

Abstract
Metabolic syndrome is a cluster of metabolic and cardiovascular symptoms: insulin resistance (IR), obesity, dyslipemia. Hypertension and vascular disorders are central to this syndrome. After a brief historical review, we discuss the role of sympathetic tone. Subsequently, we examine the link between endothelial dysfunction and IR. NO is involved in the insulin-elicited capillary vasodilatation. The insulin-signaling pathways causing NO release are different to the classical. There is a vasodilatory pathway with activation of NO synthase through Akt, and a vasoconstrictor pathway that involves the release of endothelin-1 via MAPK. IR is associated with an imbalance between both pathways in favour of the vasoconstrictor one. We also consider the link between hypertension and IR: the insulin hypothesis of hypertension. Next we discuss the importance of perivascular adipose tissue and the role of adipokines that possess vasoactive properties. Finally, animal models used in the study of vascular function of metabolic syndrome are reviewed. In particular, the Zucker fatty rat and the spontaneously hypertensive obese rat (SHROB). This one suffers macro- and microvascular malfunction due to a failure in the NO system and an abnormally high release of vasoconstrictor prostaglandins, all this alleviated with glitazones used for metabolic syndrome therapy.
1. Introduction
The metabolic syndrome is a cluster of metabolic and cardiovascular symptoms that are strongly associated with type II diabetes mellitus. In this kind of diabetes, rather than prolonged high levels of glycemia, there is insulin resistance with secondary hyperinsulinemia, both very frequently associated with, hypertension, dyslipemia, atherosclerosis, and, most importantly, obesity (Figure 1) [1]. Vascular disorders are central to this condition. Quoting prof. Yki-Järvinen “…after all, from a clinical point of view, type II diabetes mellitus is a disease of blood vessels, not muscle.” [2]. For these reasons, it is also known as cardiometabolic syndrome [1], and hypertension plays a pivotal role. Indeed, risk estimates according to the Framingham study show that roughly 80% of essential hypertension in men and 65% in women can be directly attributed to obesity [3]. There is a clear association between body mass index and arterial pressure even in nonobese, lean people [4–6]. Still, some obese people are not hypertensive. For example, the North American Pima Indians, who have a high prevalence of obesity, but do not have corresponding high rates of hypertension [7].
Figure 1.
Two ways to conceptualize metabolic syndrome and the position hypertension and the other symptoms occupy. According to the WHO definition, insulin resistance is central to any other symptom (a). Others define metabolic syndrome as a cluster of symptoms where none has a central position (b).
The history of metabolic syndrome takes us back to the early 20th century, when two physicians, the Swedish, Kylin and the Spanish Marañón nearly simultaneously and independently published in the journal Zentralblatt für Innere Medizin two articles under almost the same title: Über Hypertonie und Zuckerkrankheit [8, 9]. In these articles, the two physicians described for the first time the coexistence of hypertension and diabetes mellitus in adults and proposed a common mechanism for the development of these disorders. In 1988, Reaven, hypothesized that insulin resistance is the common etiological factor of a group of disorders, such as high blood pressure, hyperinsulinemia, high levels of low density lipoproteins (LDL), triglycerides, and cholesterol, and low levels of high density lipoproteins (HDL). Reaven named this collection of disorders “syndrome X” [1]. A year later, Kaplan added to the pathologies described by Reaven a very important factor, central adiposity (increase in splanchnic and subcutaneous fat depots in the abdominal region) [10]. Since then, abdominal obesity has been considered one of the typical components of the syndrome.
Both type 2 diabetes mellitus and metabolic syndrome are reaching epidemic proportions. Considering that 220 million people worldwide are diabetic, this disease has become a serious epidemiological problem [11]. The problem is not only the size of the figures but also the alarming increase in only a few decades (46% in the 1990s). Metabolic syndrome is, probably, the most important challenge for health authorities in developed and developing countries [11, 12]. In Europe there is a clear North-South gradient in almost all cardiovascular risk factors related with metabolic syndrome. For example, mortality from coronary heart disease, expressed as a mortality ratio, presented in men aged 30–69 the following geographical indices: 8.2 Iceland; 5.1 England; 2.2 Italy; 1.8 Spain; and 0.9 Portugal [13]. However, there is no doubt that the paradigm of overdevelopment-overweight is the United States. With the turn of the century, 61% of Americans were sufficiently overweight to suffer health problems directly derived from this condition [14]. A diet that is as excessive as inadequate has yielded these epidemiological figures in less than 20 years: between 1977 and 1995 daily caloric intake rose by 200 calories. This is the equivalent to an increment of 10 calories per year [14].
2. Role of the Sympathetic Nervous System
There are 3 conditions, typical of metabolic syndrome, that may cause an exacerbation of sympathetic tone. Namely, hyperinsulinemia, hyperleptinemia, and hyperlipidemia. In 1981, it was reported that hyperinsulinemia, independently of changes in glycemia, caused a substantial increase in circulating noradrenaline concentration accompanied by an increase in blood pressure [15]. These sympathoexcitatory effects of insulin appear to be centrally mediated, since they are apparent only during systemic insulin infusion but not local infusion [16]. In addition, high levels of insulin increase sodium reabsorption [17] favouring expansion of extracellular fluid volume, which may predispose to hypertension [18]. Furthermore, obesity impairs renal-pressure natriuresis and causes sodium retention. Obese subjects require increased arterial pressure to maintain sodium balance, indicating impaired renal-pressure natriuresis [19].
In addition to insulin, leptin can also be a link between obesity and increased sympathetic activity. Besides its effect on appetite and metabolism, leptin acts in the hypothalamus to increase blood pressure through activation of the sympathetic nervous system [20]. High circulating levels of leptin are reported to explain much of the increase in the renal sympathetic tone observed in obese human subjects [21]. Leptin-induced increases in renal sympathetic activity and blood pressure are mediated by the ventromedial and dorsomedial hypothalamus [22].
Finally, high circulating levels of free fatty acids in visceral obese individuals may participate in the activation of the sympathetic nervous system. The increased release of free fatty acids into the portal vein from lipolysis in visceral fat depots could explain the strong association between visceral obesity and increased sympathetic nerve outflow [23].
3. Role of Insulin
3.1. Insulin Resistance and Endothelial Dysfunction
In 1939, Himsworth postulated that type 2 diabetes mellitus was not only an insulin deficiency state but also a disease in which cells are unresponsive to insulin. Thus, Himsworth's work gave birth to the concept of insulin resistance [24, 25]. Insulin resistance is clinically defined as the inability of a known quantity of insulin (exogenous or endogenous) to increase glucose uptake and utilization in an individual as much as it does in a normal population [26]. There is a clear link between endothelial dysfunction and insulin resistance [27, 28] but the mechanism by which insulin resistance leads to endothelial dysfunction is complex and involves the action of mediators of inflammation in the visceral fat, liver, and muscle [29]. It is well known that insulin resistance and compensatory hyperinsulinaemia, besides activating the mechanisms mentioned above, have also a vascular toxicity effect, mainly at the endothelial level. This, partly because insulin resistance impairs the production of NO, favors the production of endothelin-1 and the vasoconstrictive and mitogenic responses on the vascular wall [30].
3.1.1. Role of NO in Insulin Resistance
King and Johnson reported in 1985 that the endothelial cell membrane displays insulin receptors [31]. Functional studies indicate that endothelium-derived NO is involved in the insulin-elicited increase in blood flow and recruitment of capillaries that physiologically links hemodynamics to the metabolic action of insulin on the tissues [32–34]. Insulin resistance is associated with impaired NO synthase activity [35] and an abnormal basal NO-mediated dilation in the forearm arterial bed [36]. The insulin-induced increase of microvascular endothelium-dependent vasodilation is abolished in insulin resistance conditions such as obesity [37]. Moreover, insulin has been shown to constrict rather than dilate forearm resistance arteries in obese patients [38]. On the other hand, inhibition of NO synthesis or endothelium removal reveals a vasoconstrictor effect of insulin on isolated arterioles [39]. Definitive proof of the relationship between NO and insulin sensitivity has been provided by knock-out mice that are homozygous null for the eNOS gene. These peculiar animals display an expected hemodynamic phenotype of increased basal blood pressure but also are insulin resistant [40]. Therefore, insulin has indeed a hemodynamic component, albeit small compared to the metabolic one. But both are coupled in such a manner that endothelial dysfunction can cause insulin resistance, and this, in a vicious circle, aggravates endothelial function.
Interestingly, insulin-signaling pathways in vascular endothelium leading to the activation of endothelial NO synthase are completely independent and distinct from classical calcium-dependent mechanisms used by G-protein-coupled receptors, such as the acetylcholine receptor [34]. The messenger pathway that is activated when insulin binds insulin receptor appears to be as follows [41]: insulin binds insulin receptor (INS-R) which is at the same time a tyrosine kinase and this undergoes autophosphorylation of tyrosine residues. INS-R phosphorylates insulin receptor substrate-1 (IRS-1). The signalling pathway from insulin branches at IRS-1. One of the branches involves the activation of phosphoinositide 3 kinase (PI-3K), leading to phosphatidylinositol-3,4,5-triphosphate as well as to phosphorylation and activation of phosphoinositide-dependent kinase 1 (PDK-1). Both products, in turn, phosphorylate and activate Akt (also called protein kinase B, PKB). Akt directly phosphorylates eNOS at Ser1177, resulting in increased eNOS activity and NO production [42]. Remarkably, the vascular actions of insulin that stimulate the production of NO possess remarkable similarities to metabolic insulin-signaling pathways. For instance, activation of Akt is also a common step for glycogen synthase kinase inhibition and GLUT-4 transporter translocation [41].
3.1.2. Role of Endothelin-1 in Insulin Resistance
In 1991, Oliver et al. demonstrated that insulin was able to stimulate endothelin-1 (ET-1, a very strong vasoconstrictor) gene expression in endothelial cells [43]. Later, it was shown that insulin can modulate circulating ET-1 levels [44] and increased plasma levels of ET-1 were observed in type II diabetic patients [45]. An additional work in the skeletal muscle circulation reported that insulin stimulates both NO activity (already known as we showed before) and ET-1 [46].
The authors then suggested that an imbalance between the release of both substances may be involved in pathophysiology of hypertension and atherosclerosis in insulin-resistant states associated with endothelial dysfunction [46]. Following research has shown that insulin induces endothelin-mediated vasoconstriction only when NO synthase or phosphatidylinositol-3 kinase (PI3K) is inhibited [47]. In a paper elegantly entitled “Endothelin antagonism uncovers insulin-mediated vasorelaxation in vitro and in vivo” [48], Verma et al. demonstrated that insulin-mediated vasorelaxation is only well patent when antagonizing ET-1 receptors. This proved previous proposals that insulin exhibits a dual and opposite action on blood vessels: NO-mediated vasodilation and ET-1-mediated vasoconstriction. It is known that MAPK activation by IRS-1 causes the release of endothelin-1, which promotes insulin resistance (by reducing blood supply to the skeletal muscle), increases oxidative stress, reduces the bioavailability of NO, and promotes a proatherogenic state [49].
3.2. Hyperglycaemia and Vascular Function
Regardless of the evidence linking the vascular dysfunction of type II diabetes mellitus with failures in the vascular biology of insulin, there are many reports that attribute these dysfunctions to the very fact of the existing hyperglycaemia. We wish to draw attention to the functional effects of the acute excess in glucose occurring in a particular moment. In this regard, it has been reported that glucose favours vasoconstriction [50] and impairs vasodilation [51]. In arteries of diabetic rats, Taylor et al. demonstrated that hyperglycaemia reduces the tonic release of NO [52] and established a central role for glucose in the development of vascular functional changes associated with experimental diabetes [50]. Most interesting is the finding that in healthy subjects, acute hyperglycaemia impairs endothelium-dependent vasodilation in both the microcirculation and the macrocirculation when assessed in the brachial artery [53]. More precise data on the mechanisms involved in hyperglycaemia was released by Sobrevia et al. [54] who showed that exposure of endothelial cells to elevated glucose was associated with stimulation of L-arginine transport paralleled by an increase in basal release of NO and prostacyclin. This would be good news if they did not find as well that insulin treatment downregulated the elevated activity of the L-arginine transport system and that of NO synthase in the cells exposed to hyperglycaemia. They concluded that the modulation of the human endothelial cell L-arginine-NO pathway by insulin is influenced by predisposing hyperglycaemic clinical conditions [54]. In a later study, Renaudin et al. demonstrated that the vasodilatory effect of insulin disappears when hyperglycaemia exists, perhaps blunted by the vasoconstrictive effect of glucose [55].
3.3. Insulin Actions on Blood Pressure: The Insulin Hypothesis of Hypertension
So far we have focused on the cardiovascular effects of insulin at a local level. However, it cannot be forgotten that insulin has systemic actions affecting the sympathetic nervous system and kidney. The surge of epidemiological reports relating insulin resistance and hyperinsulinemia has fueled the idea of the so-called insulin hypothesis of hypertension. There is no question that insulin resistance is epidemiologically linked with hypertension [1]. The insulin hypothesis of hypertension proposes that the compensatory hyperinsulinemia that occurs with insulin resistance increases sodium reabsorption and sympathetic activity, which combine to cause elevated arterial pressure. Support for this hypothesis comes from various lines of evidence. First, the correlation between insulin resistance and high blood pressure [56], which is emphasized by the fact that, even lean individuals with essential hypertension, display insulin resistance and hyperinsulinemia. Some go a step further asserting that essential hypertension is “per se” an insulin resistance state [57]. Second, as explained before, insulin has multiple actions on the sympathetic nervous system, the kidney, and the vasculature which can lead to hypertension. Third, the observation that drugs which improve insulin resistance and decrease hyperinsulinemia, are reported to be antihypertensive. For instance, Landin et al. reported that oral administration of metformin to insulin-resistant, hypertensive men increased insulin sensitivity and significantly decreased arterial pressure [58]. Another remarkable example is the well-known blood pressure lowering effects of insulin sensitizers glitazones [59]. For review, see [60]. Fourth and finally, the observation that some antihypertensives, such as angiotensin II converting enzyme inhibitors [61] or angiotensin II receptor antagonists [62], increase insulin sensitivity as well. Despite the size of the support in favour of the insulin hypothesis of hypertension, there is also important evidence against. For instance, the eminent physiologist Hall and his collaborators failed to find a correlation between insulin and hypertension in a well-controlled model in dogs [63].
4. Role of Adipokines
Traditionally, adipocytes were considered energy reservoirs that store triglycerides during feeding and deliver fatty acids during fasting. However, it has become quite clear that adipose tissue does much more than this and is responsible for the synthesis and secretion of numerous proteins. The first protein described was adipsin [64]. Later, the secretion of cytokines such as TNF-α was described [65], thus conferring immune functions to adipocytes. Funahashi et al. named these substances adipocytokines [66]. Undoubtedly, the most relevant discovery was leptin by the Friedman group in 1994 [67]. Because the vast majority of substances produced by the adipocyte are not necessarily cytokines, Trayhurn and Wood recommended the term adipokines instead. Therefore, adipokines are defined as any substance synthesized and secreted by the adipocytes [68]. Thus, it has become quite clear that adipose tissue is indeed an endocrine organ. In fact, it can be the largest organ in the body. This is physiologically and pathophysiologically important because the total amount of secreted adipokines are enormous and may affect the whole body economy, especially considering that every adipocyte is connected to the vascular network [69]. It is well known that dysregulation of the production and secretion of adipokines is involved in the development of metabolic and cardiovascular diseases. In metabolic syndrome, intra-abdominal visceral fat accumulation has been shown to play a key role in the development of a variety of metabolic and circulatory disorders through the dysregulation of adipokine secretion [70].
4.1. Perivascular Adipose Tissue and Vascular Function
The function of adipose tissue as an endocrine organ has important implications in the understanding of the pathophysiological relationships between excess body fat and hypertension. Almost all the systemic arteries are surrounded by a layer of perivascular adipose tissue (PVAT). In the majority of myographic studies, PVAT is removed on a routine basis. This is a custom based on the assumption that PVAT can prevent the diffusion of vasoactive substances. This is perhaps the reason that, despite the ubiquity of PVAT, very little is known about its function in vascular biology. Perivascular fat certainly has a modulator action on vascular contractility. This was described by Soltis and Cassis in a study published in 1991 [71]. This work has often been misinterpreted as the first postulator of a supposed prorelaxing role of PVAT. These researchers describe a decrease in the sensitivity to noradrenaline when aortic segments remain with PVAT. They demonstrate that this is due to the uptake and elimination of this catecholamine by adipose tissue. They postulate that the nerve endings within PVAT recapt and remove noradrenaline within the synaptic gap. This obviously results in a buffered effect of this neurotransmitter, but it is not postulated that PVAT releases any anticontractile factor.
In more recent years, several groups have dealt with the possible vasoactive role of PVAT. The group of González et al. has been especially interested in the vasoactive properties of the tunica adventitia, to which they attribute a role in the contractile ability of the responses modulated by the endothelium [72]. Later on, Gao et al. as well as Rey et al. claimed that PVAT promotes the vasoconstrictor response to electrical stimulation [73] and impaired endothelial function [74] via reactive oxygen species generated by NADPH oxidase. On the other hand stands the work pursued by Gollasch's group and initiated by Löhn et al. who claim to have found a diffusible factor derived from PVAT, which they called “adventitium-derived relaxing factor” or ADRF [75]. In a following paper, the “A” standed for “adipocyte” instead [76]. A relevant amount of literature has confirmed the existence of this anticontractile diffusible substance (see [77] for review). Still, there is no unanimity regarding the nature and mechanism of action of ADRF. For Verlohren and coworkers, it is independent from the endothelium [76], but not for Gao et al. [78]. What seems clear is that the vasodilatory effect of ADRF is mediated by the opening of different K+ channels on vascular smooth muscle cells [75, 76, 78–80]. Endocrine and vascular paracrine functions of a variety of adipokines are shown in Table 1. We shall focus on those with particular vasoactive actions, namely, leptin, adiponectin, TNF-α, prostaglandins, angiotensin II, and endothelin-1.
Table 1.
Endocrine and vascular paracrine functions of some adipokines.
| Adipokine | General effects | Vascular effects | References |
|---|---|---|---|
| Leptin | Satiating factor Physiological regulation of feeding behaviour through hypothalamic receptors Levels correlate with amount of body fat |
Endothelial dysfunction Endothelium-dependent and independent relaxation |
[67, 85, 160–165] |
|
| |||
| Resistin | Relates obesity to diabetes by inducing insulin resistance | Impairs endothelial function due to an increase in ET-1 production and a decrease in NO production | [166, 167] |
|
| |||
| Adiponectin | Levels inversely correlate with obesity | NO-dependent vasorelaxation mediated by K v channels | [91, 93, 94, 168] |
|
| |||
| Visfatin | Expression correlates with obesity degree Similar effects to insulin in cell culture |
NO-dependent vasorelaxation | [169–171] |
|
| |||
| TNFα | Links inflammation with obesity Increase in TNFα expression induces ROS production Reduces adiponectin production |
Endothelium-dependent and -independent vasodilatation Triggers ET-1 and Ang II-induced vasoconstriction Impairs endothelium-dependent vasodilatation due to increased ROS production or decreased NO production Less vasodilatory effect of PAT due to ROS production |
[94, 99, 172–178] |
|
| |||
| Interleukin-6 | Contributes to systemic inflammation and insulin resistance | Endothelium-independent vasodilatation Endothelial dysfunction due to an increase in ROS production and decreased NO production |
[94, 179–182] |
|
| |||
| Prostanoids | See vascular effects Hemostasis Numerous biological functions |
Vasoconstriction or vasodilatation depending on which prostanoid | [183, 184] |
|
| |||
| Angiotensin II | See vascular effects Na+ and water homeostasis Renal function |
Vasoconstriction | [185, 186] |
|
| |||
| Endothelin-1 | See vascular effects | Vasoconstriction | [187] |
|
| |||
| Reactive oxygen species | Numerous biological effects Ageing |
Vasoconstriction through Ca2+ sensitization Decrease in NO bioavailability |
[73, 188, 189] |
|
| |||
| Adventitial derived relaxing factor | See vascular effects | Vasorelaxation through opening different K+ channels | [75, 76, 78–80] |
4.2. Leptin
The discovery that the endothelium expresses the leptin receptor OB-Rb [81], converted endothelial cells, just like those of the hypothalamus, in a target for this hormone. The presence of leptin receptors in the vascular endothelium and not only in the central nervous system is important because it allows to find a link between leptin and altered vascular function in obesity [82]. Leptin is an NO-dependent vasodilator but also increases peripheral vascular resistance and sympathetic nerve activity [83]. The concentration of plasma leptin is correlated with adiposity, and hyperleptinemia is indeed considered an independent cardiovascular disease risk factor [84]. There are two theories that relate leptin's cardiovascular effects to obesity. One of them proposes that leptin is involved in the control of vascular tone simultaneously causing a neurogenic pressor action and an opposite depressor effect mediated by NO [85]. Another theory, based on experiments performed in coronary arterioles [86], proposes that, paradoxically, leptin causes itself NO-dependent vasodilation and, at the same time, its very presence impairs endothelium-dependent relaxations, that is, produces endothelial dysfunction. The problem with this interesting theory is that leptin-induced relaxation occurs at concentrations well above those found in very obese subjects. Physiological (lean) or pathophysiological concentrations (obese) of leptin have, however, little direct effect on vascular tone. Possibly, the most relevant aspect of this theory is that leptin concentrations actually existing in obese patients do elicit endothelial dysfunction [86].
4.3. Adiponectin
Adiponectin is the secretory protein produced in largest amounts by adipocytes and present in high and stable concentration in the plasma. In healthy subjects, adiponectin carries out its roles preventing the development of vascular changes and has been reported to be associated with lipid metabolism [87], glucose metabolism [88], and insulin resistance [89]. Unlike leptin, plasma adiponectin levels are negatively correlated with body mass index. This negative correlation is stronger between adiponectin levels and visceral adiposity than between the protein and subcutaneous adiposity [90]. Also, there is a close relationship between low concentrations of adiponectin in the blood, insulin resistance, and hyperinsulinemia. It has been suggested that the decrease in plasma adiponectin concentration contributes to the metabolic complications associated with obesity [91]. Adiponectin improves NO-dependent vasodilation by opening voltage-dependent potassium channels [92–94].
Some reports suggest that adiponectin plays an important role in insulin actions and hypoadiponectinemia may result in insulin resistance and diabetes mellitus. In fact, Lindsay et al. demonstrated that plasma levels of adiponectin were lower in Pima Indians, a unique cohort with high prevalence of obesity [95]. They also demonstrated that plasma levels of adiponectin are strongly correlated with insulin sensitivity evaluated by glucose disposal rate [96]. The study of the Pima Indian population demonstrates that adiponectin may play a crucial role in the development of diabetes mellitus and that high adiponectin levels should protect from the deterioration of glucose metabolism. Thus, hypoadiponectinemia could be a significant background of vascular changes and metabolic disorders, including insulin resistance and, possibly, a background for hypertension as well. Indeed, some studies show that hypertensive subjects have lower levels of plasma adiponectin [97].
4.4. Tumor Necrosis Factor-α (TNF-α)
Since Hotamisligil's group reported that adipose tissue expresses TNF-α, one of the candidate molecules inducing insulin resistance adipokines [98], this factor has been recognized as one of the most important adipokine. Adipocytes secrete TNF-α, and the expression of this factor is increased in the hypertrophied adipocytes of obese subjects. TNF-α is the molecule linking inflammation with obesity [99]. We will further discuss this adipokine in the diet-induced hypertension section.
4.5. Prostaglandins (Adipocyte Derived)
Prostaglandins, together with angiotensin II and endothelin-1, are the most vasoactive substances generated by adipocytes. Adipocytes produce prostaglandins in response to sympathetic stimulation. Lipolytic hormones, like adrenaline, are linked to the hypertensive status and obesity-associated hypertension. These hormones target membrane adipocyte β receptors and in turn activate hormone sensitive lipase. This stimulus induces lipolysis, release of fatty acids, and prostaglandins, especially PGE2 and PGI2, which are also fatty acids in origin. Antilipolytic stimuli, insulin, for example, reduce the release of prostaglandins [100] such as prostacyclin (PGI2). On the basis that insulin decreases the production of this strong vasodilator, Parker and coworkers suggested that hypertension associated with insulin resistance and hyperinsulinemia (i.e., metabolic syndrome) would be due partly caused by the lack of proper PGI2 release [69]. It appears that PGI2 production by the adipocytes results from the cooperation of adipocytes and vascular endothelial cells. Parker and coworkers proved that adipocytes are a source of the original fatty acid component of prostaglandins, arachidonic acid, that is converted into prostaglandins by the closely located vascular endothelial cells. Adipocytes provide arachidonic acid but lack the required cyclooxygenase which is provided by adjacent endothelial cells [69]. However, adipocytes do express cyclooxygenase [101], and according to Richelsen et al., adipocytes can synthesize prostaglandins, but still provide endothelial cells with adipocyte-derived arachidonic acid to further generate prostaglandins [100].
4.6. Angiotensin II (Adipocyte Derived)
The first to propose PVAT as a source of angiotensin II were our previously quoted Soltis and Cassis who suggested that adipocyte-derived angiotensin II would favor vasoconstriction [71]. This effect could be due to the fact that the angiotensin II action prevents PI3K activation, resulting in a loss of stimulation of NO synthesis by this route [102], as discussed in the section related to endothelin-1. Plasma renin activity and thus the production of angiotensin II are high in obese individuals [5, 19]. Three possible explanations have been proposed to explain this phenomenon: (1) obesity may raise renin secretion by increasing loop of Henle sodium chloride reabsorption and reduce sodium chloride delivery to the macula densa [19]; (2) obesity may stimulate renin secretion by activation of the sympathetic nervous system [19]. Finally, (3) the existence of a high renin activity in the hypertrophied adipocytes causing an increased angiotensin II release [103–106]. Today, we know that adipocytes possess the whole enzymatic machinery involved in the renin-angiotensin system [103] and, in fact, they do synthesize angiotensin II [105, 107]. Importantly, angiotensinogen gene expression is higher in intra-abdominal fat than in other fat depots or nonadipose tissues [108]. Indeed, increased production of angiotensinogen by intra-abdominal fat appears to explain the high circulating levels of this peptide observed in dietary obesity [104]. Closely related with the physiology of angiotensin II is aldosterone. The levels of this corticoid are elevated in some obese hypertensives, especially patients with visceral obesity [109]. Furthermore, it has been recently discovered that adipocytes also produce aldosterone (actually in response to angiotensin II) [106]. In this regard, the adipocyte may be considered a miniature renin-angiotensin-aldosterone system.
It is noteworthy that adipose cells also secrete mineralocorticoid-releasing factors with important effects on aldosterone release from adrenocortical cells [110]. These are called adipogensins or aldosterone-releasing factors (ARF) [111] but are not well characterized as yet. There is a lot of data that suggests a close relationship between an excess in released aldosterone and insulin resistance. Aldosterone promotes insulin resistance through mineralocorticoid receptors activation (independently of gene transcription) in a large number of tissues [112]. On the other hand, hyperinsulinaemia induces increase in aldosterone levels [113, 114] thus creating another positive feedback cycle between hyperaldosteronism and hyperinsulinemia, with important pathophysiological effects in subjects with insulin resistance and a potential mechanism for the development of complications in obese hypertensive patients.
4.7. Endothelin-1 (Adipocyte Derived)
As stated in previous lines, endothelin-1 is a vasoconstrictor protein normally produced by the endothelial cells but qualifies as adipokine as well [115]. Indeed, the levels of endothelin-1 increase in obesity and type II diabetes [116, 117]. In studies of experimental obesity, an increase in endothelin-1 gene and protein expression has been detected within the cardiovascular system [118]. Harmelen et al. found that obese adipose tissue releases 2.5 times more endothelin-1 than the adipose tissue of lean individuals. Furthermore, this ET-1 generates insulin resistance specifically in visceral, but not in subcutaneous, adipose tissue [119]. This links directly endothelin-1 with insulin resistance and obesity.
5. Animal Models of Metabolic Syndrome: Vascular Function
5.1. The Zucker Obese Rat
The Zucker rat is probably the most commonly used rat model for metabolic syndrome. In 1961, L. M. Zucker and T. F. Zucker discovered that an autosomal recessive mutation in the fatty gene (fa) resulted in obesity [120]. The homozygotes for the mutation (fa/fa) develop obesity because of a defective leptin receptor [121, 122]. Zucker rats develop insulin resistance in addition to obesity, but glycemia remains normal, and they do not develop diabetes [123]. In this aspect, the Zucker rat shares similarities with some of the obese subjects, those who are obese and insulin resistant but are not diabetic. However, the Zucker fatty rat does not mimic the cardiovascular, renal, and neurohumoral changes found in obese humans. For example, this rat has decreased plasma renin activity [124], whereas obese humans often reveal increased renin activity [5]. Also, increased sympathetic activity appears to play a significant role in causing hypertension in obese humans [125], but not in Zucker fatty rats [124]. In addition, conflicting results about whether obese Zucker rats are hypertensive or not compared with their lean controls have been repetitively reported [126]. In a carefully performed study by Hall's group, it was shown that obese Zucker rats suffer no more than 14 mmHg higher than the lean counterparts and that this depends in part on angiotensin II [124].
Regarding vascular responses, much work has been performed in aorta [127–131] and in resistance arteries [132–137] of Zucker rats. Endothelial function assessed in aortic preparations appears to be preserved, or even increased, in young Zucker obese rats compared to the lean rats [128–131]. Andrews et al. use the term endothelial hyperreactivity [130] to emphasize the superior endothelial function of Zucker obese rats [131]. For Auguet et al. the increased influence of endothelium in Zucker rats would be related to the absence of atherosclerosis (despite hypercholesterolemia) of these rats. As for resistance arteries, the majority of studies indicate impaired endothelial dysfunction [134–136] and impaired NO-dependent vasodilation [133, 137] in Zucker obese rat arterioles compared to the lean counterpart. By contrast, one study finds equal endothelial function [132].
5.2. The Spontaneously Hypertensive Obese (SHROB) Rat
The obese spontaneously hypertensive rat (SHROB), also known as Koletsky rat, is a rat strain of spontaneous hypertension breeding origin that suffers a nonsense mutation of the leptin receptor gene [138]. This animal was obtained by mating a female SHR of the Wistar-Kyoto strain with a normotensive Sprague-Dawley male. The resulting hybrid offspring was inbred and the obese rat appeared after several generations. The obesity mutation is a recessive trait, designated fa k, which is a nonsense mutation of the leptin receptor gene resulting in a premature stop codon in the leptin receptor extracellular domain. The SHROB ratcarries two fa k alleles; it is leptin resistant and has circulating leptin levels 30 times higher that the lean counterpart. This mutation makes SHROB rats unable to respond to leptin [139, 140]. This strain arose spontaneously in 1969 in Koletsky's laboratory in Case Western Reserve University School of Medicine (Ohio) [141]. The rat displays obesity, hypertension (although milder than that of their SHR ancestor), hyperinsulinaemia, hyperlipidaemia, and nephropathy, all superimposed on a hypertensive background. Thus, these rats exhibit all the symptoms of metabolic syndrome and are generally regarded as an adequate animal model of this disease [126].
Cardiovascular and renal function has been hardly explored in the SHROB rat. Still, it is known that SHROB rats develop a pronounced diabetic retinopathy. This makes them of special interest for the study of the microvascular complications associated with metabolic syndrome. Huang and coworkers noted that already at 3 months of age they displayed very mild microvascular alterations and did not develop diabetic retinopathy until 10 months of age. Interestingly, control lean SHROB rats also develop diabetic retinopathy [142].
The effect of diet on blood pressure changes has also been studied in these animals. Ernsberger and coworkers observed that drastic fluctuations in the supply of nutrients are not beneficial for blood pressure in these animals. They show that restrictive diet followed by feedback cycles produces blood pressure elevations caused by sympathetic activation and cardiac hypertrophy [143].
Regarding renal and cardiovascular function, it is known that specific binding sites for angiotensin II are decreased in SHROB rats with early glomerular sclerosis, suggesting that angiotensin receptors may be regulated by pathogenic processes in kidneys of these animals [144].
Recently, our group has characterized the macrovascular and microvascular function of this rat strain and the effects of a kind of antidiabetic drugs, glitazones, used in the handling of metabolic syndrome [145]. The SHROB rat clearly suffers macrovascular and most especially microvascular dysfunction (Figures 2(a) and 2(b)). Mesenteric resistance arteries of SHROB rats display a severely impaired endothelium-dependent relaxation due to a failure in the NO system and an abnormally high release of vasoconstrictive prostanoids. These rats also exhibit a dramatic loss in endothelium-independent relaxation, specifically to exogenous NO, suggesting a malfunction of guanylate cyclase. We also showed that drugs used for metabolic syndrome therapy, glitazones, have salutary effects on the endothelial dysfunction of these rats.
Figure 2.
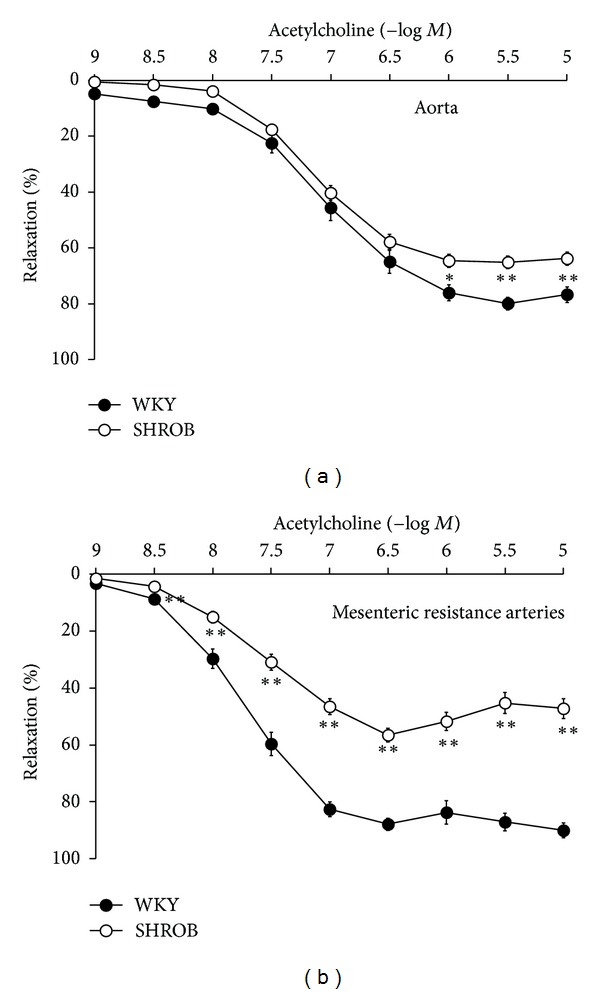
Endothelial function tested by means of acetylcholine responses in aorta (a) and resistance arteries (b) of normotensive (WKY) and metabolic syndrome rats (SHROB). Modified from Mendizábal et al. [145].
5.3. The JCR-LA-cp Corpulent Rat
The JCR-LA-cp corpulent rat is another rat model used to study metabolic syndrome. This rat is homozygous for the autosomal recessive cp gene (cp/cp) and is obese, hyperphagic, insulin resistant, hyperinsulinemic, and hypertriglyceridemic [146]. In addition, male JCR-LA-cp rats develop atherosclerosis and myocardial ischemia. Vascular responses and endothelial function were studied by O'Brien and coworkers [146] rendering similar results as for micro- versus macrovascular endothelial dysfunction as those of SHROB rats, although the latter displayed a more intense impairment of acetylcholine responses.
5.4. Diet-Induced Obesity
Stricto sensu, this model of obesity cannot be always categorized as an animal model of metabolic syndrome because dieting an animal with high fat chow rarely causes the complete cardiovascular and metabolic disease. In some cases, obesity-induced hypertension is achieved [147, 148], but this is not commonplace and most research papers do not report blood pressure values. Other metabolic syndrome symptoms are irregularly reported. For example, hyperinsulinemia or hyperglycemia is found in some studies [149, 150] but not in others [151, 152]. Dyslipemia takes place in some [149, 151] but not all the studies [152]. Hyperleptinemia seems to be common to all [150–152].
However, keeping in mind the enormous epidemiological dimension of overweight, obesity, and obesity-associated cardiovascular problems (i.e., cardiometabolic syndrome), much research and effort have been performed in these kind of rat or mouse models regardless of whether the animal develops or not a complete metabolic syndrome. Another factor in favor of diet-induced obesity animal models is that they are more human-like models, where the obesity is based on an excess intake of calories, whilst genetic models deficient in the leptin receptor or leptin synthesis are not representative of the human pathophysiology of obesity. Obesity in rodents can also be induced with the so-called cafeteria diet. In this model, animals have a choice of various energy-dense foods. The advantage to this approach is that the diet is palatable and the propensity to overeat is larger than that for the high-fat chow diet. Needless to say is that this is the most similar to the human dietary situation [153].
Regarding vascular function, the vast majority of studies have reported alterations. Endothelial function, assessed by acetylcholine responses, has been found altered in most cases. For example, in a cafeteria diet model reported by Naderali et al., a negative association between plasma lipid levels and reduction in acetylcholine-induced vasorelaxation was found [151]. Furthermore, a study in obese people showed that weight loss improves endothelial function together with various metabolic syndrome symptoms [154]. Hypercontractility, albeit less studied, has been reported in rats made hypertensive through the diet [147, 148]. In recent times, a large amount of studies have been focused on the effects that the local adiposity surrounding blood vessels (the so called PVAT) has on smooth muscle cell contractility and endothelial function [77]. Adipose tissue specifically located close to blood vessels exhibits a proinflammatory phenotype compared to other depots such as the subcutaneous one [155]. This phenotype is aggravated after a high fat feeding suggesting that PVAT is very sensitive to the effects of excess dietary fat [155]. In obese rats, including diet-induced obesity rats, it has been repetitively shown that PVAT causes endothelial dysfunction via proinflammatory cytokines such as TNFα [156] or monocyte chemotactic protein-1 [157] as well as through oxidative stress [148, 157]. Actually, Dobrian et al. report on a rat model of obesity-induced hypertension that this increase in vascular oxidative stress is associated with an increase in vascular NO production and NO synthase activity [148]. Furthermore, Jebelovszki et al. demonstrated that diet-induced obesity increases vascular smooth muscle sensitivity to NO through an activation of guanylate cyclase [149]. To have a whole picture of the biology of NO in obesity it is important to consider also that adipocytes can express NO synthase and that this expression is upregulated in obesity [158]. The adipocytic upregulation of NO synthase contrasts with the endothelial downregulation of this enzyme described by Ma et al. in diet-induced obesity rats in which this downregulation finely correlates with the vascular dysfunction they find in their own experiments [159] and, in general, in those of others [152–157]. This apparent contradiction can be explained as follows. In nonobese individuals, PVAT would have a vascular protective and beneficial role [152]. During the onset of obesity, several adaptive mechanisms within the vessel wall [149] and within PVAT itself [152] are activated. Regarding the latter, Gil-Ortega et al. have published interesting data showing the existence of an adaptive NO overproduction by PVAT during early diet-induced obesity and propose that, at some time point during obesity development, PVAT switches from a vascular protective influence to a deleterious one [152].
6. Future Directions
The pathophysiology of metabolic syndrome has become very complex. We have reviewed some of the pathophysiological aspects that affect vascular function: insulin, sympathetic system, endothelium, perivascular fat, and adipokines. The animal models in use have important limitations that need to be compensated with clinical studies. Translational research, in which animal studies are designed and carried out together with clinical investigation, is of special value. It is also highly important to merit basic science studies designed to unravel specific pathways, messengers, and intermediates of metabolic syndrome. While the era of endothelium and endothelium-derived substances has passed its summit, the age of perivascular adipocytes and adipokines is coming with a strong impulse.
Acknowledgments
This work was supported by Fondo de Investigaciones Sanitarias (ref. PI080473) and Consejería de Educación y Ciencia de Castilla-La Mancha (ref. PII1I09-0166-3114). Y. Mendizábal was supported by FISCAM (MOV 2007-JI/10).
References
- 1.Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37(12):1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 2.Yki-Järvinen H, Utriainen T. Insulin-induced vasodilatation: physiology or pharmacology? Diabetologia. 1998;41(4):369–379. doi: 10.1007/s001250050919. [DOI] [PubMed] [Google Scholar]
- 3.Garrison RJ, Kannel WB, Stokes J, III, Castelli WP. Incidence and precursors of hypertension in young adults: the Framingham offspring study. Preventive Medicine. 1987;16(2):235–251. doi: 10.1016/0091-7435(87)90087-9. [DOI] [PubMed] [Google Scholar]
- 4.Alexander J, Dustan HP, Sims EAH, Tarazi R. Report of the Hypertension Task Force. Washington, DC, USA: US Government Printing Office; 1979. (US Department of Health, Education, and Welfare Publication No. 70-1631 (NIH)). [Google Scholar]
- 5.Tuck ML, Sowers J, Dornfeld L. The effect of weight reduction on blood pressure, plasma renin activity, and plasma aldosterone levels in obese patients. The New England Journal of Medicine. 1981;304(16):930–933. doi: 10.1056/NEJM198104163041602. [DOI] [PubMed] [Google Scholar]
- 6.Reisin E, Abel R, Modan M, et al. Effect of weight loss without salt restriction on the reduction of blood pressure in overweight hypertensive patients. The New England Journal of Medicine. 1978;298(1):1–6. doi: 10.1056/NEJM197801052980101. [DOI] [PubMed] [Google Scholar]
- 7.Berchtold P, Joergens V, Finke C, Berger M. Epidemiology of obesity and hypertension. International Journal of Obesity. 1981;5(supplement 1):1–7. [PubMed] [Google Scholar]
- 8.Kylin E. Hypertonie and zuckerkrankheit. Zentralblatt für Innere Medizin. 1921;42:873–877. [Google Scholar]
- 9.Marañón G. Über hypertonie and zuckerkrankheit. Zentralblatt für Innere Medizin. 1922;43:169–176. [Google Scholar]
- 10.Kaplan NM. The deadly quartet. Upper-body obesity, glucose intolerance, hypertriglyceridemia, and hypertension. Archives of Internal Medicine. 1989;149(7):1514–1520. doi: 10.1001/archinte.149.7.1514. [DOI] [PubMed] [Google Scholar]
- 11.Zimmet P, Alberti KGMM, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414(6865):782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 12.Turtle JR. The economic burden of insulin resistance. International Journal of Clinical Practice. 2000;(113):23–28. [PubMed] [Google Scholar]
- 13.Uemura K, Pisa Z. Trends in cardiovascular disease mortality in industrialized countries since 1950. World Health Statistics Quarterly. 1988;41(3-4):155–178. [PubMed] [Google Scholar]
- 14.Critser G. Fat Land: How Americans Became the Fattest People in the World. Mariner Books; 2004. [Google Scholar]
- 15.Rowe JW, Young JB, Minaker KL, et al. Effect of insulin and glucose infusions on sympathetic nervous system activity in normal man. Diabetes. 1981;30(3):219–225. doi: 10.2337/diab.30.3.219. [DOI] [PubMed] [Google Scholar]
- 16.Lembo G, Napoli R, Capaldo B, et al. Abnormal sympathetic overactivity evoked by insulin in the skeletal muscle of patients with essential hypertension. The Journal of Clinical Investigation. 1992;90(1):24–29. doi: 10.1172/JCI115842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ter Maaten JC, Voorburg A, Heine RJ, Ter Wee PM, Donker AJM, Gans ROB. Renal handling of urate and sodium during acute physiological hyperinsulinaemia in healthy subjects. Clinical Science. 1997;92(1):51–58. doi: 10.1042/cs0920051. [DOI] [PubMed] [Google Scholar]
- 18.Vierhapper H. Effect of exogenous insulin on blood pressure regulation in healthy and diabetic subjects. Hypertension. 1985;7(6, part 2):II49–II53. doi: 10.1161/01.hyp.7.6_pt_2.ii49. [DOI] [PubMed] [Google Scholar]
- 19.Hall JE. Mechanisms of abnormal renal sodium handling in obesity hypertension. American Journal of Hypertension. 1997;10(5, part 2):49S–55S. [PubMed] [Google Scholar]
- 20.Carlyle M, Jones OB, Kuo JJ, Hall JE. Chronic cardiovascular and renal actions of leptin: role of adrenergic activity. Hypertension. 2002;39(2):496–501. doi: 10.1161/hy0202.104398. [DOI] [PubMed] [Google Scholar]
- 21.Eikelis N, Schlaich M, Aggarwal A, Kaye D, Esler M. Interactions between leptin and the human sympathetic nervous system. Hypertension. 2003;41(5):1072–1079. doi: 10.1161/01.HYP.0000066289.17754.49. [DOI] [PubMed] [Google Scholar]
- 22.Marsh AJ, Fontes MAP, Killinger S, Pawlak DB, Polson JW, Dampney RAL. Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension. 2003;42(4):488–493. doi: 10.1161/01.HYP.0000090097.22678.0A. [DOI] [PubMed] [Google Scholar]
- 23.Alvarez GE, Beske SD, Ballard TP, Davy KP. Sympathetic neural activation in visceral obesity. Circulation. 2002;106(20):2533–2536. doi: 10.1161/01.cir.0000041244.79165.25. [DOI] [PubMed] [Google Scholar]
- 24.Himsworth HP. Diabetes mellitus. its differentiation into insulin-sensitive and insulin-insensitive types. The Lancet. 1936;227(5864):127–130. [Google Scholar]
- 25.Himsworth HP, Kerr R. Insulin-sensitive and insulin-insensitive diabetes mellitus. Clinical Science. 1939;4:199–152. [Google Scholar]
- 26.Lebovitz HE. Insulin resistance: definition and consequences. Experimental and Clinical Endocrinology and Diabetes. 2001;109(supplement 2):S135–S148. doi: 10.1055/s-2001-18576. [DOI] [PubMed] [Google Scholar]
- 27.Arcaro G, Zamboni M, Rossi L, et al. Body fat distribution predicts the degree of endothelial dysfunction in uncomplicated obesity. International Journal of Obesity and Related Metabolic Disorders. 1999;23(9):936–942. doi: 10.1038/sj.ijo.0801022. [DOI] [PubMed] [Google Scholar]
- 28.Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction: implications for the syndrome of insulin resistance. The Journal of Clinical Investigation. 1996;97(11):2601–2610. doi: 10.1172/JCI118709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarkun I, Arslan BC, Cantürk Z, Türemen E, Şahin T, Duman C. Endothelial dysfunction in young women with polycystic ovary syndrome: relationship with insulin resistance and low-grade chronic inflammation. Journal of Clinical Endocrinology and Metabolism. 2004;89(11):5592–5596. doi: 10.1210/jc.2004-0751. [DOI] [PubMed] [Google Scholar]
- 30.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocrine Reviews. 2007;28(5):463–491. doi: 10.1210/er.2007-0006. [DOI] [PubMed] [Google Scholar]
- 31.King GL, Johnson SM. Receptor-mediated transport of insulin across endothelial cells. Science. 1985;227(4694):1583–1586. doi: 10.1126/science.3883490. [DOI] [PubMed] [Google Scholar]
- 32.Scherrer U, Randin D, Vollenweider P, Vollenweider L, Nicod P. Nitric oxide release accounts for insulin’s vascular effects in humans. The Journal of Clinical Investigation. 1994;94(6):2511–2515. doi: 10.1172/JCI117621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. The Journal of Clinical Investigation. 1994;94(3):1172–1179. doi: 10.1172/JCI117433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Govers R, Rabelink TJ. Cellular regulation of endothelial nitric oxide synthase. American Journal of Physiology. 2001;280(2):F193–F206. doi: 10.1152/ajprenal.2001.280.2.F193. [DOI] [PubMed] [Google Scholar]
- 35.Kashyap SR, Roman LJ, Lamont J, et al. Insulin resistance is associated with impaired nitric oxide synthase activity in skeletal muscle of type 2 diabetic subjects. Journal of Clinical Endocrinology and Metabolism. 2005;90(2):1100–1105. doi: 10.1210/jc.2004-0745. [DOI] [PubMed] [Google Scholar]
- 36.Calver A, Collier J, Vallance P. Inhibition and stimulation of nitric oxide synthesis in the human forearm arterial bed of patients with insulin-dependent diabetes. The Journal of Clinical Investigation. 1992;90(6):2548–2554. doi: 10.1172/JCI116149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Jongh RT, Serné EH, Ijzerman RG, De Vries G, Stehouwer CDA. Impaired microvascular function in obesity: implications for obesity-associated microangiopathy, hypertension, and insulin resistance. Circulation. 2004;109(21):2529–2535. doi: 10.1161/01.CIR.0000129772.26647.6F. [DOI] [PubMed] [Google Scholar]
- 38.Gudbjörnsdottir S, Elam M, Sellgren J, Anderson EA. Insulin increases forearm vascular resistance in obese, insulin-resistant hypertensives. Journal of Hypertension. 1996;14(1):91–97. [PubMed] [Google Scholar]
- 39.Schroeder CA, Chen YL, Messina EJ. Inhibition of NO synthesis or endothelium removal reveals a vasoconstrictor effect of insulin on isolated arterioles. American Journal of Physiology. 1999;276(3):H815–H820. doi: 10.1152/ajpheart.1999.276.3.H815. [DOI] [PubMed] [Google Scholar]
- 40.Shankar RR, Wu Y, Shen HQ, Zhu JS, Baron AD. Mice with gene disruption of both endothelial and neuronal nitric oxide synthase exhibit insulin resistance. Diabetes. 2000;49(5):684–687. doi: 10.2337/diabetes.49.5.684. [DOI] [PubMed] [Google Scholar]
- 41.Nava E, Llorens S, Mendizabal Y. Vascular function in diabetes mellitus. In: Holcroft R, editor. Treatment Strategies-Diabetes. Vol. 2. Cambridge Research Centre; 2010. pp. 209–225. [Google Scholar]
- 42.Montagnani M, Chen H, Barr VA, Quon MJ. Insulin-stimulated activation of eNOS is Independent of Ca2+ but requires phosphorylation by Akt at Ser1179 . Journal of Biological Chemistry. 2001;276(32):30392–30398. doi: 10.1074/jbc.M103702200. [DOI] [PubMed] [Google Scholar]
- 43.Oliver FJ, De la Rubia G, Feener EP, et al. Stimulation of endothelin-1 gene expression by insulin in endothelial cells. Journal of Biological Chemistry. 1991;266(34):23251–23256. [PubMed] [Google Scholar]
- 44.Wolpert HA, Steen SN, Istfan NW, Simonson DC. Insulin modulates circulating endothelin-1 levels in humans. Metabolism. 1993;42(8):1027–1030. doi: 10.1016/0026-0495(93)90018-j. [DOI] [PubMed] [Google Scholar]
- 45.Ferri C, Pittoni V, Piccoli A, et al. Insulin stimulates endothelin-1 secretion from human endothelial cells and modulates its circulating levels in vivo. Journal of Clinical Endocrinology and Metabolism. 1995;80(3):829–835. doi: 10.1210/jcem.80.3.7883838. [DOI] [PubMed] [Google Scholar]
- 46.Cardillo C, Nambi SS, Kilcoyne CM, et al. Insulin stimulates both endothelin and nitric oxide activity in the human forearm. Circulation. 1999;100(8):820–825. doi: 10.1161/01.cir.100.8.820. [DOI] [PubMed] [Google Scholar]
- 47.Eringa EC, Stehouwer CDA, Merlijn T, Westerhof N, Sipkema P. Physiological concentrations of insulin induce endothelin-mediated vasoconstriction during inhibition of NOS or PI3-kinase in skeletal muscle arterioles. Cardiovascular Research. 2002;56(3):464–471. doi: 10.1016/s0008-6363(02)00593-x. [DOI] [PubMed] [Google Scholar]
- 48.Verma S, Yao L, Stewart DJ, Dumont AS, Anderson TJ, McNeill JH. Endothelin antagonism uncovers insulin-mediated vasorelaxation in vitro and in vivo. Hypertension. 2001;37(2):328–333. doi: 10.1161/01.hyp.37.2.328. [DOI] [PubMed] [Google Scholar]
- 49.Tesauro M, Iantorno M, Schinzari F, Cardillo C. Vascular effects of insulin and their relation to endothelial dysfunction, insulin resistance and hypertension. Current Hypertension Reviews. 2009;5(4):251–261. [Google Scholar]
- 50.Taylor PD, Poston L. The effect of hyperglycaemia on function of rat isolated mesenteric resistance artery. British Journal of Pharmacology. 1994;113(3):801–808. doi: 10.1111/j.1476-5381.1994.tb17064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bohlen HG, Lash JM. Topical hyperglycemia rapidly suppresses EDRF-mediated vasodilation of normal rat arterioles. American Journal of Physiology. 1993;265(1, part 2):H219–H225. doi: 10.1152/ajpheart.1993.265.1.H219. [DOI] [PubMed] [Google Scholar]
- 52.Taylor PD, McCarthy AL, Thomas CR, Poston L. Endothelium-dependent relaxation and noradrenaline sensitivity in mesenteric resistance arteries of streptozotocin-induced diabetic rats. British Journal of Pharmacology. 1992;107(2):393–399. doi: 10.1111/j.1476-5381.1992.tb12757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Akbari CM, Saouaf R, Barnhill DF, Newman PA, LoGerfo FW, Veves A. Endothelium-dependent vasodilatation is impaired in both microcirculation and macrocirculation during acute hyperglycemia. Journal of Vascular Surgery. 1998;28(4):687–694. doi: 10.1016/s0741-5214(98)70095-3. [DOI] [PubMed] [Google Scholar]
- 54.Sobrevia L, Nadal A, Yudilevich DL, Mann GE. Activation of L-arginine transport (system y+) and nitric oxide synthase by elevated glucose and insulin in human endothelial cells. Journal of Physiology. 1996;490(3):775–781. doi: 10.1113/jphysiol.1996.sp021185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Renaudin C, Michoud E, Rapin JR, Lagarde M, Wiernsperger N. Hyperglycaemia modifies the reaction of microvessels to insulin in rat skeletal muscle. Diabetologia. 1998;41(1):26–33. doi: 10.1007/s001250050862. [DOI] [PubMed] [Google Scholar]
- 56.Modan M, Halkin H, Almog S. Hyperinsulinemia. A link between hypertension obesity and glucose intolerance. The Journal of Clinical Investigation. 1985;75(3):809–817. doi: 10.1172/JCI111776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferrannini E, Buzzigoli G, Bonadonna R. Insulin resistance in essential hypertension. The New England Journal of Medicine. 1987;317(6):350–357. doi: 10.1056/NEJM198708063170605. [DOI] [PubMed] [Google Scholar]
- 58.Landin K, Tengborn L, Smith U. Treating insulin resistance inhypertension with metformin reduces both blood pressure and metabolic risk factors. Journal of Internal Medicine. 1991;229(2):181–187. doi: 10.1111/j.1365-2796.1991.tb00328.x. [DOI] [PubMed] [Google Scholar]
- 59.Ogihara T, Rakugi H, Ikegami H, Mikami H, Masuo K. Enhancement of insulin sensitivity by troglitazone lowers blood pressure in diabetic hypertensives. American Journal of Hypertension. 1995;8(3):316–320. doi: 10.1016/0895-7061(95)96214-5. [DOI] [PubMed] [Google Scholar]
- 60.Martens FMAC, Visseren FLJ, Lemay J, De Koning EJP, Rabelink TJ. Metabolic and additional vascular effects of thiazolidinediones. Drugs. 2002;62(10):1463–1480. doi: 10.2165/00003495-200262100-00004. [DOI] [PubMed] [Google Scholar]
- 61.Sanchez RA, Masnatta LD, Pesiney C, Fischer P, Ramirez AJ. Telmisartan improves insulin resistance in high renin nonmodulating salt-sensitive hypertensives. Journal of Hypertension. 2008;26(12):2393–2398. doi: 10.1097/HJH.0b013e328312677e. [DOI] [PubMed] [Google Scholar]
- 62.Umeda M, Kanda T, Murakami M. Effects of angiotensin II receptor antagonists on insulin resistance syndrome and leptin in sucrose-fed spontaneously hypertensive rats. Hypertension Research. 2003;26(6):485–492. doi: 10.1291/hypres.26.485. [DOI] [PubMed] [Google Scholar]
- 63.Hall JE, Summers RL, Brands MW, Keen H, Alonso-Galicia M. Resistance to metabolic actions of insulin and its role in hypertension. American Journal of Hypertension. 1994;7(8):772–788. doi: 10.1093/ajh/7.8.772. [DOI] [PubMed] [Google Scholar]
- 64.Cook KS, Min HY, Johnson D, et al. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science. 1987;237(4813):402–405. doi: 10.1126/science.3299705. [DOI] [PubMed] [Google Scholar]
- 65.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 66.Funahashi T, Nakamura T, Shimomura I, et al. Role of adipocytokines on the pathogenesis of atherosclerosis in visceral obesity. Internal Medicine. 1999;38(2):202–206. doi: 10.2169/internalmedicine.38.202. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 68.Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. British Journal of Nutrition. 2004;92(3):347–355. doi: 10.1079/bjn20041213. [DOI] [PubMed] [Google Scholar]
- 69.Parker J, Lane J, Axelrod L. Cooperation of adipocytes and endothelial cells required for catecholamine stimulation of PGI2 production by rat adipose tissue. Diabetes. 1989;38(9):1123–1132. doi: 10.2337/diab.38.9.1123. [DOI] [PubMed] [Google Scholar]
- 70.Matsuzawa Y. The metabolic syndrome and adipocytokines. FEBS Letters. 2006;580(12):2917–2921. doi: 10.1016/j.febslet.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 71.Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clinical and Experimental Hypertension A. 1991;13(2):277–296. doi: 10.3109/10641969109042063. [DOI] [PubMed] [Google Scholar]
- 72.González MC, Arribas SM, Molero F, Fernández-Alfonso MS. Effect of removal of adventitia on vascular smooth muscle contraction and relaxation. American Journal of Physiology. 2001;280(6):H2876–H2881. doi: 10.1152/ajpheart.2001.280.6.H2876. [DOI] [PubMed] [Google Scholar]
- 73.Gao YJ, Takemori K, Su LY, et al. Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovascular Research. 2006;71(2):363–373. doi: 10.1016/j.cardiores.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 74.Rey FE, Li XC, Carretero OA, Garvin JL, Pagano PJ. Perivascular superoxide anion contributes to impairment of endothelium-dependent relaxation role of gp91phox. Circulation. 2002;106(19):2497–2502. doi: 10.1161/01.cir.0000038108.71560.70. [DOI] [PubMed] [Google Scholar]
- 75.Löhn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM. Periadventitial fat releases a vascular relaxing factor. The FASEB Journal. 2002;16(9):1057–1063. doi: 10.1096/fj.02-0024com. [DOI] [PubMed] [Google Scholar]
- 76.Verlohren S, Dubrovska G, Tsang SY, et al. Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension. 2004;44(3):271–276. doi: 10.1161/01.HYP.0000140058.28994.ec. [DOI] [PubMed] [Google Scholar]
- 77.Maenhaut N, Van deVoorde J. Regulation of vascular tone by adipocytes. BMC Medicine. 2011;9, article 25 doi: 10.1186/1741-7015-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gao YJ, Lu C, Su LY, Sharma AM, Lee RMKW. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. British Journal of Pharmacology. 2007;151(3):323–331. doi: 10.1038/sj.bjp.0707228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao YJ, Zeng ZH, Teoh K, et al. Perivascular adipose tissue modulates vascular function in the human internal thoracic artery. Journal of Thoracic and Cardiovascular Surgery. 2005;130(4):1130–1136. doi: 10.1016/j.jtcvs.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 80.Schleifenbaum J, Köhn C, Voblova N, et al. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. Journal of Hypertension. 2010;28(9):1875–1882. doi: 10.1097/HJH.0b013e32833c20d5. [DOI] [PubMed] [Google Scholar]
- 81.Sierra-Honigmann MR, Nath AK, Murakami C, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281(5383):1683–1686. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- 82.Winters B, Mo Z, Brooks-Asplund E, et al. Reduction of obesity, as induced by leptin, reverses endothelial dysfunction in obese (Lep(ob)) mice. Journal of Applied Physiology. 2000;89(6):2382–2390. doi: 10.1152/jappl.2000.89.6.2382. [DOI] [PubMed] [Google Scholar]
- 83.Shirasaka T, Takasaki M, Kannan H. Cardiovascular effects of leptin and orexins. American Journal of Physiology. 2003;284(3):R639–R651. doi: 10.1152/ajpregu.00359.2002. [DOI] [PubMed] [Google Scholar]
- 84.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. The New England Journal of Medicine. 1996;334(5):292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 85.Frühbeck G. Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes. 1999;48(4):903–908. doi: 10.2337/diabetes.48.4.903. [DOI] [PubMed] [Google Scholar]
- 86.Knudson JD, Dincer UD, Zhang C, et al. Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction. American Journal of Physiology. 2005;289(1):H48–H56. doi: 10.1152/ajpheart.01159.2004. [DOI] [PubMed] [Google Scholar]
- 87.Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nature Medicine. 2001;7(8):941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 88.Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nature Medicine. 2002;8(11):1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 89.Kondo H, Shimomura L, Matsukawa Y, et al. Association of adiponectin mutation with type 2 diabetes: a candidate gene for the insulin resistance syndrome. Diabetes. 2002;51(7):2325–2328. doi: 10.2337/diabetes.51.7.2325. [DOI] [PubMed] [Google Scholar]
- 90.Takahashi M, Funahashi T, Shimomura I, Miyaoka K, Matsuzawa Y. Plasma leptin levels and body fat distribution. Hormone and Metabolic Research. 1996;28(12):751–752. doi: 10.1055/s-2007-979893. [DOI] [PubMed] [Google Scholar]
- 91.Asayama K, Hayashibe H, Dobashi K, et al. Decrease in serum adiponectin level due to obesity and visceral fat accumulation in children. Obesity Research. 2003;11(9):1072–1079. doi: 10.1038/oby.2003.147. [DOI] [PubMed] [Google Scholar]
- 92.Xi W, Satoh H, Kase H, Suzuki K, Hattori Y. Stimulated HSP90 binding to eNOS and activation of the PI3-Akt pathway contribute to globular adiponectin-induced NO production: vasorelaxation in response to globular adiponectin. Biochemical and Biophysical Research Communications. 2005;332(1):200–205. doi: 10.1016/j.bbrc.2005.04.111. [DOI] [PubMed] [Google Scholar]
- 93.Fésüs G, Dubrovska G, Gorzelniak K, et al. Adiponectin is a novel humoral vasodilator. Cardiovascular Research. 2007;75(4):719–727. doi: 10.1016/j.cardiores.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 94.Greenstein AS, Khavandi K, Withers SB, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119(12):1661–1670. doi: 10.1161/CIRCULATIONAHA.108.821181. [DOI] [PubMed] [Google Scholar]
- 95.Lindsay RS, Funahashi T, Hanson RL, et al. Adiponectin and development of type 2 diabetes in the Pima Indian population. The Lancet. 2002;360(9326):57–58. doi: 10.1016/S0140-6736(02)09335-2. [DOI] [PubMed] [Google Scholar]
- 96.Stefan N, Vozarova B, Funahashi T, et al. Plasma adiponectin concentration is associated with skeletal muscle insulin receptor tyrosine phosphorylation, and low plasma concentration precedes a decrease in whole-body insulin sensitivity in humans. Diabetes. 2002;51(6):1884–1888. doi: 10.2337/diabetes.51.6.1884. [DOI] [PubMed] [Google Scholar]
- 97.Mallamaci F, Zoccali C, Cuzzola F, et al. Adiponectin in essential hypertension. Journal of Nephrology. 2002;15(5):507–511. [PubMed] [Google Scholar]
- 98.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF- α function. Nature. 1997;389(6651):610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 99.Bastard JP, Maachi M, Lagathu C, et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. European Cytokine Network. 2006;17(1):4–12. [PubMed] [Google Scholar]
- 100.Richelsen B, Borglum JD, Sorensen SS. Biosynthetic capacity and regulatory aspects of prostaglandin E2 formation in adipocytes. Molecular and Cellular Endocrinology. 1992;85(1-2):73–81. doi: 10.1016/0303-7207(92)90126-q. [DOI] [PubMed] [Google Scholar]
- 101.Fain JN, Leffler CW, Cowan GSM, Jr., Buffington C, Pouncey L, Bahouth SW. Stimulation of leptin release by arachidonic acid and prostaglandin E2 in adipose tissue from obese humans. Metabolism. 2001;50(8):921–928. doi: 10.1053/meta.2001.24927. [DOI] [PubMed] [Google Scholar]
- 102.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113(15):1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 103.Engeli S, Schling P, Gorzelniak K, et al. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? International Journal of Biochemistry and Cell Biology. 2003;35(6):807–825. doi: 10.1016/s1357-2725(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 104.Boustany CM, Bharadwaj K, Daugherty A, Brown DR, Randall DC, Cassis LA. Activation of the systemic and adipose renin-angiotensin system in rats with diet-induced obesity and hypertension. American Journal of Physiology. 2004;287(4):R943–R949. doi: 10.1152/ajpregu.00265.2004. [DOI] [PubMed] [Google Scholar]
- 105.Gálvez-Prieto B, Bolbrinker J, Stucchi P, et al. Comparative expression analysis of the renin—angiotensin system components between white and brown perivascular adipose tissue. Journal of Endocrinology. 2008;197(1):55–64. doi: 10.1677/JOE-07-0284. [DOI] [PubMed] [Google Scholar]
- 106.Briones AM, Nguyen Dinh Cat A, Callera GE, et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways. Hypertension. 2012;59(5):1069–1078. doi: 10.1161/HYPERTENSIONAHA.111.190223. [DOI] [PubMed] [Google Scholar]
- 107.Frederich RC, Jr., Kahn BB, Peach MJ, Flier JS. Tissue-specific nutritional regulation of angiotensinogen in adipose tissue. Hypertension. 1992;19(4):339–344. doi: 10.1161/01.hyp.19.4.339. [DOI] [PubMed] [Google Scholar]
- 108.Rahmouni K, Mark AL, Haynes WG, Sigmund CD. Adipose depot-specific modulation of angiotensinogen gene expression in diet-induced obesity. American Journal of Physiology. 2004;286(6):E891–E895. doi: 10.1152/ajpendo.00551.2003. [DOI] [PubMed] [Google Scholar]
- 109.Goodfriend TL, Calhoun DA. Resistant hypertension, obesity, sleep apnea, and aldosterone: theory and therapy. Hypertension. 2004;43(3):518–524. doi: 10.1161/01.HYP.0000116223.97436.e5. [DOI] [PubMed] [Google Scholar]
- 110.Ehrhart-Bornstein M, Lamounier-Zepter V, Schraven A, et al. Human adipocytes secrete mineralocorticoid-releasing factors. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(2):14211–14216. doi: 10.1073/pnas.2336140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Krug AW, Ehrhart-Bornstein M. Newly discovered endocrine functions of white adipose tissue: possible relevance in obesity-related diseases. Cellular and Molecular Life Sciences. 2005;62(12):1359–1362. doi: 10.1007/s00018-005-4555-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Connell JMC, Davies E. The new biology of aldosterone. Journal of Endocrinology. 2005;186(1):1–20. doi: 10.1677/joe.1.06017. [DOI] [PubMed] [Google Scholar]
- 113.Rocchini AP, Moorehead C, DeRemer S, Goodfriend TL, Ball DL. Hyperinsulinemia and the aldosterone and pressor responses to angiotensin II. Hypertension. 1990;15(6, part 2):861–866. doi: 10.1161/01.hyp.15.6.861. [DOI] [PubMed] [Google Scholar]
- 114.Petrasek D, Jensen G, Tuck M, Stern N. In vitro effects of insulin on aldosterone production in rat zona glomerulosa cells. Life Sciences. 1992;50(23):1781–1787. doi: 10.1016/0024-3205(92)90062-t. [DOI] [PubMed] [Google Scholar]
- 115.Fain JN, Tagele BM, Cheema P, Madan AK, Tichansky DS. Release of 12 adipokines by adipose tissue, nonfat cells, and fat cells from obese women. Obesity. 2010;18(5):890–896. doi: 10.1038/oby.2009.335. [DOI] [PubMed] [Google Scholar]
- 116.Morise T, Takeuchi Y, Kawano M, Koni I, Takeda R. Increased plasma levels of immunoreactive endothelin and von Willebrand factor in NIDDM patients. Diabetes Care. 1995;18(1):87–89. doi: 10.2337/diacare.18.1.87. [DOI] [PubMed] [Google Scholar]
- 117.Da Silva AA, Kuo JJ, Tallam LS, Hall JE. Role of endothelin-1 in blood pressure regulation in a rat model of visceral obesity and hypertension. Hypertension. 2004;43(2):383–387. doi: 10.1161/01.HYP.0000111139.94378.74. [DOI] [PubMed] [Google Scholar]
- 118.Barton M, Carmona R, Ortmann J, Krieger JE, Traupe T. Obesity-associated activation of angiotensin and endothelin in the cardiovascular system. International Journal of Biochemistry and Cell Biology. 2003;35(6):826–837. doi: 10.1016/s1357-2725(02)00307-2. [DOI] [PubMed] [Google Scholar]
- 119.Van Harmelen V, Eriksson A, Åström G, et al. Vascular peptide endothelin-1 links fat accumulation with alterations of visceral adipocyte lipolysis. Diabetes. 2008;57(2):378–386. doi: 10.2337/db07-0893. [DOI] [PubMed] [Google Scholar]
- 120.Zucker LM, Zucker TF. Fatty, a new mutation in the rat. Journal of Heredity. 1961;52(6):275–278. [Google Scholar]
- 121.Chua SC, Jr., Chung WK, Wu-Peng XS, et al. Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science. 1996;271(5251):994–996. doi: 10.1126/science.271.5251.994. [DOI] [PubMed] [Google Scholar]
- 122.Phillips MS, Liu Q, Hammond HA, et al. Leptin receptor missense mutation in the fatty Zucker rat. Nature Genetics. 1996;13(1):18–19. doi: 10.1038/ng0596-18. [DOI] [PubMed] [Google Scholar]
- 123.Bray GA. The Zucker fatty rat: a review. Federation Proceedings. 1977;36(2):148–153. [PubMed] [Google Scholar]
- 124.Alonso-Galicia M, Brands MW, Zappe DH, Hall JE. Hypertension in obese Zucker rats: role of angiotensin II and adrenergic activity. Hypertension. 1996;28(6):1047–1054. doi: 10.1161/01.hyp.28.6.1047. [DOI] [PubMed] [Google Scholar]
- 125.Landsberg L, Krieger DR. Obesity, metabolism, and the sympathetic nervous system. American Journal of Hypertension. 1989;2(3, part 2):125S–132S. doi: 10.1093/ajh/2.3.125s. [DOI] [PubMed] [Google Scholar]
- 126.Aleixandre A, Miguel M. Experimental rat models to study the metabolic syndrome. British Journal of Nutrition. 2009;102(9):1246–1253. doi: 10.1017/S0007114509990729. [DOI] [PubMed] [Google Scholar]
- 127.Auguet M, Delaflotte S, Braquet P. Increased influence of endothelium in obese zucker rat aorta. Journal of Pharmacy and Pharmacology. 1989;41(12):861–864. doi: 10.1111/j.2042-7158.1989.tb06389.x. [DOI] [PubMed] [Google Scholar]
- 128.Cox RH, Kikta DC. Age-related changes in thoracic aorta of obese Zucker rats. American Journal of Physiology. 1992;262(5, part 2):H1548–H1556. doi: 10.1152/ajpheart.1992.262.5.H1548. [DOI] [PubMed] [Google Scholar]
- 129.Sexl V, Mancusi G, Raberger G, Schutz W. Age-related changes in vascular reactivity in genetically diabetic rats. Pharmacology. 1995;50(4):238–246. doi: 10.1159/000139288. [DOI] [PubMed] [Google Scholar]
- 130.Andrews TJ, Laicht DW, Änggård EE, Carrier MJ. Investigation of endothelial hyperreactivity in the obese Zucker rat in-situ: reversal by vitamin E. Journal of Pharmacy and Pharmacology. 2000;52(1):83–86. doi: 10.1211/0022357001773544. [DOI] [PubMed] [Google Scholar]
- 131.Laight DW, Änggård EE, Carrier MJ. Investigation of basal endothelial function in the obese Zucker rat in vitro. General Pharmacology. 2000;35(6):303–309. doi: 10.1016/s0306-3623(01)00120-3. [DOI] [PubMed] [Google Scholar]
- 132.Bohlen HG, Lash JM. Endothelial-dependent vasodilation is preserved in non-insulin-dependent Zucker fatty diabetic rats. American Journal of Physiology. 1995;268(6, part 2):H2366–H2374. doi: 10.1152/ajpheart.1995.268.6.H2366. [DOI] [PubMed] [Google Scholar]
- 133.Frisbee JC, Stepp DW. Impaired NO-dependent dilation of skeletal muscle arterioles in hypertensive diabetic obese Zucker rats. American Journal of Physiology. 2001;281(3):H1304–H1311. doi: 10.1152/ajpheart.2001.281.3.H1304. [DOI] [PubMed] [Google Scholar]
- 134.Xiang L, Naik JS, Hodnett BL, Hester RL. Altered arachidonic acid metabolism impairs functional vasodilation in metabolic syndrome. American Journal of Physiology. 2006;290(1):R134–R138. doi: 10.1152/ajpregu.00295.2005. [DOI] [PubMed] [Google Scholar]
- 135.Xiang L, Dearman J, Abram SR, Carter C, Hester RL. Insulin resistance and impaired functional vasodilation in obese Zucker rats. American Journal of Physiology. 2008;294(4):H1658–H1666. doi: 10.1152/ajpheart.01206.2007. [DOI] [PubMed] [Google Scholar]
- 136.Goodwill AG, James ME, Frisbee JC. Increased vascular thromboxane generation impairs dilation of skeletal muscle arterioles of obese Zucker rats with reduced oxygen tension. American Journal of Physiology. 2008;295(4):H1522–H1528. doi: 10.1152/ajpheart.00596.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Frisbee JC. Reduced nitric oxide bioavailability contributes to skeletal muscle microvessel rarefaction in the metabolic syndrome. American Journal of Physiology. 2005;289(2):R307–R316. doi: 10.1152/ajpregu.00114.2005. [DOI] [PubMed] [Google Scholar]
- 138.Ernsberger P, Koletsky RJ, Friedman JE. Molecular pathology in the obese spontaneous hypertensive Koletsky rat: a model of Syndrome X. Annals of the New York Academy of Sciences. 1999;892:272–288. doi: 10.1111/j.1749-6632.1999.tb07801.x. [DOI] [PubMed] [Google Scholar]
- 139.Takaya K, Ogawa Y, Hiraoka J, et al. Nonsense mutation of leptin receptor in the obese spontaneously hypertensive Koletsky rat. Nature Genetics. 1996;14(2):130–131. doi: 10.1038/ng1096-130. [DOI] [PubMed] [Google Scholar]
- 140.Hiraoka J, Hosoda K, Ogawa Y, et al. Augmentation of obese (ob) gene expression and leptin secretion in obese spontaneously hypertensive rats (Obese SHR or Koletsky rats) Biochemical and Biophysical Research Communications. 1997;231(3):582–585. doi: 10.1006/bbrc.1997.6145. [DOI] [PubMed] [Google Scholar]
- 141.Koletsky S. Obese spontaneously hypertensive rats: a model for study of atherosclerosis. Experimental and Molecular Pathology. 1973;19(1):53–60. doi: 10.1016/0014-4800(73)90040-3. [DOI] [PubMed] [Google Scholar]
- 142.Huang SS, Khosrof SA, Koletsky RJ, Benetz BA, Ernsberger P. Characterization of retinal vascular abnormalities in lean and obese spontaneously hypertensive rats. Clinical and Experimental Pharmacology and Physiology. 1995;22(1):S129–S131. doi: 10.1111/j.1440-1681.1995.tb02850.x. [DOI] [PubMed] [Google Scholar]
- 143.Ernsberger P, Koletsky RJ, Baskin JS, Foley M. Refeeding hypertension in obese spontaneously hypertensive rats. Hypertension. 1994;24(6):699–705. doi: 10.1161/01.hyp.24.6.699. [DOI] [PubMed] [Google Scholar]
- 144.Ernsberger P, Koletsky RJ, Collins LA, Douglas JG. Renal angiotensin receptor mapping in obese spontaneously hypertensive rats. Hypertension. 1993;21(6, part 2):1039–1045. doi: 10.1161/01.hyp.21.6.1039. [DOI] [PubMed] [Google Scholar]
- 145.Mendizábal Y, Llorens S, Nava E. Reactivity of the aorta and mesenteric resistance arteries from the obese spontaneously hypertensive rat: effects of glitazones. American Journal of Physiology. 2011;301(4):H1319–H1330. doi: 10.1152/ajpheart.01280.2010. [DOI] [PubMed] [Google Scholar]
- 146.O’Brien SF, McKendrick JD, Radomski MW, Davidge ST, Russell JC. Vascular wall reactivity in conductance and resistance arteries: differential effects of insulin resistance. Canadian Journal of Physiology and Pharmacology. 1998;76(1):72–76. [PubMed] [Google Scholar]
- 147.Boustany-Kari CM, Gong M, Akers WS, Guo Z, Cassis LA. Enhanced vascular contractility and diminished coronary artery flow in rats made hypertensive from diet-induced obesity. International Journal of Obesity. 2007;31(11):1652–1659. doi: 10.1038/sj.ijo.0803426. [DOI] [PubMed] [Google Scholar]
- 148.Dobrian AD, Davies MJ, Schriver SD, Lauterio TJ, Prewitt RL. Oxidative stress in a rat model of obesity-induced hypertension. Hypertension. 2001;37(2, part 2):554–560. doi: 10.1161/01.hyp.37.2.554. [DOI] [PubMed] [Google Scholar]
- 149.Jebelovszki E, Kiraly C, Erdei N, et al. High-fat diet-induced obesity leads to increased NO sensitivity of rat coronary arterioles: role of soluble guanylate cyclase activation. American Journal of Physiology. 2008;294(6):H2558–H2564. doi: 10.1152/ajpheart.01198.2007. [DOI] [PubMed] [Google Scholar]
- 150.Haddock RE, Grayson TH, Morris MJ, Howitt L, Chadha PS, Sandow SL. Diet-induced obesity impairs endothelium-derived hyperpolarization via altered potassium channel signaling mechanisms. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016423.e16423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Naderali EK, Pickavance LC, Wilding JPH, Williams G. Diet-induced endothelial dysfunction in the rat is independent of the degree of increase in total body weight. Clinical Science. 2001;100(6):635–641. doi: 10.1042/cs1000635. [DOI] [PubMed] [Google Scholar]
- 152.Gil-Ortega M, Stucchi P, Guzmán-Ruiz R, et al. Adaptative nitric oxide overproduction in perivascular adipose tissue during early diet-induced obesity. Endocrinology. 2010;151(7):3299–3306. doi: 10.1210/en.2009-1464. [DOI] [PubMed] [Google Scholar]
- 153.Nilsson C, Raun K, Yan FF, Larsen MO, Tang-Christensen M. Laboratory animals as surrogate models of human obesity. Acta Pharmacologica Sinica. 2012;33(2):173–181. doi: 10.1038/aps.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mavri A, Poredoš P, Šuran D, Gaborit B, Juhan-Vague I, Poredoš P. Effect of diet-induced weight loss on endothelial dysfunction: early improvement after the first week of dieting. Heart and Vessels. 2011;26(1):31–38. doi: 10.1007/s00380-010-0016-1. [DOI] [PubMed] [Google Scholar]
- 155.Chatterjee TK, Stoll LL, Denning GM, et al. Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circulation Research. 2009;104(4):541–549. doi: 10.1161/CIRCRESAHA.108.182998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Donato AJ, Henson GD, Morgan RG, Enz RA, Walker AE, Lesniewski LA. TNF-alpha impairs endothelial function in adipose tissue resistance arteries of mice with diet-induced obesity. American Journal of Physiology. 2012;303(6):H672–H679. doi: 10.1152/ajpheart.00271.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ketonen J, Shi J, Martonen E, Mervaala E. Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese C57BL/6 mice. Circulation Journal. 2010;74(7):1479–1487. doi: 10.1253/circj.cj-09-0661. [DOI] [PubMed] [Google Scholar]
- 158.Elizalde M, Rydén M, Van Harmelen V, et al. Expression of nitric oxide synthases in subcutaneous adipose tissue of nonobese and obese humans. Journal of Lipid Research. 2000;41(8):1244–1251. [PubMed] [Google Scholar]
- 159.Ma L, Ma S, He H, et al. Perivascular fat-mediated vascular dysfunction and remodeling through the AMPK/mTOR pathway in high-fat diet-induced obese rats. Hypertension Research. 2010;33(5):446–453. doi: 10.1038/hr.2010.11. [DOI] [PubMed] [Google Scholar]
- 160.Kimura K, Tsuda K, Baba A, et al. Involvement of nitric oxide in endothelium-dependent arterial relaxation by leptin. Biochemical and Biophysical Research Communications. 2000;273(2):745–749. doi: 10.1006/bbrc.2000.3005. [DOI] [PubMed] [Google Scholar]
- 161.Lembo G, Vecchione C, Fratta L, et al. Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes. 2000;49(2):293–297. doi: 10.2337/diabetes.49.2.293. [DOI] [PubMed] [Google Scholar]
- 162.Vecchione C, Maffei A, Colella S, et al. Leptin effect on endothelial nitric oxide is mediated through Akt-endothelial nitric oxide synthase phosphorylation pathway. Diabetes. 2002;51(1):168–173. doi: 10.2337/diabetes.51.1.168. [DOI] [PubMed] [Google Scholar]
- 163.Matsuda K, Teragawa H, Fukuda Y, Nakagawa K, Higashi Y, Chayama K. Leptin causes nitric-oxide independent coronary artery vasolidation in humans. Hypertension Research. 2003;26(2):147–152. doi: 10.1291/hypres.26.147. [DOI] [PubMed] [Google Scholar]
- 164.Momin AU, Melikian N, Shah AM, et al. Leptin is an endothelial-independent vasodilator in humans with coronary artery disease: evidence for tissue specificity of leptin resistance. European Heart Journal. 2006;27(19):2294–2299. doi: 10.1093/eurheartj/ehi831. [DOI] [PubMed] [Google Scholar]
- 165.Fortuño A, Rodríguez A, Gómez-Ambrosi J, et al. Leptin inhibits angiotensin II-induced intracellular calcium increase and vasoconstriction in the rat aorta. Endocrinology. 2002;143(9):3555–3560. doi: 10.1210/en.2002-220075. [DOI] [PubMed] [Google Scholar]
- 166.Gentile MT, Vecchione C, Marino G, et al. Resistin impairs insulin-evoked vasodilation. Diabetes. 2008;57(3):577–583. doi: 10.2337/db07-0557. [DOI] [PubMed] [Google Scholar]
- 167.Chen C, Jiang J, Lü JM, et al. Resistin decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. American Journal of Physiology. 2010;299(1):H193–H201. doi: 10.1152/ajpheart.00431.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Xi W, Satoh H, Kase H, Suzuki K, Hattori Y. Stimulated HSP90 binding to eNOS and activation of the PI3-Akt pathway contribute to globular adiponectin-induced NO production: vasorelaxation in response to globular adiponectin. Biochemical and Biophysical Research Communications. 2005;332(1):200–205. doi: 10.1016/j.bbrc.2005.04.111. [DOI] [PubMed] [Google Scholar]
- 169.Yamawaki H, Hara N, Okada M, Hara Y. Visfatin causes endothelium-dependent relaxation in isolated blood vessels. Biochemical and Biophysical Research Communications. 2009;383(4):503–508. doi: 10.1016/j.bbrc.2009.04.074. [DOI] [PubMed] [Google Scholar]
- 170.Varma V, Yao-Borengasser A, Rasouli N, et al. Human visfatin expression: relationship to insulin sensitivity, intramyocellular lipids, and inflammation. Journal of Clinical Endocrinology and Metabolism. 2007;92(2):666–672. doi: 10.1210/jc.2006-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yang CC, Deng SJ, Hsu CC, et al. Visfatin regulates genes related to lipid metabolism in porcine adipocytes. Journal of Animal Science. 2010;88(10):3233–3241. doi: 10.2527/jas.2010-2799. [DOI] [PubMed] [Google Scholar]
- 172.Zhang H, Park Y, Wu J, et al. Role of TNF-α in vascular dysfunction. Clinical Science. 2009;116(3):219–230. doi: 10.1042/CS20080196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hector J, Schwarzloh B, Goehring J, et al. TNF-α alters visfatin and adiponectin levels in human fat. Hormone and Metabolic Research. 2007;39(4):250–255. doi: 10.1055/s-2007-973075. [DOI] [PubMed] [Google Scholar]
- 174.Zhang DX, Yi FX, Zou AP, Li PL. Role of ceramide in TNF-α-induced impairment of endothelium-dependent vasorelaxation in coronary arteries. American Journal of Physiology. 2002;283(5):H1785–H1794. doi: 10.1152/ajpheart.00318.2002. [DOI] [PubMed] [Google Scholar]
- 175.Brian JE, Faraci FM. Tumor necrosis factor-α-induced dilatation of cerebral arterioles. Stroke. 1998;29(2):509–515. doi: 10.1161/01.str.29.2.509. [DOI] [PubMed] [Google Scholar]
- 176.Johns DG, Webb RC. TNF-α-induced endothelium-independent vasodilation: a role for phospholipase A2-dependent ceramide signaling. American Journal of Physiology. 1998;275(5):H1592–H1598. doi: 10.1152/ajpheart.1998.275.5.H1592. [DOI] [PubMed] [Google Scholar]
- 177.Wort SJ, Ito M, Chou PC, et al. Synergistic induction of endothelin-1 by tumor necrosis factor α and interferon γ is due to enhanced NF-κB binding and histone acetylation at specific κB sites. Journal of Biological Chemistry. 2009;284(36):24297–24305. doi: 10.1074/jbc.M109.032524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Brasier AR, Li J, Wimbish KA. Tumor necrosis factor activates angiotensinogen gene expression by the Rel A transactivator. Hypertension. 1996;27(4):1009–1017. doi: 10.1161/01.hyp.27.4.1009. [DOI] [PubMed] [Google Scholar]
- 179.Ohkawa F, Ikeda U, Kawasaki K, Kusano E, Igarashi M, Shimada K. Inhibitory effect of interleukin-6 on vascular smooth muscle contraction. American Journal of Physiology. 1994;266(3):H898–H902. doi: 10.1152/ajpheart.1994.266.3.H898. [DOI] [PubMed] [Google Scholar]
- 180.Minghini A, Britt LD, Hill MA. Interleukin-1 and interleukin-6 mediated skeletal muscle arteriolar vasodilation: in vitro versus in vivo studies. Shock. 1998;9(3):210–215. doi: 10.1097/00024382-199803000-00009. [DOI] [PubMed] [Google Scholar]
- 181.Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II-induced endothelial dysfunction and hypertrophy. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27(12):2576–2581. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 182.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. The Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Moncada S. Adventures in vascular biology: a tale of two mediators. Philosophical Transactions of the Royal Society B. 2006;361(1469):735–759. doi: 10.1098/rstb.2005.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Feletou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. British Journal of Pharmacology. 2011;164(3):894–912. doi: 10.1111/j.1476-5381.2011.01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang C, Hein TW, Wang W, Kuo L. Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circulation Research. 2003;92(3):322–329. doi: 10.1161/01.res.0000056759.53828.2c. [DOI] [PubMed] [Google Scholar]
- 186.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. American Journal of Physiology. 2007;292(1):C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 187.Taddei S, Virdis A, Ghiadoni L, Salvetti A. Vascular effects of endothelin-1 in essential hypertension: relationship with cyclooxygenase-derived endothelium-dependent contracting factors and nitric oxide. Journal of Cardiovascular Pharmacology. 2000;35:S37–S40. doi: 10.1097/00005344-200000002-00009. [DOI] [PubMed] [Google Scholar]
- 188.Knock GA, Snetkov VA, Shaifta Y, et al. Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca2+ sensitization. Free Radical Biology and Medicine. 2009;46(5):633–642. doi: 10.1016/j.freeradbiomed.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gryglewski RJ, Palmer RMJ, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320(6061):454–456. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]