasible, but potentially a potent form of anticancer therapy. To identify BRM-restoring compounds, we developed a cell-based luciferase assay. In this review, we discuss some of the challenges we encountered, issues related to this type of drug discovery, and our future ambitions. We hope this review will provide insight to this type of endeavor and lead to more investigations pursuing this type of drug research.
Keywords: Brahma, Anticancer, Tumor suppressor, Epigenetic suppression, SWI/SNF, High throughput screening
A NEW TARGET: ACTIVATION OF TUMOR SUPPRESSOR
Like oncogenes or kinases, which are targets for inhibition by small molecular inhibitors, tumor suppressors could also be targeted for therapy. Tumor suppressors are almost always mutated or deleted in cancer, making the restoration of their expression nearly impossible. However, there are some tumor suppressor proteins that, for as-yet unknown reasons, are not altered but rather are epigenetically suppressed. It is possible to restore the expression of these proteins with small molecular inhibitors (SMIs). One such target for therapy is the BRM tumor suppressor protein, which is an essential catalytic component of the SWI/SNF chromatin remodeling complex and is known to be involved in cancer development [1]. Re-expression of BRM in BRM-deficient cells almost always causes growth inhibition. Moreover, BRM is lost in 15–20% of all cancer types, suggesting that loss of BRM affects a significant number of cancer patients [2]. Intriguingly, it has never been found to be mutated but rather is always reversibly suppressed [2–4].
We and other investigators initially found that histone deacetylase (HDAC) inhibitors can induce BRM, but as these compounds act broadly to inhibit most of the known HDACs, they unfortunately cause the inactivation of BRM by acetylating it at specific residues in the C-terminus [4]. Changing these residues has been shown to block this inhibition, indicating that the acetylation of BRM inhibits it. We further investigated this and found that inhibition of HDAC1/2 results in the acetylation of BRM, while inhibition of HDAC3 induces a functional BRM (unpublished data). Hence, because BRM inhibition and BRM induction are governed by different HDACs, BRM can be selectively and functionally induced. Further, BRM is essentially never mutated, has clear-cut anticancer (e.g. growth inhibition, differentiation) activities, is inappropriately silenced in a significant number tumors, and there exists a straightforward assay to screen for compounds that can restore its expression in vitro. Thus, the BRM tumor suppressor protein has many qualities that would make it a strong target for therapy.
BRM’s Role in Cancer
Though SWI/SNF contains a multitude of subunits, the function of this complex requires at least one functional catalytic subunit, either BRM or its homologue BGR1. The concomitant loss of these subunits, which has been documented to occur in several cancer types, including lung cancer, results in the complete abrogation of SWI/SNF complex activity. Both SWI/SNF and BRM associate with and often are required for the function of many key anticancer proteins—for example, both p53 and Rb growth inhibition actions are SWI/SNF-dependent [5–11]. Introduction of a constitutively active isoform of Rb induces growth inhibition in most cell types [10]. In cell lines that lack BRM and BRG1 (and thus lack an active SWI/SNF complex), such as SW13, inducing either p16 (an upstream activator of Rb) or the constituently active isoform of Rb does not inhibit growth. However, if BRM is introduced along with the constituent isoform of Rb, growth inhibition ensues [11]. If BRM alone is introduced into BRM-deficient cell lines, growth inhibition ensues over several days [12, 13]. In addition to growth control, there are a number of reasons why loss of BRM and inactivation of the SWI/SNF complex could contribute to cancer development. As SWI/SNF is known to facilitate and be involved in DNA repair, the loss of this complex, like many other proteins involved in DNA repair, could enhance cancer development and progression [14–16]. BRM also facilitates the expression of a variety of cell adhesion proteins, such as CD44, E-cadherin, Ceacam1, and a number of integrins, such that the loss of BRM might facilitate tumor spread and tissue tropism [11, 17, 18]. To this end, we have done both proteomic and microarray experiments examining the spectrum of the genes regulated in mammalian BRM cell lines and found that it regulates several hundred genes (unpublished data). These findings are in accord with the literature in yeast, where SWI/SNF up- and downregulates 5–7% of yeast genes [19, 20].
To determine the role of a suspected tumor suppressor gene in cancer, researchers will often inactivate the gene in mice to see if tumors develop [21, 22]. The knockout of BRM does not by itself yield tumors; however, it does subtly impact the phenotype of the resultant mice. These mice are 10–15% bigger than their wild type littermates, and examination of the livers from these mice demonstrates an increase in the number of mitotic nuclei [23]. Also, murine embryonic fibroblasts isolated from these mice demonstrate abnormalities in cell cycle control. For example, BRM null fibroblasts do not undergo normal contact inhibition and do not undergo growth arrest in response to irradiation [23]. Moreover, when exposed to carcinogens, BRM null mice develop 10-fold more as well as larger tumors than their wild type littermates [2]. Hence, BRM appears to have a role in growth control but does not appear to be a tumor suppressor. Rather, BRM appears to be a tumor susceptibility gene whose loss would enhance tumor development based data derived from murine models.
BRM Polymorphic Sites Correlate with BRM Loss: Potential Biomarkers for antiBRM Treatments
In an effort to understand how BRM was silenced, we sequenced the promoter region of BRM and found two polymorphic sites consisting of 6–7bp insertions located at −741 and −1321 upstream of the transcriptional start site. We sequenced samples from 160 healthy Caucasian subjects to determine the frequency of these so-called insertion/deletion polymorphisms (IDP) and found both polymorphisms in roughly 20%, 50%, and 30% of the samples for the homozygous state (with), heterozygous state, and wild type (without), respectively [24]. These sites appear to highly similar to binding sites for the transcription family MEF2 [25]—which is interesting, because these transcription factors are known to recruit HDACs and result in the silencing of the target genes [26–28]. Interestly, we and others have found that HDAC inhibitors rapidly reverse BRM suppression in BRM-deficient cancer cells, indicating that HDACs underlie the silencing of this gene [2–4]. We have recently found that knocking down HDAC3 with shRNAi induces BRM (unpublished data). These data suggest that these BRM promoter polymorphisms are likely binding sites for MEF2 transcription factors, which recruit HDACs and in turn silence BRM.
Regardless of the potential mechanism, we observed that homozygous variant (present of the polymorphic site) were found at much higher-than-expect frequencies in twelve BRM-negative cell lines but were almost absent in twelve BRM-positive cell lines. The BRM positive cell lines contained mostly wild type (without) or heterozygous variants [24]. Hence, we found that there was an association between these polymorphic sites and the loss of BRM expression. To verify and validate this association, we analyzed 10 BRM-negative lung tumors and 12 BRM-positive lung cancers. Like the cancer cell lines we tested, in BRM-negative lung tumor cells, we found that both polymorphic sites were homozgyously present, while the distribution of these sites in BRM-positive tumors was approximately the same as that seen in the population we analyzed (unpublished data). We also notice four case of LOH in the BRM-postive tumors where the normal tissue with heterozygous and the tumor was wildtype. But because the BRM-negative patients case where homozygous in the normal tissue, we did not observed any LOH [24]. However, we have detected 7/12 case of LOH in BRM-negative cell lines suggesting that LOH may indeed play a role in the silencing of BRM (unpublished data). Based on this, we therefore conclude that these BRM polymorphisms correlate with BRM loss in both cancer cell lines and primary lung cancers. Further research will be needed to determine whether this correlation occurs in other tumor types and in other ethnic groups and population types. Most importantly, these polymorphisms will potentially serve as a biomarker that can be used to efficiently direct treatment only to those patients whose tumors show loss of BRM expression.
As noted before, BRM null mice develop many more tumors when exposed to carinogens, indicating that BRM is a cancer susceptibility gene, and thus it is not surprising that we found that these BRM polymorphic sites correlate with cancer risk [24]. Our findings suggest that these polymorphic sites can be used to risk stratify patients for cancer screening. Since most specific genetic changes that contribute to cancer development occur in only a fraction of cancer patients (5–35%) and cancers themselves are heterozygous, a key to effective drug treatment is using specific biomarkers to target the most responsive patient populations. Because these polymorphisms correlate so tightly with BRM loss, we may be able to tell which tumors are likely to lack BRM expression by simple genotyping. By genotyping, we could target patients for treatment with BRM-reactivating agents if their tumors were analyzed to be homozygous for both polymorphic sites. We would not even need access to the biopsy to detemine if BRM is lost in a patient’s tumor. Of course, this hypothesis must be validated, but this example illustrates the tandem approach that one must utilize in order to increase success in the development of new therapies. Because we do know that BRM is lost in 10–20% of a variety of solid tumor types, including colon, prostate, breast, esophageal, head/neck, bladder, ovarian, pancreatic, liver and renal cell cancers [2], BRM loss is likely a general phenomenon. If true, reactivation of BRM could benefit a large number of patients.
BRM Assay
Published data with HDAC inhibitors has clearly shown that BRM expression is inducible and that HDACs are functionally linked to the loss of BRM expression. Because first generation HDAC inhibitors (TSA, butyrate, MS-275 etc.) have a broad spectrum of activity and inhibit the majority of HDACs, they cause the BRM to become acetylated and inactivated, in addition to restoring BRM expression [2]. As such, these compounds cannot be used to clinically restore BRM function. While HDACs might be linked to the regulation of BRM, the precise genetic change that takes place to silence BRM is not known. Because we do not know the mechanism to reactivate BRM, we decided to develop an assay to measure the function of BRM and screen compound libraries to find agents that could not only restore BRM expression but its function as well. To this end, we utilized the fact that the glucocorticoid receptor (as well all steroid receptors) is functionally dependent on the SWI/SNF complex and functions poorly or not all in the absence of this complex [29–31]. Because BRM is a key catalytic subunit required for the function of SWI/SNF, in a BRM-deficient cell line it must be re-expressed and be functional for SWI/SNF also to be functional and facilitate steroid receptor-mediated transcription. We stably introduced the glucocorticoid-responsive promoter MMTV linked to luciferase into a BRM/BRG1-deficient cell line, SW13 [2]. In this cell line, luciferase activity becomes upregulated if and only if a glucocorticoid agonist (dexamethasone) and BRM are added or induced (Fig. 1). This occurs because application of agonist (dexamethasone) binds to and activates the glucocorticoid receptor; this receptor in turn binds to glucorticoid response elements in the MMTV promoter and thereby induces transcription of the luciferase gene. It is important that we used a functional assay, since compounds such as HDAC inhibitors when applied to BRM-deficient cancer cells can readily induce BRM expression but cannot functionally activate BRM. This functional screen as designed will thus not target compounds such HDAC inhibitors though they can readily induce BRM. To detect BRM function, hence SWI/SNF function, we used the fact that the glucocorticoid receptor must interact with this complex to induce gene expression.
Fig. 1.
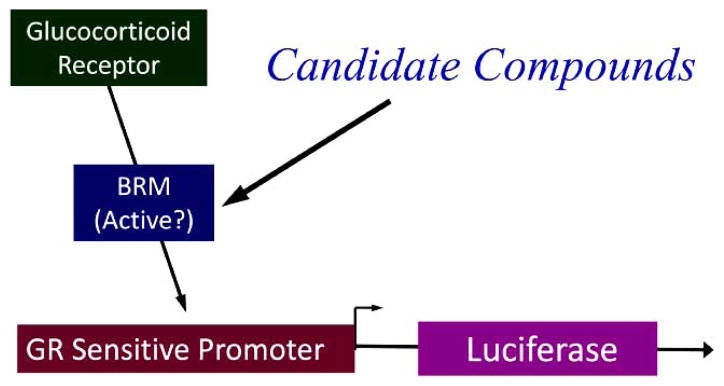
The diagram show the relationship of the glucocorticoid receptor and BRM to the reporter construct.
When dexamethasone is added, GR is activated and then binds to BRM, which in turn binds to the MMTV promoter and induces luciferase.
To test this reporter cell line, we transfected BRG1, BRM, dominant negative BRG1 or dominant negative BRM into the cell line and found robust luciferase activity when either BRG1 or BRM (since they are needed for SWI/SNF activity and are absent in the SW13 cell line) were reintroduced [2]. But we did not see this activity when the dominant negative forms were ectopically expressed in this cell line. To show that HDAC inhibitors cannot activate this assay, we applied dexamethasone and an HDAC inhibitor, either TSA, MS-275, butyrate, or C1-994, for 48–72 hrs to induce BRM and then assayed for luciferase activity. In each case, essentially no luciferase activity was seen [2]. This was not surprising, since a prior publication showed that HDAC inhibitors inhibit BRM by causing BRM to become acetylated [4]. We conducted a number of time course experiments showing that this induction of BRM is transient; these experiments showed that after removal of the HDAC inhibitor, BRM levels, though robustly induced, return to baseline levels in 4–5 days after removal of the HDAC inhibitor [2]. We reasoned that while the HDAC inhibitor is present all BRM proteins become inactivated, but the BRM proteins produced after the HDAC is removed but before BRM is shut off would theoretically be functional. Hence, we tested whether we could observe endogenous BRM function by using our reporter cell line and measuring luciferase activity after the HDAC inhibitor was removed. These experiments showed that regardless of which HDAC inhibitor was used to induce BRM, peak luciferase activity was observed a couple of days after each HDAC inhibitor was removed [1, 2]. Similarly, we tested the induction BRM-dependent genes (CD44, Sparc) as surrogate markers for BRM function during a short period after HDAC removal [2]. For each HDAC, we observed robust induction of both CD44 and Sparc post-HDAC inhibitor treatment but not while these HDAC inhibitors were present [2].
These preliminary experiments demonstrated that our luciferase-based assay robustly detects endogenous induction and ectopic expression of BRM. To proceed with our high throughput screen, however, we needed positive controls. We therefore initially screened about 5000 compounds, finding two in particular, indoprofen and 4 methoxyflavone, that appeared to robustly induce this assay. To optimize our assay, we conducted several different experiments using methoxyflavone and indoprofen, including time-course and dose-response experiments, to determine the optimal dose and time interval between application of the compounds and assaying for luciferase (Fig. 2). We found, using western blotting, that between 36–48 hours, BRM is maximally induced with these compounds, like HDAC inhibitors. We also treated BRM-negative cells (SW13) with different concentrations of these compounds and found by western blotting that 6–10 um of methoxyflavone and 200 nM of indoprofen were optimal for inducing BRM (Fig. 4). Using these optimal parameters, we found with our reporter system that maximal induction of luciferase activity was about 3–4 fold with either 10 um 4 methoxyflavone or 200 nM indoprofen, with incubation periods of 72 hrs Fig. (2). We tried these conditions in a 96-well-plated format and found that we were able to routinely get a z-score of greater than 0.5. We also conducted a series of experiments to determine the optimal number of cells to seed in 96 well plates, observing 7000–8000 cells would continue to grow but not reach confluence or exhaust the media. This point is important, because we found that prolonged incubation at high density would often induce our gene of interest, BRM, thereby potentially giving us false positive results. To control for this, we ran on each plate a number of wells with DMSO and then with and without dexamethasone, establishing a baseline for luciferase activity that was well below that of the positive control (Fig. 3).
Fig. 2.

Luciferase induction. MG213 cells (7500 cells/well) were incubated in the presence of 0.1 uM dexamethasone and various concentrations of methoxyflavone or indoprofen. After 72 hours, luciferase expression was measured using the Steady Glo assay (Promega).
Fig. 4.
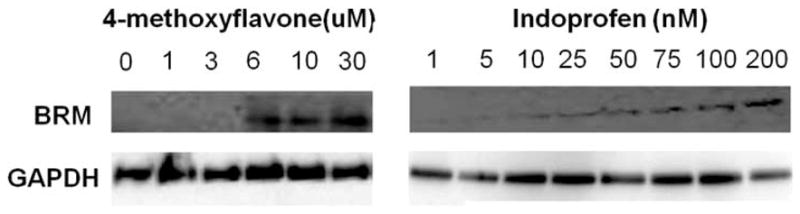
Methoxyflavone and indoprofen were added at various concentrations, and after three days of treatment, the total protein was collected and BRM protein expression was assessed. Indoprofen maximally induced BRM at 100–200 nM, while methoxyflavone maximally induced BRM at 6–10 um.
Fig. 3.
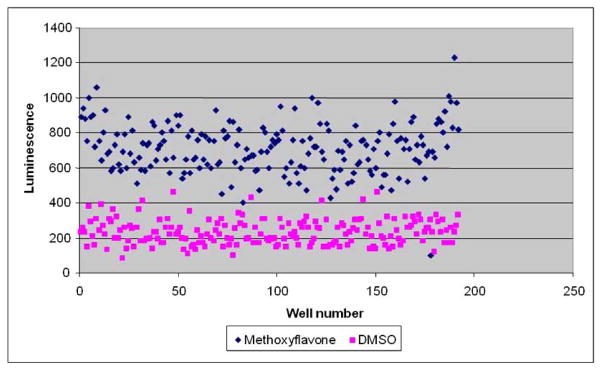
Assay reproducibility. MG213 cells (7500 cells/well) were treated with 0.1 uM dexamethasone then either indoprofen (0.20 uM) or DMSO for 3 days. Luciferase signal was measured using the Steady Glo assay.
Conducting the Screen
Using this assay, we screened ~125,000 compound libraries at the University of Michigan Life Science Institute (LSI) and the Michigan High-throughput Screening Facility in Kalamazoo, Michigan. All compounds were solubilized in 100% DMSO and were delivered to cell plates. The DMSO was maintained at 0.4% for all single dose screens. The compounds were screened at a final concentration of 12 uM. For all experiments, the 4-methoxyflavone was solubilized in DMSO and used at a final concentration of 10 uM. The reporter cell line MGR13, a subline of the BRG1/BRM-deficient cell line SW13 that contains a stably integrated glucocorticoid-responsive MMTV-luciferase reporter, was used for high throughput screening purposes and was maintained in RPMI with 5% bovine serum albumen, 1% L-Glutamine and penicillin/streptomycin, and 500 ug/ml G418. Cells were maintained at 5% CO2 and 37°C. Using this assay, we observed an initial hit rate between 1% to 2%, which we narrowed to 500 compounds by using dose-response screening and arbitarily selecting hits as those with greater than 75% of the activity seen with the positive controls. A lower threshold level would have of course increased the number of hits. The z-score for this assay was variable, but we most often got a value of over 0.5, which is good for a cell-based assay such as this one. This z-score finding is important because it demonstrates the reliability of this cell-based assay. To calculate the z-score, repeated measurement for methoxyflavone compared to control (DMSO and 0.1 uM dexamethasone) was done and is show in Fig. (3).
Though we have to yet to conduct our proposed counter screens on the vast majority of these compounds, we have tested two compounds, which we termed RH and GK. Using 10 uM concentrations of RH and GK in this screen, these compounds were found to induce luciferase 3–4 fold. Further dose response studies of these two compounds showed a maximum induction of BRM mRNA of 10–20 fold by quantitative PCR with 50 uM RH and 200 uM GK in both C33a and SW13 cell lines [24]. Using these two concentrations, we observed that these two compounds induced growth inhibition over 90% after 5 days of treatment in two different BRM-deficient cell lines (SW13 and C33A). This growth inhibition was mainly due to BRM induction, as these cell lines infected with three different antiBRM shRNAi (sufficient to block induction of BRM) did not demonstrate any significant growth inhibition when either compound was applied for the same 5-day test period [24]. Western blotting confirmed that both compounds induce BRM protein only in C33A and SW13 cell lines and not the infected antiBRM shRNAi daughter cell lines [24]. To further show that the RH and GK induce functional BRM, we used quantitative PCR to see if specific BRM-dependent genes were induced when RH and GK were applied to the parental C33A and SW13 but not the daughter cell lines. We observed a 5–10 fold induction of two BRM-dependent genes P8 and GPR56 in each of these two cell lines [24]. These findings show that BRM can be targeted and pharmacologically reactivated.
TROUBLESHOOTING
We addressed a number of potential problems as we developed this assay. We first focused on cell number. We needed to establish optimal levels of luciferase activity to differentiate potential “hits” from background in this assay. We started optimizing at the 96-well-plate level, plating different cell densities from 10,000–30,000 cells and eventually selected an optimal density of 30,000 cells/well. After establishing the appropriate cell density in the 96-well format, we transitioned to the 384-well format and tested cell numbers ranging from 6,000–12,000 cell/well, with the optimal density being 7,500 cells/well. As we continued to run the experiment over time, the cells we were using began to grow faster, causing them to reach a higher density. To ameliorate this problem, we tried both reducing the cell number by roughly 30% and reducing the serum from 5% to 2% with the latter experimental change being the most effective. Thus, for the duration of these screens, it was important to monitor for changes in final cell density over time, as it could affect overall assay sensitivity.
Another important consideration for high-throughput screening was the selection of a positive and a negative control, which is essential for the purpose of accurately determining potential hits. Because high throughput screening entails assaying thousands of compounds measured as single samples, the positive and negative controls must be well defined. The assay should analyze the distribution of positive and negative controls relative to the single potential hit measurement to determine if the unknown is significantly different from the negative control and to ascertain whether the value is real or occurred merely by chance (Zhang, 1999) [32, 33]. Ultimately, as a quality assessment of assay conditions, the z score is calculated and has been defined by Zhang et al. [32].
During the optimization process, identifying the appropriate positive control for this assay proved to be more difficult than anticipated. In our initial pilot studies, we transfected MGR13 cells with the BRM gene. We had previously shown BRM transfection as an effective method of activating luciferase. Yet, for high throughput screening purposes, this approach was problematic, as BRM causes growth inhibition and reduced adherence, making it difficult to replate transfected cells in the 384-well format. Yet, because our assay was based on identifying compounds that activate rather than inhibit BRM, we initially based our assay on the negative control. Specifically, the negative control was DMSO and dexamethasone (0.1 uM)-treated cells, which consistently provide a relative luciferase value of ~250 (Fig. 3). Thus, in our initial chemical library screen, we designated an artificial “positive” hit as anything at least 2-fold higher than the negative control. From this screen, we identified 4-methoxyflavone as the compound that demonstrated the highest levels of relative luciferase activity, which was roughly ~3-fold greater than the negative control. We therefore chose to use this compound in every subsequent assay as a positive control. We also conducted western blot in BRM-negative cell lines with 4-methoxyflavone and indoprofen showing that these compound can indeed induce BRM expresson (Fig. 4). Next, we established the concentration of 4-methoxyflavone that would provide the optimal luciferase activity. Performing dose response studies ranging from 0–30 uM, we found that 10 uM of 4-methoxyflavone provided the optimum luciferase activation Fig. (2), and this dose was therefore used as the positive control concentration for all subsequent high throughput screening assays. The third parameter was determining the time period. A simple time course experiment showed us that about 48 hrs was sufficient to induce BRM without controls. However, we used 72 hrs, as we reasoned that other proteins may require a longer time to activate BRM. Next, we replicated our investigations and developed a z-score that fostered confidence in our study. Typically, we established a z-score of 0.5–0.6. A thorough discussion of the validity of this parameter can be found elsewhere [34]. Finally, we discovered that plating the cells on the bench rather than in a tissue culture hood did not result in contamination. Nevertheless, one has to be concerned that contamination may occur if long incubation periods are used.
Identifying the Targets
As functional screens are not target-specific, they readily identify drugs that activate the target but fail to distinguish which protein is impacted by a given compound. By having multiple possible targets that elicit the same response, it is feasible that multiple drug classes of drugs could be developed that have the same assay endpoint. For example, if one were to screen compounds for their ability to kill bacteria, then one could potentially identify multiple different antiboitics, each of them having value in different situation. Similarly, there are probably several different ways test compounds that could activate BRM. To proceed with drug development, one must know or be able to identify the targets of interest. The other question that arises is which is the best target protein, if multiple target proteins exist? Though there maybe multiple drug targets that activate BRM, it would be prudent to focus on those that become dysregulated or dysfunctional and thus silence BRM. As the effectiveness of drug therapy hinges on minimizing untoward effects on normal cells, developing theraupetic drugs that selectively impact oncogenic changes may be better tolerated by reducing side-effects and overall toxicity. By this reasoning, one must identify as many of the target proteins as possible and then test each of the potential targets to see if they are mutated, altered, over- or underexpressed, or otherwise dysfunctional, thereby causing the silencing of the anticancer gene—in this case BRM.
How does one identify target proteins involving in the regulation of genes? Typically, one could undertake a complex molecular approach and tease out which proteins are vital to the suppression of the target gene. Alternatively, one could take advantage of the same screening assay to help identify specific targets. To identify these potential targets, we can use our new assay again but in a slightly different way. We activated BRM expression by inhibiting any number of potential target proteins using oligo shRNAi. Thus, instead of screening with compounds, we will screen with an oligonucleotide sRNAi library purchased from Dharmacon. This sRNAi will knock down some 18,000 different genes, and those tied to the regulation of BRM will be restored if this protein can be detected by induction of luciferase. By identifying which proteins when inhibited upregulate BRM, we can then match them with the compounds identified in our high-throughput screening. In this way, one could conceivably develop a series of novel classes of targeted therapies. Thus, functional high throughput screening assays are ideally done in parallel with shRNAi screens to identify as many target proteins and pathways as possible.
SUMMARY
Because cancer is driven both by the gain of cancer-causing genes and the loss of growth-control genes, it seems reasonable that any method that attempts to restore the homeostatic state of normal cells would be sound to use in the battle against it. In the last 10 years, the inhibition of growth-promoting kinases such as EGFR or HER2 has been increasingly exploited. But the development of drugs that activate anticancer proteins such as BRM is novel. One reason for the lag in development of drug to activate anticancer proteins comes from the fact that most anticancer proteins are mutated or irreversibly altered, thereby limiting the application of this type of approach. However, as more and more anticancer proteins are identified, a subset of these proteins will likely be epigenetically-and therefore reversibly-silenced. In our pursuit to develop a drug to activate BRM, we have determined some basic principles for drug discovery unique to anticancer proteins. Because the regulatory mechanisms of most anticancer proteins are likely to be complex, there will be multiple targets that when inhibited activate the gene in question. Defining the various proteins involved in the regulation of a given anticancer protein will therefore most likely be a prerequisite to any effective drug development scheme. As such, oligio RNAi screens will be needed to be employed whenever possible, and we have outlined a series of parameters that should be maximized or minimized in order to increase the efficiency of a screen. Together, these observations should encourage other investigators to pursue this line of drug discovery.
References
- 1.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28(14):1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 2.Glaros S, Cirrincione GM, Muchardt C, Kleer CG, Michael CW, Reisman D. The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene. 2007;26(49):7058–7066. doi: 10.1038/sj.onc.1210514. [DOI] [PubMed] [Google Scholar]
- 3.Yamamichi N, Yamamichi-Nishina M, Mizutani T, Watanabe H, Minoguchi S, Kobayashi N, Kimura S, Ito T, Yahagi N, Ichinose M, Omata M, Iba H. The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene. 2005;24(35):5471–5481. doi: 10.1038/sj.onc.1208716. [DOI] [PubMed] [Google Scholar]
- 4.Bourachot B, Yaniv M, Muchardt C. Growth inhibition by the mammalian SWI-SNF subunit Brm is regulated by acetylation. EMBO J. 2003;22(24):6505–6515. doi: 10.1093/emboj/cdg621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oh J, Sohn DH, Ko M, Chung H, Jeon SH, Seong RH. BAF60a interacts with p53 to recruit the SWI/SNF complex. J Biol Chem. 2008;283(18):11924–11934. doi: 10.1074/jbc.M705401200. [DOI] [PubMed] [Google Scholar]
- 6.Naidu SR, Love IM, Imbalzano AN, Grossman SR, Androphy EJ. The SWI/SNF chromatin remodeling subunit BRG1 is a critical regulator of p53 necessary for proliferation of malignant cells. Oncogene. 2009;28(27):2492–2501. doi: 10.1038/onc.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee JH, Lee JY, Chang SH, Kang MJ, Kwon H. Effects of Ser2 and Tyr6 mutants of BAF53 on cell growth and p53-dependent transcription. Mol Cells. 2005;19(2):289–293. [PubMed] [Google Scholar]
- 8.Lee D, Kim JW, Seo T, Hwang SG, Choi EJ, Choe J. SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. J Biol Chem. 2002;277(25):22330–22337. doi: 10.1074/jbc.M111987200. [DOI] [PubMed] [Google Scholar]
- 9.Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101(1):79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- 10.Strobeck MW, Knudsen KE, Fribourg AF, DeCristofaro MF, Weissman BE, Imbalzano AN, Knudsen ES. BRG-1 is required for RB-mediated cell cycle arrest. Proc Natl Acad Sci U S A. 2000;97(14):7748–7753. doi: 10.1073/pnas.97.14.7748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reisman DN, Strobeck MW, Betz BL, Sciariotta J, Funkhouser W, Jr, Murchardt C, Yaniv M, Sherman LS, Knudsen ES, Weissman BE. Concomitant down-regulation of BRM and BRG1 in human tumor cell lines: differential effects on RB-mediated growth arrest vs CD44 expression. Oncogene. 2002;21(8):1196–1207. doi: 10.1038/sj.onc.1205188. [DOI] [PubMed] [Google Scholar]
- 12.Strober BE, Dunaief JL, Guha Goff SP. Functional interactions between the hBRM/hBRG1 transcriptional activators the pRB family of proteins. Mol Cell Biol. 1996;16(4):1576–1583. doi: 10.1128/mcb.16.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, Begemann M, Crabtree GR, Goff SP. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994;79(1):119–130. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- 14.Park JH, Park EJ, Lee HS, Kim SJ, Hur SK, Imbalzano AN, Kwon J. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J. 2006;25(17):3986–3997. doi: 10.1038/sj.emboj.7601291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park JH, Park EJ, Hur SK, Kim S, Kwon J. Mammalian SWI/SNF chromatin remodeling complexes are required to prevent apoptosis after DNA damage. DNA Repair (Amst) 2009;8(1):29–39. doi: 10.1016/j.dnarep.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Gong F, Fahy D, Liu H, Wang W, Smerdon MJ. Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle. 2008;7(8):1067–1074. doi: 10.4161/cc.7.8.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Batsche E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol. 2006;13(1):22–29. doi: 10.1038/nsmb1030. [DOI] [PubMed] [Google Scholar]
- 18.Strobeck MW, DeCristofaro MF, Banine F, Weissman BE, Sherman LS, Knudsen ES. The BRG-1 subunit of the SWI/SNF complex regulates CD44 expression. J Biol Chem. 2001;276(12):9273–9278. doi: 10.1074/jbc.M009747200. [DOI] [PubMed] [Google Scholar]
- 19.Zraly CB, Middleton FA, Dingwall AK. Hormone-response genes are direct in vivo regulatory targets of Brahma (SWI/SNF) complex function. J Biol Chem. 2006;281(46):35305–35315. doi: 10.1074/jbc.M607806200. [DOI] [PubMed] [Google Scholar]
- 20.Sudarsanam P, Iyer VR, Brown PO, Winston F. Whole-genome expression analysis of snf/swi mutants of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2000;97(7):3364–3369. doi: 10.1073/pnas.050407197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim CF, Jackson EL, Kirsch DG, Grimm J, Shaw AT, Lane K, Kissil J, Olive KP, Sweet-Cordero A, Weissleder R, Jacks T. Mouse models of human non-small-cell lung cancer: raising the bar. Cold Spring Harb Symp Quant Biol. 2005;70:241–250. doi: 10.1101/sqb.2005.70.037. [DOI] [PubMed] [Google Scholar]
- 22.Jacks T. Lessons from the p53 mutant mouse. J Cancer Res Clin Oncol. 1996;122(6):319–327. doi: 10.1007/BF01220798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha) EMBO J. 1998;17(23):6979–6991. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu G, Rogers CB, Munoz D, Gordon V, Cheng D, Kalam A, Mirshams M, Xu W, Shepherd F, Tsao M, Reisman D. Two novel functional BRM insertion promoter polymorphisms, loss of BRM expression, and lung cancer risk. Oncogene. 2010 doi: 10.1038/onc.2011.81. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fickett JW. Quantitative discrimination of MEF2 sites. Mol Cell Biol. 1996;16(1):437–441. doi: 10.1128/mcb.16.1.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nebbioso A, Manzo F, Miceli M, Conte M, Manente L, Baldi A, De Luca A, Rotili D, Valente S, Mai A, Usiello A, Gronemeyer H, Altucci L. Selective class II HDAC inhibitors impair myogenesis by modulating the stability and activity of HDAC-MEF2 complexes. EMBO Rep. 2009;10(7):776–782. doi: 10.1038/embor.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gregoire S, Xiao L, Nie J, Zhang X, Xu M, Li J, Wong J, Seto E, Yang XJ. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol Cell Biol. 2007;27(4):1280–1295. doi: 10.1128/MCB.00882-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bertos NR, Wang AH, Yang XJ. Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol. 2001;79(3):243–252. [PubMed] [Google Scholar]
- 29.Inoue H, Furukawa T, Giannakopoulos S, Zhou S, King DS, Tanese N. Largest subunits of the human SWI/SNF chromatin-remodeling complex promote transcriptional activation by steroid hormone receptors. J Biol Chem. 2002;277(44):41674–41685. doi: 10.1074/jbc.M205961200. [DOI] [PubMed] [Google Scholar]
- 30.Fryer CJ, Archer TK. Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature. 1998;393(6680):88–91. doi: 10.1038/30032. [DOI] [PubMed] [Google Scholar]
- 31.Hsiao PW, Fryer CJ, Trotter KW, Wang W, Archer TK. BAF60a mediates critical interactions between nuclear receptors and the BRG1 chromatin-remodeling complex for transactivation. Mol Cell Biol. 2003;23(17):6210–6220. doi: 10.1128/MCB.23.17.6210-6220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 33.Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5(2):73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rabow AA, Shoemaker RH, Sausville EA, Covell DG. Mining the National Cancer Institute’s tumor-screening database: identification of compounds with similar cellular activities. J Med Chem. 2002;45(4):818–840. doi: 10.1021/jm010385b. [DOI] [PubMed] [Google Scholar]