

Abstract
Cancer metabolism and epigenetics are two relatively new areas of cancer research. Recent years have seen an explosion of studies implicating either altered tumor metabolism or epigenetic mechanisms in the pathogenesis or maintenance of brain tumors. A new paradigm is emerging in cancer biology that represents a convergence of these themes, the metabolic regulation of epigenetics. We discuss this interrelationship in the context of two metabolic enzymes that can influence the pathogenesis of gliomas by altering the epigenetic state. The first of these enzymes is isocitrate dehydrogenase 1 (IDH1), which is mutated in secondary glioblastomas and ∼70% of grade II/III astrocytomas and oligodendrogliomas. Mutant IDH1 results in the production of a metabolite 2‐hydroxyglutarate (2‐HG) that can inhibit DNA and histone demethylating enzymes resulting in the glioma‐CpG island phenotype (G‐CIMP) and increased histone methylation marks. Pyruvate kinase M2 (PKM2), an enzyme that plays a critical role in the glycolytic pathway, is a second example of a metabolic enzyme that can affect histone modifications. In epidermal growth factor receptor (EGFR)‐driven glioblastoma, PKM2 translocates to the nucleus and phosphorylates histone 3 at threonine 11 (H3‐T11). This causes dissociation of HDAC3 from the CCND1 (Cyclin D1) and c‐MYC promoters and subsequent histone acetylation, leading to transcription of Cyclin‐D1 and c‐MYC, and subsequent cell proliferation. Modification of the epigenetic state by alterations in metabolic enzymes is a novel phenomenon that contributes to the pathogenesis of gliomas and may help in the identification of new therapeutic targets.
Keywords: epigenetics, glioma, IDH1, metabolism, PKM2.
Introduction
Gliomas are infiltrative brain tumors consisting primarily of astrocytomas and oligodendrogliomas classified into low‐grade [World Health Organization (WHO) grades I and II] and high‐grade (WHO grades III and IV) tumors. Glioblastomas (grade IV astrocytic tumors) are the most lethal and neurologically destructive of gliomas. Despite several decades of intensive research, the prognosis for glioblastomas and high‐grade gliomas in general remains dismal. This underscores the importance of elucidating the pathogenesis of these tumors to effectively combat them. Recent years have uncovered many aspects of glioma biology including novel genetic and molecular alterations, leading to classification of gliomas into various subgroups. It is becoming increasingly evident that at least some of these genetic and molecular alterations result in changes in cellular metabolism.
Glioblastomas frequently exhibit increased glucose consumption and lactate production in the presence of oxygen, the Warburg effect 43. Activation of PI3K/AKT in glioblastoma cell lines leads to increased glucose uptake and glycolysis 5, 17, 43. Primary glioblastomas show various genetic alterations such epidermal growth factor receptor (EGFR) amplification, phosphatase and tensin homolog (PTEN) loss and platelet‐derived growth factor receptor type A (PDGFRA) amplification resulting in enhanced signaling of receptor tyrosine kinases and deregulation of the PI3K/AKT pathway 1, 22, thus stimulating glucose uptake and aerobic glycolysis 5, 17. More recently, the NADP+‐dependent enzyme isocitrate dehydrogenase 1 (IDH1) was found to be mutated in ∼70% of grade II and grade III astrocytomas and oligodendrogliomas, and secondary glioblastomas 4, 35, 50. IDH1 catalyzes oxidative decarboxylation of isocitrate to α‐ketoglutarate (α‐KG) in the cytosol. Mutant IDH1 alters cellular metabolism by generating the oncometabolite, 2‐hydroxyglutarate (2‐HG), from α‐KG that can accumulate to millimolar concentrations 13, 45.
Epigenetics encompasses heritable changes in DNA and DNA‐associated proteins that are often accompanied by changes in gene expression. DNA methylation represents the most extensively studied epigenetic phenomenon in glioblastomas [recently reviewed in 15, 26, 33]. In contrast, very little is known about histone modifications in gliomas. Histones are proteins around which DNA is organized into nucleosomes. Each nucleosome consists of ∼147 DNA base pairs wrapped around a histone octomer composed of H2A, H2B, H3 and H4. Histones have amino acid tails that can undergo a variety of post‐translational modifications such as acetylation, methylation, phosphorylation, ubiquitination and SUMOylation of arginine (R) and lysine (K) residues 38. This results in changes in DNA function and transcription by regulating accessibility to cellular transcriptional machinery [reviewed in 3]. To date, the most widely studied histone modifications are acetylation and methylation, which affect transcription differently. Histone acetylation of lysine residues usually activates transcription, while methylation can be an activator or repressor of transcription, depending on the histone residue that is methylated 3. For instance, methylation of H3K9, H3K27 and H4K20 is thought to be associated with silencing of transcription, and methylation of H3K4, H3K36 and H3K79 seems to be associated with activation of transcription [reviewed in 3, 9]. Methylation of histone lysine residues is a complex phenomenon and is regulated by a variety of histone lysine methyltransferases (KMTs) and demethylases (KDMs) 3.
Recent studies have emerged implicating histone modifications in adult glioblastomas 16, 32, 51 and mutations in genes encoding histone proteins in pediatric glioblastomas 25, 36, 40, 48. This article focuses on the molecular connections between metabolism and epigenetics in gliomas using the two examples of IDH1 mutations in intermediate‐grade gliomas and secondary glioblastomas, and PKM2 alterations in EGFR‐driven primary glioblastomas to illustrate this phenomenon.
Mutations in IDH1 Result in the Glioma‐CpG Island Methylator Phenotype (G‐CIMP)
Profiling of DNA promoter methylation in glioblastomas reveals a unique subset of cases collectively referred to as G‐CIMP within the proneuronal subtype of glioblastomas that exhibit hypermethylation at a large number of loci 34. Glioblastomas and intermediate‐grade gliomas (grade II and grade III) with G‐CIMP are strongly associated with mutations in IDH1 11, 16, 27, 34, 42. IDH1 mutations are the most common (specifically IDH1 R132H) of the IDH mutations in gliomas and account for more than 95% of cases 2, 6, 21. Astrocytic cell lines transfected with mutant IDH1 R132H and colon cancer cell lines with knock‐in mutant IDH1 R132H show G‐CIMP phenotypes very similar to IDH1 mutant gliomas 16, 42.
How mutations in IDH1 result in DNA methylation in gliomas is not entirely known. One hypothesis is that 2‐HG inhibits α‐KG‐dependent enzymes as 2‐HG is structurally similar to α‐KG 10, 49. α‐KG‐dependent dioxygenases form a large family of enzymes influencing various functions in the cell such as carnitine synthesis, hypoxic sensing, collagen modifications and histone and DNA demethylation [reviewed in 29]. Profiling experiments in samples from patients with acute myeloid leukemia showed mutual exclusivity between IDH1/2 and TET2 mutations 19. TET2 belongs to the TET family of enzymes that is dependent on α‐KG to catalyze cytosine 5‐hydroxymethylation (5hmC) and subsequent DNA demethylation 23, 41. Further astrocytic cell lines expressing mutant IDH1 R132H inhibit TET2‐dependent 5hmC 42. However, no mutations in the TET family of proteins are reported in gliomas. Another factor to be considered is that histone methylation can promote DNA methylation and vice versa 18. In astrocytic cell lines expressing mutant IDH1 R132H, trimethylation of H3K9 occurs prior to DNA methylation, suggesting that histone methylation may contribute to DNA methylation and may provide an additional mechanism by which mutant IDH can contribute to G‐CIMP 31.
IDH1 Mutations Are Associated with Increased Histone Methylation Marks in Gliomas
Immortalized human astrocytic cell lines or murine neurosphere cultures transfected with mutant IDH1 R132H show increases in H3K27me3, H3K9me3 32, 42 and H3K36me3 42 compared with cells overexpressing wild‐type IDH1. Similarly, U87MG cells transfected with mutant IDH1 R132H show increased H3K9me2, H3K4me3, H3K27me2 and H3K79me2 compared with wild‐type controls 49. In addition, heterozygous knock‐in of IDH1 R132H in HCT116 colon cancer cell lines results in increased H3K9me3, H3K27me3 and H3K4me3 compared with the controls 16. The structural analogy between 2‐HG and α‐KG also comes into play in regulating histone methylation. The Jumonji C family of KDMs uses α‐KG, Fe (II) and oxygen as cofactors to demethylate histone lysine residues [reviewed in 29]. 2‐HG inhibits these enzymes 10, 49 and inhibition of KDM4C by 2‐HG decreases histone demethylation, resulting in increased methylation marks on H3K9 and H3K27 32.
H3K9me3 is one of the methylation marks that showed a striking increase in cells expressing mutant IDH1 R132H 32. Methylation of H3K9 is thought to influence cell differentiation 8. Conditional deletion of the H3K9‐specific KMT Setdb1 causes lowered H3K9me3 and results in upregulation of lineage‐specific differentiation markers 30. Conversely, increased H3K9me3 after knockdown of the KDM JmjD2A prevents neuronal crest cell induction 39. Cells with mutant IDH1 show suppressed glial differentiation. Neurospheres transfected with mutant IDH1 R132H show increased H3K9me3 accompanied by a decrease in glial fibrillary acidic protein (GFAP) and an increase in nestin expression compared with their IDH1 wild‐type expressing counterparts 32, 42. Interestingly, in human glioma samples, the relationship between H3K9me3 and IDH1 R132H mutations varied between glioma subtypes and grades as assessed by immunohistochemistry 44. A robust relationship was observed between H3K9me3 staining and IDH1 mutations in all grades of oligodendrogliomas. However, grade III astrocytomas and glioblastomas showed positivity for H3K9me3 but did not show a significant relationship with IDH1 mutations 44. These data suggest that the roles played by IDH1 mutations and IDH1 mutant‐derived 2‐HG in H3K9 trimethylation may be context dependent and can vary between oligodendrogliomas and astrocytomas. While future experiments will address differences in the role played by IDH1 mutations in oligodendrogliomas vs. astrocytomas, the current working model of how IDH1 mutations and 2‐HG can affect both DNA and histone methylation is presented in Figure 1.
Figure 1.
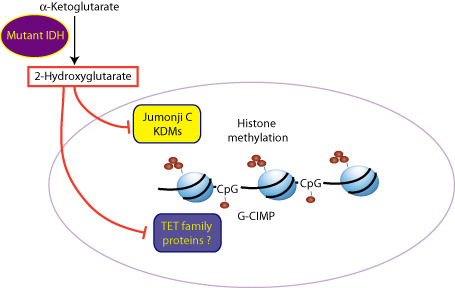
Isocitrate dehydrogenase (IDH) mutations result in glioma‐CpG methylation phenotype (G‐CIMP) and histone methylation in gliomas. Mutant IDH1 catalyzes the production of 2‐hydroxyglutarate (2‐HG) from α‐ketoglutarate (α‐KG). 2‐HG is thought to inhibit α‐KG‐dependent dioxygenase enzymes as 2‐HG is structurally similar to α‐KG. The Jumonji C family of histone lysine demethylases (KDMs) and the TET group of DNA hydroxylases are α‐KG‐dependent dioxygenases. Inhibition of these enzymes may result in increased histone methylation marks and DNA methylation contributing to G‐CIMP.
Pyruvate Kinase M2 Regulates Histone Modifications in Gliomas
Pyruvate kinase is a key enzyme in the glycolytic pathway that catalyzes the conversion of phosphoenolpyruvate (PEP) to pyruvate. Four isoforms of pyruvate kinase exist (PKM1, PKM2, L and R). PKM2 is the main splice variant during brain development, while the major isoform in the adult brain is PKM1 47. Cancer cells including glioblastoma cell lines show upregulated PKM2 12, 24. Replacing PKM2 with PKM1 in lung cancer cell lines results in decreased lactate production and increased oxygen consumption, essentially reversing the Warburg effect and decreased xenograft proliferation in nude mice 12. Furthermore, PKM2 was identified as essential for survival of glioma stem‐like cells using an unbiased RNAi screen 20. Knockdown of PKM2 in glioblastoma cell lines decreases survival and proliferation while lowering ATP levels 24. The effects of PKM2 on metabolic reprogramming in cancer cells are manifold and have been discussed elsewhere 7. More relevant to this review is the effect of PKM2 on histone modifications in EGFR‐driven glioblastomas 51.
Upon EGFR activation, PKM2 translocates to the nucleus and phosphorylates histone 3 at threonine 11 (H3‐T11) 51. This, in turn, leads to removal of the histone deacetylase 3 (HDAC3) from the CCND1 (Cyclin D1) and c‐MYC promoter regions 51. PKM2 then forms a complex with β‐catenin, which binds to the CCND1 and c‐MYC promoter regions, leading to acetylation of H3K9 and activation of transcription 51, 52 (Figure 2). Reconstitution of cells with a mutant form of H3 [where threonine is replaced with an alanine residue (H3‐T11A) that cannot be phosphorylated] in place of wild‐type H3 prevented HDAC3 removal from the CCND1 and c‐MYC promoters. Cells expressing mutant H3‐T11A implanted into the mouse brains fail to form xenografts compared with their wild‐type controls. Furthermore, phosphorylated EGFR correlated with both nuclear PKM2 and phosphorylated H3T11 levels in human glioma samples. Phosphorylated H3T11 levels were higher in 45 grade IV glioblastomas when compared to 30 diffused astrocytoma cases 51. These data suggest that PKM2 may play an important role as a regulator of histone phosphorylation and acetylation in EGFR‐driven gliomas.
Figure 2.
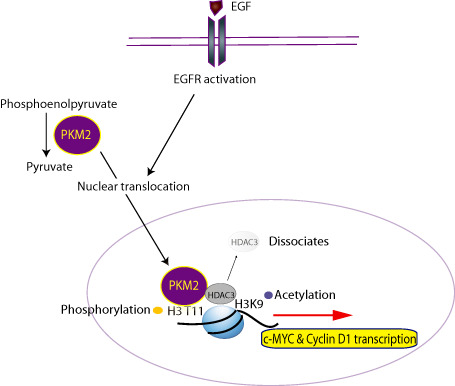
Pyruvate kinase M2 (PKM2) regulates histone modifications in epidermal growth factor receptor (EGFR)‐driven glioblastomas. PKM2 catalyzes the conversion of phosphoenolpyruvate (PEP) to pyruvate in the glycolytic pathway. EGFR activation results in PKM2 translocation to the nucleus and phosphorylation of histone 3 at the threonine 11 residue (H3‐T11). This leads to removal of the histone deacetylase 3 (HDAC3) from the CCND1 (Cyclin D1) and c‐MYC promoter regions, acetylation of H3K9 and activation of transcription of Cyclin D1 and c‐MYC.
Summary
The complex interplay between the metabolic state of a cancerous cell and its epigenetic machinery represents a novel mechanism by which the normal control of cell proliferation/differentiation can be disrupted. Epigenetic marks can integrate metabolism with nuclear transcription to allow cancer cells to coordinate their response to intrinsic and extracellular signals. Recent studies of IDH1 mutations in intermediate‐grade gliomas and secondary glioblastomas, and PKM2 in EGFR‐driven glioblastomas illustrate disruptions of this process. These tumorigenic events are not restricted to gliomas and other metabolic pathways likely contribute to the regulation of the epigenetic machinery. For example, the enzyme ATP‐citrate lyase (ACL) drives nuclear/cytosolic acetyl‐CoA synthesis and a significant defect in histone acetylation is seen when ACL is knocked down in cell lines 46. Sir2 belonging to the NAD+‐dependent sirtuin family of histone deacetylases extends lifespan in yeast on calorie restriction 28. Deacetylation of histone 4 at the K14 residue contributes to this effect on yeast lifespan 14. In mouse embryonic stem cells, threonine contributes to the synthesis of s‐adenosylmethionine, which functions as a substrate for histone methylation reactions. Depletion of threonine or threonine dehydrogenase reduced trimethylation of H3K4 37. While it remains to be determined if these pathways contribute to glioma pathology, these examples underscore the variety and complexity of mechanisms that link metabolism to epigenetics. Unraveling the dynamics and intricacies of these processes may help us develop a better understanding of gliomas to enable the development of novel therapeutic targets to effectively combat these highly aggressive tumors.
Acknowledgments
We thank Dennis Pozega, Chao Lu, Patrick Ward and Tullia Lindsten for critical reading of the manuscript. CBT is co‐founder of Agios Pharmaceuticals and has financial interest in Agios.
References
- 1. Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Balss J, Meyer J, Mueller W, Korshunov A, von Hartmann C, Deimling A (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116:597–602. [DOI] [PubMed] [Google Scholar]
- 3. Berger SL (2007) The complex language of chromatin regulation during transcription. Nature 447:407–412. [DOI] [PubMed] [Google Scholar]
- 4. Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP et al (2009) IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high‐grade gliomas but not in other solid tumors. Hum Mutat 30:7–11. [DOI] [PubMed] [Google Scholar]
- 5. Buzzai M, Bauer DE, Jones RG, Deberardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB (2005) The glucose dependence of Akt‐transformed cells can be reversed by pharmacologic activation of fatty acid beta‐oxidation. Oncogene 24:4165–4173. [DOI] [PubMed] [Google Scholar]
- 6. Capper D, Zentgraf H, Balss J, von Hartmann C, Deimling A (2009) Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol 118:599–601. [DOI] [PubMed] [Google Scholar]
- 7. Chaneton B, Gottlieb E (2012) Rocking cell metabolism: revised functions of the key glycolytic regulator PKM2 in cancer. Trends Biochem Sci 37:309–316. [DOI] [PubMed] [Google Scholar]
- 8. Chen J, Liu H, Liu J, Qi J, Wei B, Yang J et al (2013) H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat Genet 45:34–42. [DOI] [PubMed] [Google Scholar]
- 9. Chi P, Allis CD, Wang GG (2010) Covalent histone modifications—miswritten, misinterpreted and mis‐erased in human cancers. Nat Rev Cancer 10:457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR et al (2011) The oncometabolite 2‐hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12:463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Christensen BC, Smith AA, Zheng S, Koestler DC, Houseman EA, Marsit CJ et al (2011) DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J Natl Cancer Inst 103:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R et al (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452:230–233. [DOI] [PubMed] [Google Scholar]
- 13. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al (2010) Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature 465:966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A et al (2009) Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459:802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dubuc AM, Mack S, Unterberger A, Northcott PA, Taylor MD (2012) The epigenetics of brain tumors. Methods Mol Biol 863:139–153. [DOI] [PubMed] [Google Scholar]
- 16. Duncan CG, Barwick BG, Jin G, Rago C, Kapoor‐Vazirani P, Powell DR et al (2012) A heterozygous IDH1R132H/WT mutation induces genome‐wide alterations in DNA methylation. Genome Res 12:2339–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR et al (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64:3892–3899. [DOI] [PubMed] [Google Scholar]
- 18. Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR et al (2006) Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 20:3089–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Figueroa ME, Abdel‐Wahab O, Lu C, Ward PS, Patel J, Shih A et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goidts V, Bageritz J, Puccio L, Nakata S, Zapatka M, Barbus S et al (2012) RNAi screening in glioma stem‐like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene 31:3235–3243. [DOI] [PubMed] [Google Scholar]
- 21. Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A et al (2009) Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol 118:469–474. [DOI] [PubMed] [Google Scholar]
- 22. Huse JT, Holland EC (2010) Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer 10:319–331. [DOI] [PubMed] [Google Scholar]
- 23. Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES‐cell self‐renewal and inner cell mass specification. Nature 466:1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kefas B, Comeau L, Erdle N, Montgomery E, Amos S, Purow B (2010) Pyruvate kinase M2 is a target of the tumor‐suppressive microRNA‐326 and regulates the survival of glioma cells. Neuro Oncol 12:1102–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khuong‐Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E et al (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kreth S, Thon N, Kreth FW (2012) Epigenetics in human gliomas. Cancer Lett [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 27. Laffaire J, Everhard S, Idbaih A, Criniere E, de Marie Y, Reynies A et al (2011) Methylation profiling identifies 2 groups of gliomas according to their tumorigenesis. Neuro Oncol 13:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin SJ, Defossez PA, Guarente L (2000) Requirement of NAD and SIR2 for life‐span extension by calorie restriction in Saccharomyces cerevisiae . Science 289:2126–2128. [DOI] [PubMed] [Google Scholar]
- 29. Loenarz C, Schofield CJ (2008) Expanding chemical biology of 2‐oxoglutarate oxygenases. Nat Chem Biol 4:152–156. [DOI] [PubMed] [Google Scholar]
- 30. Lohmann F, Loureiro J, Su H, Fang Q, Lei H, Lewis T et al (2010) KMT1E mediated H3K9 methylation is required for the maintenance of embryonic stem cells by repressing trophectoderm differentiation. Stem Cells 28:201–212. [DOI] [PubMed] [Google Scholar]
- 31. Lu C, Thompson CB (2012) Metabolic regulation of epigenetics. Cell Metab 16:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel‐Wahab O et al (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483:474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martinez R, Esteller M (2010) The DNA methylome of glioblastoma multiforme. Neurobiol Dis 39:40–46. [DOI] [PubMed] [Google Scholar]
- 34. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP et al (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231. [DOI] [PubMed] [Google Scholar]
- 37. Shyh‐Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S et al (2013) Influence of threonine metabolism on S‐adenosylmethionine and histone methylation. Science 339:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403:41–45. [DOI] [PubMed] [Google Scholar]
- 39. Strobl‐Mazzulla PH, Sauka‐Spengler T, Bronner‐Fraser M (2010) Histone demethylase JmjD2A regulates neural crest specification. Dev Cell 19:460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sturm D, Witt H, Hovestadt V, Khuong‐Quang DA, Jones DT, Konermann C et al (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. [DOI] [PubMed] [Google Scholar]
- 41. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y et al (2009) Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E et al (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483:479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Venneti S, Felicella MM, Coyne T, Phillips JJ, Gorovets D, Huse JT et al (2012) Histone 3 lysine 9 trimethylation (H3K9me3) is differentially associated with isocitrate dehydrogenase 1 (IDH1) R132H mutations in oligodendrogliomas and high‐grade astrocytomas. (2013) Journal of Neuropathology & Experimental Neurology: (In Press). [DOI] [PMC free article] [PubMed]
- 45. Ward PS, Patel J, Wise DR, Abdel‐Wahab O, Bennett BD, Coller HA et al (2010) The common feature of leukemia‐associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha‐ketoglutarate to 2‐hydroxyglutarate. Cancer Cell 17:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB (2009) ATP‐citrate lyase links cellular metabolism to histone acetylation. Science 324:1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wolf A, Agnihotri S, Munoz D, Guha A (2011) Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol Dis 44:84–91. [DOI] [PubMed] [Google Scholar]
- 48. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non‐brainstem glioblastomas. Nat Genet 44:251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH et al (2011) Oncometabolite 2‐hydroxyglutarate is a competitive inhibitor of alpha‐ketoglutarate‐dependent dioxygenases. Cancer Cell 19:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D et al (2012) PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W et al (2011) Nuclear PKM2 regulates beta‐catenin transactivation upon EGFR activation. Nature 480:118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]