

Abstract
The family of insulin receptor substrates (IRS) consists of four proteins (IRS-1 - IRS-4), which were initially characterized as typical cytosolic adaptor proteins involved in insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) signaling. The first cloned and characterized member of the IRS family, IRS-1, has predicted molecular weight of 132 kDa, however, as a result of its extensive serine phosphorylation it separates on a SDS gel as a band of approximately 160–185 kDa. In addition to its metabolic and growth-promoting functions, IRS-1 is also suspected to play a role in malignant transformation. The mechanism by which IRS-1 supports tumor growth is not fully understood, and the argument that IRS-1 merely amplifies the signal from the IGF-1R and/or IR requires further investigation. Almost a decade ago, we reported the presence of nuclear IRS-1 in medulloblastoma clinical samples, which express viral oncoprotein, large T-antigen of human polyomavirus JC (JCV T-antigen). This first demonstration of nuclear IRS-1 was confirmed in several other laboratories. The nuclear IRS-1 was also detected by cells expressing the SV40 T-antigen, v-Src, in immortalized fibroblasts stimulated with IGF-I, in hepatocytes, 32D cells, and in an osteosarcoma cell line. More recently, nuclear IRS-1 was detected in breast cancer cells in association with estrogen receptor alpha (ERα), and in JC virus negative medulloblastoma cells expressing ERβ, further implicating nuclear IRS-1 in cellular transformation. Here, we discuss how nuclear IRS-1 acting on DNA repair fidelity, transcriptional activity, and cell growth can support tumor development and progression.
IRS-1 Structure and Function
IRS-1 was long considered an example of a typical cytosolic molecule involved in signal transduction from two prominent membrane tyrosine kinases, insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) (Myers et al., 1993). The molecular structure of IRS-1 has been well characterized revealing two conserved regions within N-terminal portion of the protein (Myers and White, 1996) and (Fig.1). The first one is designated PH due to its similarity to a pleckstrin homology (PH) domain; and the second, PTB, shows similarities to a putative phosphotyrosine-binding (PTB) domain present in Shc and other proteins (Sun et al., 1992). The PH domain contains a positively charged binding pocket that mediates interaction with phospholipids and with proteins containing an acidic motif (Burks et al., 1997; Burks et al., 1998; Myers et al., 1995). The PTB domain has the ability of recognizing phosphorylated tyrosine residues within NPXY motives, providing a mechanism of coupling IRS-1 with Tyr950 or Tyr960 in the juxtamembrane region of the IGF-IR and IR, respectively (Craparo et al., 1995; Eck et al., 1996; Wolf et al., 1995). Over 20 tyrosine phosphorylation sites on the IRS-1 docking molecule can recruit proteins equipped with src-homology (SH2) domain, and several enzymes and adapter proteins have been confirmed as partners in IRS-1 mediated signaling cascade (Fig.1). These include Grb-2 (Myers et al., 1994; Valverde et al., 2001), PI-3 kinase (Myers et al., 1992), SHP2 phosphatase (Myers et al., 1996), Fyn (Sun et al., 1996), Nck (Lee et al., 1993), and Crk (Beitner-Johnson et al., 1996). Other proteins such as polyomavirus large T-antigens (Fei et al., 1995), 14-3-3 (Ogihara et al., 1997), integrins (Reiss et al., 2001; Vuori and Ruoslahti, 1994; Wang et al., 2007), and estrogen receptors (Morelli et al., 2004; Sisci et al., 2007a; Surmacz and Bartucci, 2004; Urbanska et al., 2009) that may contribute to malignant transformation, associate with IRS-1 through interaction/s that seem to be independent from IRS-1 tyrosine phosphorylation. Interestingly, the binding and cytoplasmic retention of the DNA repair protein, Rad51, by hypo-phosphorylated IRS-1 was inhibited in cells stimulated with IGF-I, strongly implicating the IGF-IR/IRS-1 signaling axis in homologous recombination directed DNA repair (Gualco et al., 2009b; Reiss et al., 2006; Trojanek et al., 2006b; Trojanek et al., 2003).
Figure 1. Schematic diagram of mouse IRS-1 protein.
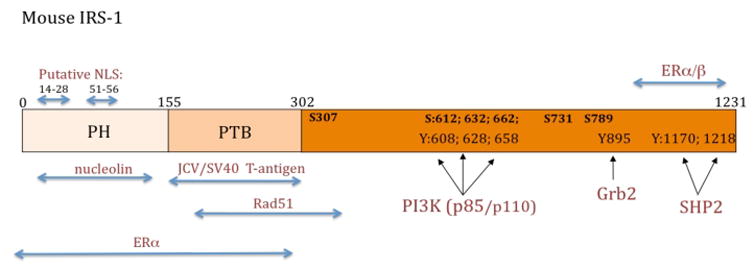
There are two major functional domains within the N-terminus portion of IRS-1: pleckstrin homology domain (PH), spanning between amino acids 0–155, and phosphotyrozine binding domain (PTB) located between amino acids 155–302. Black arrows indicate exact binding sites for PI3 kinase, Grab2 and SHP2 at indicated tyrosine residues (Y). Functionally relevant serine residues (S) and their corresponding amino acid positions are also indicated. Blue arrows indicate putative binding regions for proteins, which are suspected to translocate IRS-1 to the nucleus, including nucleolin, polyomavirus T-antigens (JCV and SV40), estrogen receptor α (ERα) and estrogen receptor β (ERβ). Other indicated sites include the binding between IRS-1 and DNA repair protein, Rad51, and positions of putative nuclear localization signals (NLS).
Another important aspect of IRS-1 regulation is its phosphorylation of serine residues, which in contrast to IRS-1 tyrosine phosphorylation is thought to inactivate some of the most relevant functions of this docking molecule (Myers and White, 1996; Sun et al., 1992), including TNFα–mediated development of insulin resistance (Hotamisligil et al., 1996; Peraldi et al., 1996). The best characterized serine residues of IRS-1 are: Ser307 (murine)/Ser312 (humans), which become phosphorylated as a result of PI3K, PKC, JNK and TNFα activation; Ser612, phosphorylated by MAPK; the cluster of serines Ser632, 662, and 731, which upon phosphorylation by Akt/mTOR pathway may lead to IRS-1 degradation; and Ser789 which following AMPK mediated phosphorylation, may result in enhanced association of IRS-1 with IR during exercise and starvation (Schmitz-Peiffer and Whitehead, 2003; Shaw, 2006; White, 2002). Interestingly, elevated serine phosphorylation of IRS-1 has been found to facilitate its interaction with integrins (Reiss et al., 2001; Wang et al., 2006; Wang et al., 2007), however, the same serine phosphorylation strongly inhibited the binding between IRS-1 and JCV T-antigen (Lassak et al., 2002).
Nuclear Localization of IRS-1
Although the mechanism by which IRS-1 translocates to the nucleus is not fully understood, IRS-1 could employ its own putative nuclear localization signals (NLS) (Fig.1). One such a possibility was discussed in the report describing nuclear translocation of IRS-1 following ectopic expression of IGF-IR and IRS-1 in 32D cells, which resulted in IL-3 independence and tumor formation by these mouse hematopoietic cells (Prisco et al., 2002). One putative NLS site stretches between amino acids 14–28 within the PH domain of the IRS-1, where 8 out of 15 amino acids are basic (Prisco et al., 2002), and the other (KKWRHK) proposed in the original paper of Keller et al. describing isolation of mouse IRS-1 cDNA, also resides within the PH domain (Keller et al., 1993). The presence of these sequences may explain translocation of IRS-1 to the nucleus following IGF-1-induced activation of IGF-IR (Tu et al., 2002), or insulin/IGF-2 - induced activation of IR, but only isoform A (IR-A) (Wu et al., 2003). Interestingly, IR-A lacks 12 amino acids from the C-terminus and is expressed preferentially in fetal and transformed tissues, where it is suspected to activate growth-promoting signals via IGF-2-mediated activation (Lawrence et al., 2007).
In addition to the described putative NLS sequences, PH domains of IRS-1 and IRS-2 interact with a nuclear acidic protein, nucleolin (Burks et al., 1998), which could imply that chaperon proteins are involved in the nuclear translocation of IRS proteins. Indeed, detection of nuclear IRS-1 in cells expressing JCV T-antigen (Lassak et al., 2002), SV40 T-antigen (Prisco et al., 2002), ERα (Morelli et al., 2004) and ERβ (Urbanska et al., 2009) indicates that IRS-1 may require other proteins equipped with NLS for its effective shuttling to the nucleus. The first characterization of the interaction between IRS-1 and JCV T-antigen, using a GST pull-down assay and a collection of the overlapping IRS-1 truncation mutants, revealed that this viral oncoprotein binds to the stretch of amino acids 212–300 within the N-terminal fragment of IRS-1 (Lassak et al., 2002), which includes a portion of PTB domain, and represents an overlap between two N-terminal fragments of IRS-1, which both are capable of pulling down JCV T-antigen. In addition, the IRS-1-JCV T-antigen interaction is independent from IRS-1 tyrosine phosphorylation and is strongly inhibited by IRS-1 serine phosphorylation (Lassak et al., 2002).
We have also identified that the region between amino acids 412–628 within the C-terminal portion of JCV T-antigen pulls down IRS-1 (Khalili et al., 2003). Although this region is far from the site where T-antigens interact with pRB proteins, it could overlap with the sites responsible for the p53 binding (Sullivan and Pipas, 2002). Therefore, in view of a potential role of nuclear IRS-1 in cellular transformation, it will be critically important to know if indeed IRS-1 interaction with T-antigens interferes with T-antigen –mediated p53 inactivation. Interestingly, Merkel cell polyomavirus, which is most firmly established as an etiological agent in Merkel cell carcinoma (Feng et al., 2008; Kassem et al., 2008; Viscidi and Shah, 2008), viral DNA is chromosomally integrated in the manner that T-antigen is disrupted. The resulting truncated T-antigen loses the ability to support viral replication, but it retains the ability of bind pRb (Harrison et al., 2011; Houben et al., 2011; Shuda et al., 2008). Further experiments are required to determine if such a truncated large T-antigen found in Merkel cell carcinoma can mediate IRS-1 nuclear translocation.
Two additional proteins with a strong nuclear affinity and with the ability of binding IRS-1 are estrogen receptor alpha (ERα) and estrogen receptor beta (ERβ). The first indication of a possible functional interplay between IRS-1 and ERα was demonstrated in the course of vitro breast cancer studies, which provide initial evidence that estrogens could regulate IRS-1 expression and stability (Morelli et al., 2003; Oesterreich et al., 2001). More recently, a direct nuclear binding between IRS-1 and ERα has been demonstrated (Morelli et al., 2004) and characterized (Sisci et al., 2007a) in breast cancer cells. In addition, molecular evaluation of the interaction revealed that ERα has two putative binding sites on the IRS-1 molecule. The first is located within the first 200 amino acids spanning entire PH and part of PTB domain, and the second is located within the C-terminal portion of IRS-1 (Sisci et al., 2007a).
In medulloblastomas, which develop in the cerebellum, and are considered the most common intracranial tumors of the childhood (Reiss, 2002), nuclear IRS-1 was found first in association with JCV T-antigen (Lassak et al., 2002), and later with ERβ (Urbanska et al., 2009). Our recent studies demonstrate that ERβ-IRS-1 interaction involves exclusively the C-terminal domain of IRS-1 between amino acids 931 and 1233 (Urbanska et al., 2009). However, in contrast to ERα-IRS-1 complex, we did not observe ERβ binding to the N-terminal portion of IRS-1, which on the other hand, represents the prominent binding site for JCV T-antigen (Lassak et al., 2002).
Nuclear Localization of IRS-2 and IRS-3
IRS-3 is a shorter version of IRS-1, and is expressed in rodents but not in humans (Bjornholm et al., 2002). The nuclear presence of IRS-3 was demonstrated in the Takahashi lab (Kabuta et al., 2002) about the same time when nuclear IRS-1 was discovered (Lassak et al., 2002). The region of IRS-3 necessary for its nuclear localization was cloned between amino acids 192 and 223, the region that represents the fragment of the PTB domain that is unique for IRS-3 (Kabuta et al., 2002). In the same experimental setting the authors did not observe nuclear IRS-1, IRS-2 or IRS-4 despite the fact that COS-7 cells used in this study are expected to express SV40 T-antigen. Recent analysis of nuclear IRS-3, by the same group, revealed that importin-β binds IRS-3, which was sufficient for nuclear translocation of the complex (Kabuta et al., 2008). Since IRS-1 and IRS-2 failed to bind importin-β, the IRS-3 - importin-β interaction seems to represent a unique mechanism of nuclear translocation within the IRS family.
IRS-2 can also translocate to the nucleus. IRS-1 was found in the nuclei of mouse embryo fibroblasts stimulated with IGF-I, however in contrast to IRS-1, IRS-2 translocation was not mediated by SV40 T-antigen, and required instead the presence of a functionally active IGF-IR (Sun et al., 2003). Inactivating mutations in the tyrosine kinase domain of the IGF-IR prevented nuclear translocation of IRS-2, which implicated tyrosine kinase activity of the IGF-IR in this process (Sun et al., 2003). In addition, nuclear IRS-2 has been shown to form nucleolar complexes with the upstream binding factor (UBF-1), a key regulator of RNA polymerase I activity involved in biosynthesis of ribosomal RNAs (Sun et al., 2003). In a different study, nuclear IRS-2 was found in association with NFκB in breast cancer cells, which supported the recruitment of IRS-2 to the cyclin D1 promoter (Wu et al., 2010). Interestingly, antisense against IRS-2 inhibited IGF-I-induced PI3K and NFkB activities and repressed proliferation of both BT20 (IRS-1 negative) and MCF-7 (IRS-1 positive) breast cancer cells (Wu et al., 2010). Since MCF-7 cells have been previously shown to possess nuclear IRS-1 in association with ERα, and nuclear IRS-1 also binds cyclin D1 promoter (Chen et al., 2005), further experiments are required to clarify a possible distinct role for nuclear IRS-1 and nuclear IRS-2 in this cellular model. In this respect, a different role for IRS-1 and IRS-2 has been already proposed by Nagle et al. (Nagle et al., 2004) who demonstrated that breast cancer cells which are IRS-1 negative but express IRS-2 are highly metastatic, however, their invasive potential is lost when IRS-2 is downregulated. In a different scenario, reintroduction of IRS-1 to IRS-1-negative prostate cancer cells, LNCaP, triggered cell aggregation and inhibited their invasiveness (Reiss et al., 2000; Reiss et al., 2001). Therefore, at least in breast and prostate cancer cells, the expression of IRS-2 seems to associate with higher, and IRS-1 with lower tumor cell invasiveness. Further studies are required to determine if nuclear translocation of IRS-1 and/or IRS-2 can contribute to the process of malignant transformation in general, and to tumor invasiveness in particular.
Nuclear IRS-1 and DNA Repair
Nuclear IRS-1 was initially detected in association with polyomavirus JC (Lassak et al., 2002; Reiss et al., 2006) and SV40 T-antigens (Tu et al., 2002), which are viral oncoproteins known to trigger abnormal cell proliferation and cause genomic instability. Polyomaviruses, including human JCV and BKV, and their simian counterpart, SV40, are small non-enveloped viruses with a single copy of double-stranded DNA. Their oncogenic potential is closely associated with the activation of so-called “early genome” of the virus, which forces infected cells to re-enter the S phase of the cell cycle, providing cellular DNA replication machinery for viral replication. The early genome of SV40 and JCV transcribes a common precursor RNA, which is differentially spliced yielding several viral products among which large tumor antigen (T-antigen), and small tumor antigen (t-antigen), predominate (Corallini et al., 1987; Khalili et al., 1999; Khalili and Stoner, 2001; Reiss and Khalili, 2003; Reiss Krzysztof, 2010; Wang et al., 2004; White et al., 2005). Polyomaviruses infect humans, monkeys, rodents, and birds with a restricted host and tissue specificity, and the infection of cells in which the virus does not fully replicate may lead to a partial expression of the viral genome (Imperiale, 2000; Imperiale, 2001). When SV40 or JCV T-antigen is expressed in the cells, it binds and inactivates two major negative regulators of the cell cycle, p53 and pRb (Kao et al., 1993; Krynska et al., 1997; Saenz-Robles et al., 2001). Although these canonical interactions with host proteins initiate DNA replication, they do not explain why cells expressing T-antigen are often characterized by genomic instability and undergo malignant transformation. Multiple studies have already demonstrated chromosomal instability with no consistent patterns and with many new karyotypes emerging at each consecutive passage of the T-antigen expressing cells (Hunter and Gurney, 1994; Kappler et al., 1999; Ramel et al., 1995; Ricciardiello et al., 2003; Woods et al., 1994). A large variety of chromosomal defects suggest that T-antigens may affect stability of the genome at a very basic level. For instance, one explanation could be unfaithful DNA repair of double strand breaks (DSBs) in cells that are actively replicating DNA. To ensure uninterrupted DNA replication and to avoid apoptosis, at least one of the two prominent DNA repair mechanisms: homologous recombination directed DNA repair (HRR) or non-homologous end joining (NHEJ), has to be activated (Hoeijmakers, 2001).
We have demonstrated that cells expressing JCV T-antigen are characterized by impaired HRR, which resulted in the accumulation of mutations in cells replicating DNA (Trojanek et al., 2006a; Trojanek et al., 2006b). In this process, JCV T-antigen did not interact directly with the HRR complex, but instead it utilized IRS-1 (Fig.2). Following T-antigen-mediated nuclear translocation (Lassak et al., 2002), IRS-1 has been found in complex with Rad51, which is the main enzymatic component of HRR (Thacker, 1999). Importantly, this T-antigen-induced inhibition of HRR did not function in cells lacking IRS-1, and was reproduced in the absence of T-antigen by the mutant IRS-1 equipped with an artificial nuclear localization signal. As a result of this interaction between nuclear IRS-1 and Rad51 HRR was significantly repressed, however, enzymatically-induced DNA strand breaks were still repaired most likely by NHEJ (Trojanek et al., 2006a). This compensatory action of NHEJ was however associated with the accumulation of spontaneous mutations detected at the sites of damaged DNA (Trojanek et al., 2006a) (Fig.2). Other examples of T-antigen-mediated interference with DNA repair include SV40 T-antigen interference with MRE11 nuclear foci formation (Digweed et al., 2002), and T-antigen binding to another DNA repair protein, Nbs1, which forms an early DNA repair complex with MRE11 and Rad50 (Wu et al., 2004). In these studies however the involvement of nuclear IRS-1 was not evaluated.
Figure 2. The IGF-IR-IRS-1-JCV T-antigen signaling interplay: effects on cell proliferation cell survival and DNA repair fidelity.
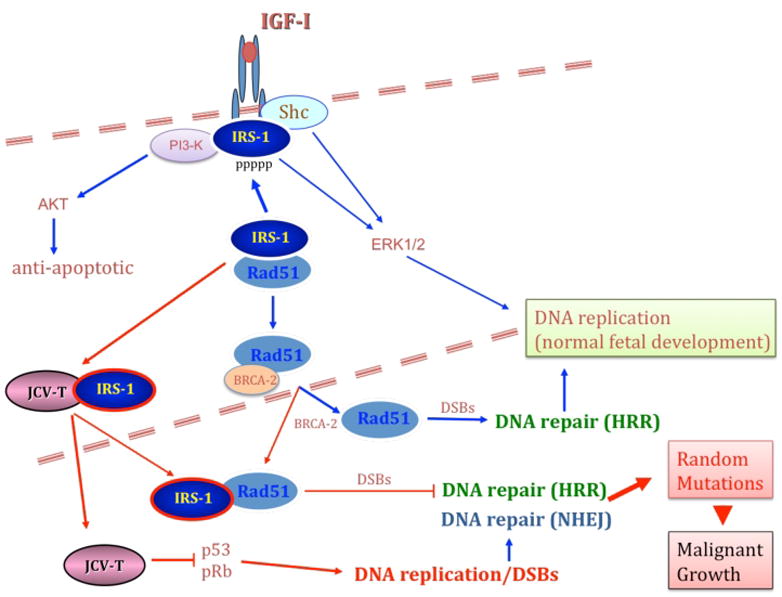
Here we propose a sequence of events in which ligand activated IGF-IR triggers multiple signaling responses leading to synchronized activation of cell proliferation (SHC or IRS-1-mediated activation of Ras-MAP kinase pathways); protection from apoptosis (IRS-1 induced activation of Akt); and DNA repair by homologous recombination. The IGF-I-mediated phosphorylation of IRS-1 seems to play a critical role in this model. In the absence of IGF-I a fraction of hypo-phosphorylated IRS-1 accumulates in the perinuclear region in complex with Rad51 (Trojanek et al., 2003). Following IGF-I stimulation, activated IGF-IR phosphorylates IRS-1 on multiple tyrosine residues, decreasing the affinity of IRS-1 to Rad51, and engages IRS-1 in multiple signaling events supporting IGF-I-induced cell proliferation and cell survival (Reiss et al., 1998; Trojanek et al., 2006b; Trojanek et al., 2003). If at the same time DNA double strands (DSBs) are formed (either naturally or by genotoxic treatment), the cell can repair them in a faithful manner, by Rad51-supported homologous recombination directed DNA repair (HRR), or less faithfully, by non-homologous end joining (NHEJ). In the presence of JCV T-antigen cells can proliferation because of p53, pRb inactivation. In parallel, JCV T-antigen translocates IRS-1 to the nucleus (Lassak et al., 2002), thus creating a condition in which IRS-1 can bind Rad51 in the subcellular compartment in which Rad51 is expected to support HRR. Therefore, if JCV T-antigen expressing cells experience extensive DNA damage, the resulting DNA double strand breaks (DSBs) can either trigger apoptosis, or if NHEJ will compensate for the impaired HRR, spontaneous mutations can accumulate in the surviving cells. These mutations when accumulate may provide the cells with growth and survival advantage, which could result in the selection of clone/s initiating tumor development and progression.
Recently, we have reported the presence of nuclear IRS-1 in medulloblastoma cells negative for JCV T-antigen in which nuclear IRS-1 was found in complex with ERβ (Urbanska et al., 2009) and (Fig.3). Following cisplatin-induced DNA damage, nuclear IRS-1 localized at the sites of damaged DNA where it interacted again with Rad51. In medulloblastoma cells, engineered to express reporter plasmid for homologous recombination (Pierce et al., 1999; Trojanek et al., 2003), the ER antagonist, ICI 182,780, and an IRS-1 mutant (931–1233) lacking the ERβ binding site, both decreased the content of nuclear IRS-1 and stimulated DNA repair by homologous recombination (Urbanska et al., 2009). These encouraging results brought however one unexpected side effect, which should be seriously considered in view of estrogen receptor-related anticancer strategies. Although our further experiments with ICI182,780 confirmed the expected decrease in the accumulation of nuclear IRS-1, the accompanied increase in DNA repair by HRR resulted in the development of cisplatin resistance in medulloblastoma cells. This could be clinically relevant since the use of ICI182,780 has already been suggested as a supplementary treatment for medulloblastoma (Belcher et al., 2009).
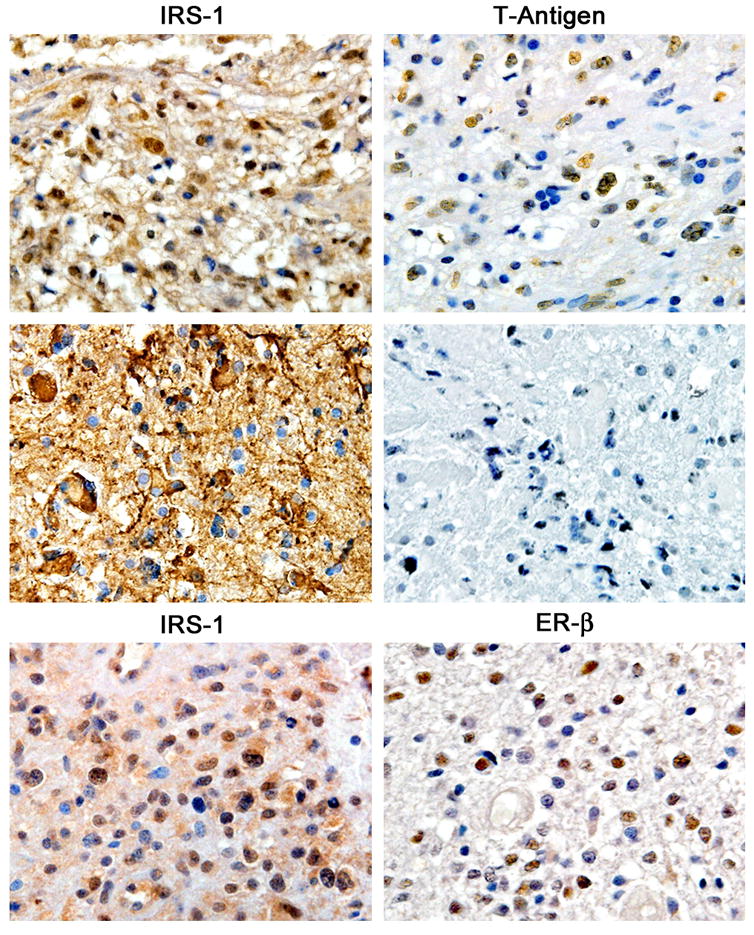
Figure 3. Nuclear translocation of IRS-1 in Medulloblastomas.

Immunohistochemistry for the IGF-IR/IR docking molecule, IRS-1, shows a prominent nuclear localization in clinical samples of medulloblastomas that express the JCV early oncoprotein, large T-antigen (upper panels). In T-antigen negative samples, IRS-1 remains in the cytoplasm of neoplastic cells (middle panels). In some tumors that lack T-antigen expression IRS-1 is still located to the nucleus in association with Estrogen Receptor beta (lower panels). Original magnification for all panels 600x.
Nuclear IRS-1 and Cell Growth
The involvement of the IGF-IR-IRS-1 signaling axis in controlling cell size has been well documented in a wide variety of organisms including Drosophila (Bohni et al., 1999) and mice (Pete et al., 1999), and was confirmed in cell culture (Valentinis et al., 2000). For example, transgenic mice knockouts for the IGF-IR gene (Baker et al., 1993; Gualco et al., 2009a), or IRS-1 knockouts (Araki et al., 1994) are characterized by severe growth retardation. In a similar manner, deletion of a Drosophila homolog of IRS-1, a protein referred as Chico, decreased size of the fly about 50% by affecting both cell size and cell number (Bohni et al., 1999). Conversely, targeted disruption of the IGF-2 receptor (IGF-2R), which is thought to sequester IGF-2 during fetal development, increased overall IGF-2 availability, which led to over-stimulation of the IGF-IR/IRS-1 signaling axis, and resulted in the development of abnormally large embryos (Ludwig et al., 1996). Despite of these multiple examples, we still do not fully understand how IGF-I or insulin-activated IRS proteins contribute to the increase in size. One explanation has been provided by the observation that nuclear/nucleolar IRS-1 and IRS-2 interact with the upstream binding factor 1 (UBF-1), which is a key regulator of RNA polymerase-I involved in biosynthesis of ribosomal RNA (rRNA) (Drakas et al., 2004; Tu et al., 2002). There are several important observations, which emerged from this finding: (i) IGF-1 stimulation induces phosphorylation of the C-terminal portion of UBF, and the presence of IRS-1 increases markedly this phosphorylation; (ii) antibody against IRS-1 precipitates UBF-1 exclusively from the nuclear fraction; (iii) both IRS-1 and IRS-2 are capable of binding UBF-1 (ref); (iv) nuclear translocation of IRS-1 in 32D cells correlated well with a marked increase in rRNA synthesis; (v) in the nucleus, IRS-1 co-precipitated with PI3-K; and (vi) IRS-1 bound PI-3K directly phosphorylated UBF-1. Collectively, these data strongly indicate that the IGF-I-mediated determination of cell size depends, at least partially, on the activation of PI3-K by nuclear IRS-1, which results in the phosphorylation-dependent activation of UBF-1 and subsequent activation of rRNA synthesis. Since the increase in cell size is also important during cell proliferation when cells must double in size between the G1 and G2 phase of the cell cycle, this could represent a new function for nuclear IRS-1 in supporting abnormal cell proliferation.
Nuclear IRS-1 and Gene Expression
Although a direct interaction between nuclear IRS-1 and double stranded DNA has not been reported, several recent publications suggest that nuclear IRS-1 could participate in modulating transcriptional activity of genes involved in cell growth and cell proliferation (Chen et al., 2005; Wu et al., 2008). The first study suggesting nuclear IRS-1 as a transcriptional modulator demonstrated its’ interaction with the upstream binding factor 1 (UBF-1), a key regulator of RNA polymerase-I involved in biosynthesis of rRNA (Tu et al., 2002). Importantly, the IRS-1/UBF-1 complex localizes preferentially in the nucleolus, which is known to contain multiple tandem copies of the ribosomal DNA (rDNA). Importantly, the UBF-1/IRS-1 nucleolar complex activated the expression from rDNA promoters, which resulted in elevated rRNA biosynthesis, leading to overall increase in protein synthesis (Chen et al., 2005; Drakas et al., 2004; Wu et al., 2005). Another example of nuclear IRS-1 working as a transcriptional modulator is its cytosolic and nuclear binding to β-catenin (Chen et al., 2005). The IRS-1/β-catenin complex has been found to interact with the c-myc and cyclin D1 promoters, and the binding was associated with elevated transcriptional activity from these two growth-control genes (Chen et al., 2005). IRS-1 has been also found to form complexes with ERα (Morelli et al., 2004) and with androgen receptor (AR) (Lanzino et al., 2009). Both complexes translocate to the nucleus, and have been shown to interact with the promoter regions containing ER and AR responsive elements, respectively. In case of ERα the binding of IRS-1 was associated with a significant decrease in transcriptional activity (Morelli et al., 2004), and the IRS-1/AR complex was found to be stimulatory (Lanzino et al., 2009). Collectively, these findings suggest a new role for nuclear IRS-1 in IGF-IR/IRS-1 signaling axis, which in addition to its canonical signaling effects on cell growth and cell proliferation, propose nuclear IRS-1 as a transcriptional modulator. In particular, the findings that nuclear IRS-1 can function as AR and ER transcriptional modulator could indicate its involvement in the development and progression of breast and prostate cancer.
Nuclear IRS-1 in Cancer Clinical Samples
Nuclear IRS-1 was detected for the first time in archival clinical samples of medulloblastoma (Lassak et al., 2002). The original study of 17 medulloblastoma biopsies revealed the presence of IRS-1 in the cytoplasm of 6 cases, while another 6 cases showed both cytosolic and prominently nuclear IRS-1. Interestingly, all cases of cytoplasmic IRS-1 were negative for T-antigen expression, while all cases in which IRS-1 was located in the nucleus were T-antigen positive. Furthermore, T-antigen and IRS-1 seem to colocalize the nuclei of tumor cells (Lassak et al., 2002). Subsequent immunohistochemical studies performed on an additional 20 cases of medulloblastoma, corroborated the nuclear localization of IRS-1 in 3 T-antigen positive cases, however 6 samples exhibiting nuclear IRS-1 were T-antigen negative. In these 8 samples, the nuclear IRS-1 was found in complex with another nuclear protein, estrogen receptor beta (ERβ), which was later confirmed in vitro (Urbanska et al., 2009) and (Fig. 3). Since nuclear IRS-1 was detected in classic, neuroblastic, and desmoplastic medulloblastomas, which differ in respect to their invasiveness and behavior (Kim et al., 2011; Rossi et al., 2008), we were not able to assign any prognostic values associated with nuclear IRS-1, ERβ or with JCV T-antigen (Lassak et al., 2002; Urbanska et al., 2009). In addition, to medulloblastoma clinical samples our preliminary observations from Glioblastomas also indicate the presence of nuclear IRS- in association with JCV T-antigen and/or with ERβ, (Fig. 4). However, more intensive studies are required to determine if nuclear IRS-1 could have any prognostic or diagnostic values for these malignant Glial tumors.
Figure 4. Nuclear translocation of IRS-1 in Glioblastomas.
In a similar patter of expression, IRS-1 is detected by immunohistochemistry in the cytoplasm and nuclei of neoplastic cells in cases of Glioblastoma multiforme that are positive for JCV T-Antigen (upper panels). However, tumors that lack T-antigen demonstrate exclusive cytoplasmic IRS-1 (middle panels). Finally, IRS-1 can be translocated to the nucleus in the presence of Estrogen Receptor beta (lower panels). All panels original magnification 600x.
In contrast to observations made with medulloblastoma, nuclear IRS-1 detected in breast cancer biopsies correlated well with more differentiated and less metastatic phenotype (Sisci et al., 2007b). In the breast cancer cells and in biopsies nuclear IRS-1 was found in association with ERα (Morelli et al., 2003; Morelli et al., 2004; Sisci et al., 2007a; Sisci et al., 2007b). Nuclear IRS-1 was detected in 1.6% of control normal mammary epithelium and in 20% of benign tumors. In ductal carcinoma, both nuclear IRS-1 and ERα negatively correlated with tumor grade, size, mitotic index and lymph node involvement (Sisci et al., 2007b). Recently, nuclear IRS-1 has been shown to be a good predictor for tamoxifen-response in patients with early breast cancer (Migliaccio et al., 2010). In this study, tissue array from over thousand patients diagnosed with stage 1 and 2 breast cancer reviled positive correlation between nuclear IRS-1 and ERα, and between nuclear IRS-1 and progesterone receptor, and nuclear IRS-1 per se showed negative correlation with lymph node involvement. On the other hand, cytosolic IRS-1 did not correlate with ERα, but showed positive correlation with tumor size and S-phase fraction. Importantly, tamoxifen-treated patients with the tumor cells showing nuclear IRS-1 had both better recurrence–free survival and overall survival (Migliaccio et al., 2010).
Conclusions
In this review, we have presented the experimental work from multiple laboratories, which indicate that IRS proteins, in addition to their canonical function as cytosolic signal transduction molecules, can be shuttled to the nucleus, and that nuclear presence of IRS-1, may contribute to the process of malignant transformation. As illustrated in Fig.5, several nuclear proteins have been implicated in the process of IRS-1 translocation. They include: JC virus T-antigen; SV40 T-antigen; v-Src; Estrogen receptor α; and Estrogen receptor β. A different mechanism has been also proposed, which involves nuclear acidic protein, nucleolin, ant its’ binding to the PH domain of IRS-1 and IRS-2. Also within the PH-domain of IRS-1 there are two putative nuclear localization signals (NLS), which possibly contributed to nuclear translocation of IRS-1 in cells stimulated with IGF-I. Finally, nuclear translocation of IRS-3 was mediated by importin-β, which has been shown binding to the unique sequence found exclusively on the PTB domain of IRS-3.
Figure 5.
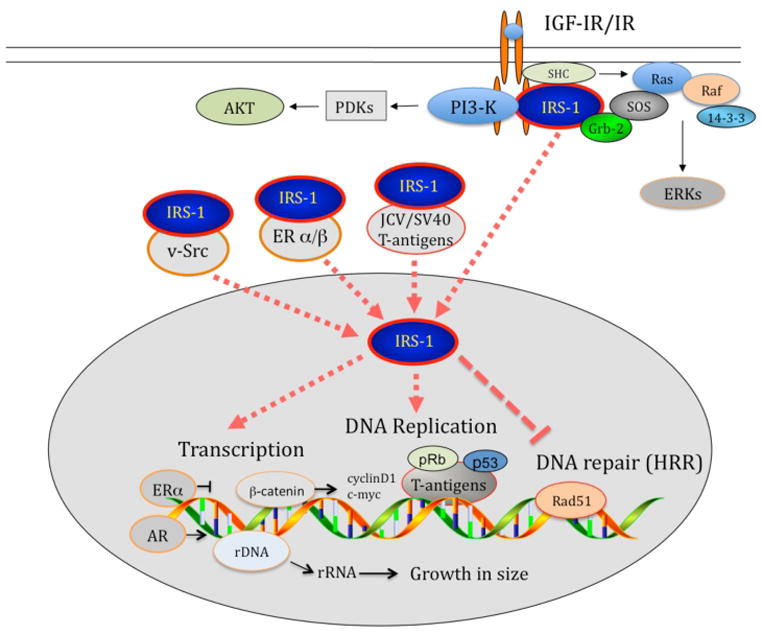
Schematic illustration summarizing mechanisms involved in IRS-1 nuclear translocation and basic cellular processes affected by nuclear IRS-1.
In the nucleus, IRS-1 affects several basic control mechanisms, which when dysregulated, may support malignant transformation (Fig.5). In particular, nuclear IRS-1 was found to inhibited homologous recombination directed DNA repair (HRR) via its direct binding with Rad51, which resulted in the accumulation of spontaneous mutations. Another potential cancer related function of nuclear IRS-1 is its’ association with UBF1, which activated ribosomal RNA biosynthesis, stimulated overall protein synthesis, and contributed to the increase in cell size during G1 – G2/M progression. Also by modulating gene transcription, nuclear IRS-1 stimulated cyclin D1 and c-myc promoter activities, in this case however nuclear IRS-1 was found in complex with β-catenin. Collectively, presented data indicate that nuclear IRS-1, by limiting DNA repair fidelity and by forcing cell growth and DNA replication, could link this signaling molecule to malignant transformation.
Acknowledgments
We would like to thank Mel McGuire for her editorial help. This work was supported by grant from NIH awarded to KR (RO1CA095518)
References
- Araki E, Lipes MA, Patti ME, Bruning JC, Haag B, 3rd, Johnson RS, Kahn CR. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372(6502):186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75(1):73–82. [PubMed] [Google Scholar]
- Beitner-Johnson D, Blakesley VA, Shen-Orr Z, Jimenez M, Stannard B, Wang LM, Pierce J, LeRoith D. The proto-oncogene product c-Crk associates with insulin receptor substrate-1 and 4PS. Modulation by insulin growth factor-I (IGF) and enhanced IGF-I signaling. J Biol Chem. 1996;271(16):9287–9290. doi: 10.1074/jbc.271.16.9287. [DOI] [PubMed] [Google Scholar]
- Belcher SM, Ma X, Le HH. Blockade of estrogen receptor signaling inhibits growth and migration of medulloblastoma. Endocrinology. 2009;150(3):1112–1121. doi: 10.1210/en.2008-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornholm M, He AR, Attersand A, Lake S, Liu SC, Lienhard GE, Taylor S, Arner P, Zierath JR. Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia. 2002;45(12):1697–1702. doi: 10.1007/s00125-002-0945-z. [DOI] [PubMed] [Google Scholar]
- Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen E. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1–4. Cell. 1999;97(7):865–875. doi: 10.1016/s0092-8674(00)80799-0. [DOI] [PubMed] [Google Scholar]
- Burks DJ, Pons S, Towery H, Smith-Hall J, Myers MG, Jr, Yenush L, White MF. Heterologous pleckstrin homology domains do not couple IRS-1 to the insulin receptor. J Biol Chem. 1997;272(44):27716–27721. doi: 10.1074/jbc.272.44.27716. [DOI] [PubMed] [Google Scholar]
- Burks DJ, Wang J, Towery H, Ishibashi O, Lowe D, Riedel H, White MF. IRS pleckstrin homology domains bind to acidic motifs in proteins. J Biol Chem. 1998;273(47):31061–31067. doi: 10.1074/jbc.273.47.31061. [DOI] [PubMed] [Google Scholar]
- Chen J, Wu A, Sun H, Drakas R, Garofalo C, Cascio S, Surmacz E, Baserga R. Functional significance of type 1 insulin-like growth factor-mediated nuclear translocation of the insulin receptor substrate-1 and beta-catenin. J Biol Chem. 2005;280(33):29912–29920. doi: 10.1074/jbc.M504516200. [DOI] [PubMed] [Google Scholar]
- Corallini A, Pagnani M, Viadana P, Silini E, Mottes M, Milanesi G, Gerna G, Vettor R, Trapella G, Silvani V, et al. Association of BK virus with human brain tumors and tumors of pancreatic islets. International journal of cancer. 1987;39(1):60–67. doi: 10.1002/ijc.2910390111. [DOI] [PubMed] [Google Scholar]
- Craparo A, O’Neill TJ, Gustafson TA. Non-SH2 domains within insulin receptor substrate-1 and SHC mediate their phosphotyrosine-dependent interaction with the NPEY motif of the insulin-like growth factor I receptor. J Biol Chem. 1995;270(26):15639–15643. doi: 10.1074/jbc.270.26.15639. [DOI] [PubMed] [Google Scholar]
- Digweed M, Demuth I, Rothe S, Scholz R, Jordan A, Grotzinger C, Schindler D, Grompe M, Sperling K. SV40 large T-antigen disturbs the formation of nuclear DNA-repair foci containing MRE11. Oncogene. 2002;21(32):4873–4878. doi: 10.1038/sj.onc.1205616. [DOI] [PubMed] [Google Scholar]
- Drakas R, Tu X, Baserga R. Control of cell size through phosphorylation of upstream binding factor 1 by nuclear phosphatidylinositol 3-kinase. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(25):9272–9276. doi: 10.1073/pnas.0403328101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eck MJ, Dhe-Paganon S, Trub T, Nolte RT, Shoelson SE. Structure of the IRS-1 PTB domain bound to the juxtamembrane region of the insulin receptor. Cell. 1996;85(5):695–705. doi: 10.1016/s0092-8674(00)81236-2. [DOI] [PubMed] [Google Scholar]
- Fei ZL, D’Ambrosio C, Li S, Surmacz E, Baserga R. Association of insulin receptor substrate 1 with simian virus 40 large T antigen. Mol Cell Biol. 1995;15(8):4232–4239. doi: 10.1128/mcb.15.8.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualco E, Urbanska K, Perez-Liz G, Sweet T, Peruzzi F, Reiss K, Del Valle L. IGF-IR dependent expression of Survivin is required for T-Antigen mediated Protection from Apoptosis and Proliferation of Neural Progenitors. Cell Growth & Differ. 2009a doi: 10.1038/cdd.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gualco E, Wang JY, Del Valle L, Urbanska K, Peruzzi F, Khalili K, Amini S, Reiss K. IGF-IR in neuroprotection and brain tumors. Front Biosci. 2009b;14:352–375. doi: 10.2741/3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison CJ, Meinke G, Kwun HJ, Rogalin H, Phelan PJ, Bullock PA, Chang Y, Moore PS, Bohm A. Asymmetric assembly of Merkel cell polyomavirus large T-antigen origin binding domains at the viral origin. J Mol Biol. 2011;409(4):529–542. doi: 10.1016/j.jmb.2011.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271(5249):665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- Houben R, Adam C, Baeurle A, Hesbacher S, Grimm J, Angermeyer S, Henzel K, Hauser S, Elling R, Brocker EB, Gaubatz S, Becker JC, Schrama D. An intact retinoblastoma protein-binding site in Merkel cell polyomavirus large T antigen is required for promoting growth of Merkel cell carcinoma cells. International journal of cancer. 2011 doi: 10.1002/ijc.26076. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Gurney EG. The genomic instability associated with integrated simian virus 40 DNA is dependent on the origin of replication and early control region. J Virol. 1994;68(2):787–796. doi: 10.1128/jvi.68.2.787-796.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperiale MJ. The human polyomaviruses, BKV and JCV: molecular pathogenesis of acute disease and potential role in cancer. Virology. 2000;267(1):1–7. doi: 10.1006/viro.1999.0092. [DOI] [PubMed] [Google Scholar]
- Imperiale MJ. In: The Human Polyoviruses: an Overview. Khalili K, Stoner GI, editors. Wiley-Liss; 2001. [Google Scholar]
- Kabuta T, Hakuno F, Asano T, Takahashi S. Insulin receptor substrate-3 functions as transcriptional activator in the nucleus. J Biol Chem. 2002;277(9):6846–6851. doi: 10.1074/jbc.M107058200. [DOI] [PubMed] [Google Scholar]
- Kabuta T, Take K, Kabuta C, Hakuno F, Takahashi S. Differential subcellular localization of insulin receptor substrates depends on C-terminal regions and importin beta. Biochem Biophys Res Commun. 2008;377(3):741–746. doi: 10.1016/j.bbrc.2008.09.106. [DOI] [PubMed] [Google Scholar]
- Kao C, Huang J, Wu SQ, Hauser P, Reznikoff CA. Role of SV40 T antigen binding to pRB and p53 in multistep transformation in vitro of human uroepithelial cells. Carcinogenesis. 1993;14(11):2297–2302. doi: 10.1093/carcin/14.11.2297. [DOI] [PubMed] [Google Scholar]
- Kappler R, Pietsch T, Weggen S, Wiestler OD, Scherthan H. Chromosomal imbalances and DNA amplifications in SV40 large T antigen-induced primitive neuroectodermal tumor cell lines of the rat. Carcinogenesis. 1999;20(8):1433–1438. doi: 10.1093/carcin/20.8.1433. [DOI] [PubMed] [Google Scholar]
- Kassem A, Schopflin A, Diaz C, Weyers W, Stickeler E, Werner M, Zur Hausen A. Frequent detection of Merkel cell polyomavirus in human Merkel cell carcinomas and identification of a unique deletion in the VP1 gene. Cancer Res. 2008;68(13):5009–5013. doi: 10.1158/0008-5472.CAN-08-0949. [DOI] [PubMed] [Google Scholar]
- Keller SR, Aebersold R, Garner CW, Lienhard GE. The insulin-elicited 160 kDa phosphotyrosine protein in mouse adipocytes is an insulin receptor substrate 1: identification by cloning. Biochim Biophys Acta. 1993;1172(3):323–326. doi: 10.1016/0167-4781(93)90222-y. [DOI] [PubMed] [Google Scholar]
- Khalili K, Del Valle L, Wang JY, Darbinian N, Lassak A, Safak M, Reiss K. T-antigen of human polyomavirus JC cooperates withIGF-IR signaling system in cerebellar tumors of the childhood-medulloblastomas. Anticancer Res. 2003;23(3A):2035–2041. [PubMed] [Google Scholar]
- Khalili K, Krynska B, Del Valle L, Katsetos CD, Croul S. Medulloblastomas and the human neurotropic polyomavirus JC virus. Lancet. 1999;353(9159):1152–1153. doi: 10.1016/s0140-6736(99)00357-8. [DOI] [PubMed] [Google Scholar]
- Khalili K, Stoner GI. Human Polyomaviruses: molecular and clinical perspectives. Wiley-Liss, Inc; 2001. [Google Scholar]
- Kim W, Choy W, Dye J, Nagasawa D, Safaee M, Fong B, Yang I. The tumor biology and molecular characteristics of medulloblastoma identifying prognostic factors associated with survival outcomes and prognosis. J Clin Neurosci. 2011;18(7):886–890. doi: 10.1016/j.jocn.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Krynska B, Gordon J, Otte J, Franks R, Knobler R, DeLuca A, Giordano A, Khalili K. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J Cell Biochem. 1997;67(2):223–230. doi: 10.1002/(sici)1097-4644(19971101)67:2<223::aid-jcb7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Lanzino M, Garofalo C, Morelli C, Le Pera M, Casaburi I, McPhaul MJ, Surmacz E, Ando S, Sisci D. Insulin receptor substrate 1 modulates the transcriptional activity and the stability of androgen receptor in breast cancer cells. Breast Cancer Res Treat. 2009;115(2):297–306. doi: 10.1007/s10549-008-0079-1. [DOI] [PubMed] [Google Scholar]
- Lassak A, Del Valle L, Peruzzi F, Wang JY, Enam S, Croul S, Khalili K, Reiss K. Insulin Receptor Substrate 1 Translocation to the Nucleus by the Human JC Virus T-antigen. J Biol Chem. 2002;277(19):17231–17238. doi: 10.1074/jbc.M110885200. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, McKern NM, Ward CW. Insulin receptor structure and its implications for the IGF-1 receptor. Curr Opin Struct Biol. 2007;17(6):699–705. doi: 10.1016/j.sbi.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Lee CH, Li W, Nishimura R, Zhou M, Batzer AG, Myers MG, Jr, White MF, Schlessinger J, Skolnik EY. Nck associates with the SH2 domain-docking protein IRS-1 in insulin-stimulated cells. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(24):11713–11717. doi: 10.1073/pnas.90.24.11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig T, Eggenschwiler J, Fisher P, D’Ercole AJ, Davenport ML, Efstratiadis A. Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in Igf2 and Igf1r null backgrounds. Dev Biol. 1996;177(2):517–535. doi: 10.1006/dbio.1996.0182. [DOI] [PubMed] [Google Scholar]
- Migliaccio I, Wu MF, Gutierrez C, Malorni L, Mohsin SK, Allred DC, Hilsenbeck SG, Osborne CK, Weiss H, Lee AV. Nuclear IRS-1 predicts tamoxifen response in patients with early breast cancer. Breast Cancer Res Treat. 2010;123(3):651–660. doi: 10.1007/s10549-009-0632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli C, Garofalo C, Bartucci M, Surmacz E. Estrogen receptor-alpha regulates the degradation of insulin receptor substrates 1 and 2 in breast cancer cells. Oncogene. 2003;22(26):4007–4016. doi: 10.1038/sj.onc.1206436. [DOI] [PubMed] [Google Scholar]
- Morelli C, Garofalo C, Sisci D, del Rincon S, Cascio S, Tu X, Vecchione A, Sauter ER, Miller WH, Jr, Surmacz E. Nuclear insulin receptor substrate 1 interacts with estrogen receptor alpha at ERE promoters. Oncogene. 2004;23(45):7517–7526. doi: 10.1038/sj.onc.1208014. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Backer JM, Sun XJ, Shoelson S, Hu P, Schlessinger J, Yoakim M, Schaffhausen B, White MF. IRS-1 activates phosphatidylinositol 3′-kinase by associating with src homology 2 domains of p85. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(21):10350–10354. doi: 10.1073/pnas.89.21.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr, Grammer TC, Brooks J, Glasheen EM, Wang LM, Sun XJ, Blenis J, Pierce JH, White MF. The pleckstrin homology domain in insulin receptor substrate-1 sensitizes insulin signaling. J Biol Chem. 1995;270(20):11715–11718. doi: 10.1074/jbc.270.20.11715. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Sun XJ, Cheatham B, Jachna BR, Glasheen EM, Backer JM, White MF. IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3′-kinase. Endocrinology. 1993;132(4):1421–1430. doi: 10.1210/endo.132.4.8384986. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Wang LM, Sun XJ, Zhang Y, Yenush L, Schlessinger J, Pierce JH, White MF. Role of IRS-1-GRB-2 complexes in insulin signaling. Mol Cell Biol. 1994;14(6):3577–3587. doi: 10.1128/mcb.14.6.3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr, White MF. Insulin signal transduction and the IRS proteins. Annu Rev Pharmacol Toxicol. 1996;36:615–658. doi: 10.1146/annurev.pa.36.040196.003151. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Zhang Y, Aldaz GA, Grammer T, Glasheen EM, Yenush L, Wang LM, Sun XJ, Blenis J, Pierce JH, White MF. YMXM motifs and signaling by an insulin receptor substrate 1 molecule without tyrosine phosphorylation sites. Mol Cell Biol. 1996;16(8):4147–4155. doi: 10.1128/mcb.16.8.4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle JA, Ma Z, Byrne MA, White MF, Shaw LM. Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol Cell Biol. 2004;24(22):9726–9735. doi: 10.1128/MCB.24.22.9726-9735.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterreich S, Zhang P, Guler RL, Sun X, Curran EM, Welshons WV, Osborne CK, Lee AV. Re-expression of estrogen receptor alpha in estrogen receptor alpha-negative MCF-7 cells restores both estrogen and insulin-like growth factor-mediated signaling and growth. Cancer Res. 2001;61(15):5771–5777. [PubMed] [Google Scholar]
- Ogihara T, Isobe T, Ichimura T, Taoka M, Funaki M, Sakoda H, Onishi Y, Inukai K, Anai M, Fukushima Y, Kikuchi M, Yazaki Y, Oka Y, Asano T. 14-3-3 protein binds to insulin receptor substrate-1, one of the binding sites of which is in the phosphotyrosine binding domain. J Biol Chem. 1997;272(40):25267–25274. doi: 10.1074/jbc.272.40.25267. [DOI] [PubMed] [Google Scholar]
- Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem. 1996;271(22):13018–13022. doi: 10.1074/jbc.271.22.13018. [DOI] [PubMed] [Google Scholar]
- Pete G, Fuller CR, Oldham JM, Smith DR, D’Ercole AJ, Kahn CR, Lund PK. Postnatal growth responses to insulin-like growth factor I in insulin receptor substrate-1-deficient mice. Endocrinology. 1999;140(12):5478–5487. doi: 10.1210/endo.140.12.7219. [DOI] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13(20):2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisco M, Santini F, Baffa R, Liu M, Drakas R, Wu A, Baserga R. Nuclear translocation of insulin receptor substrate-1 by the simian virus 40 T antigen and the activated type 1 insulin-like growth factor receptor. J Biol Chem. 2002;277(35):32078–32085. doi: 10.1074/jbc.M204658200. [DOI] [PubMed] [Google Scholar]
- Ramel S, Sanchez CA, Schimke MK, Neshat K, Cross SM, Raskind WH, Reid BJ. Inactivation of p53 and the development of tetraploidy in the elastase-SV40 T antigen transgenic mouse pancreas. Pancreas. 1995;11(3):213–222. doi: 10.1097/00006676-199510000-00001. [DOI] [PubMed] [Google Scholar]
- Reiss K. Insulin-like growth factor-I receptor - a potential therapeutic target in medulloblastomas. Expert Opin Ther Targets. 2002;6(5):539–544. doi: 10.1517/14728222.6.5.539. [DOI] [PubMed] [Google Scholar]
- Reiss K, Khalili K. Viruses and cancer: lessons from the human polyomavirus, JCV. Oncogene. 2003;22(42):6517–6523. doi: 10.1038/sj.onc.1206959. [DOI] [PubMed] [Google Scholar]
- Reiss K, Khalili K, Giordano A, Trojanek J. JC virus large T-antigen and IGF-I signaling system merge to affect DNA repair and genomic integrity. Journal of cellular physiology. 2006;206(2):295–300. doi: 10.1002/jcp.20455. [DOI] [PubMed] [Google Scholar]
- Reiss K, Valentinis B, Tu X, Xu SQ, Baserga R. Molecular markers of IGF-I-mediated mitogenesis. Exp Cell Res. 1998;242(1):361–372. doi: 10.1006/excr.1998.4113. [DOI] [PubMed] [Google Scholar]
- Reiss K, Wang JY, Romano G, Furnari FB, Cavenee WK, Morrione A, Tu X, Baserga R. IGF-I receptor signaling in a prostatic cancer cell line with a PTEN mutation. Oncogene. 2000;19(22):2687–2694. doi: 10.1038/sj.onc.1203587. [DOI] [PubMed] [Google Scholar]
- Reiss K, Wang JY, Romano G, Tu X, Peruzzi F, Baserga R. Mechanisms of regulation of cell adhesion and motility by insulin receptor substrate-1 in prostate cancer cells. Oncogene. 2001;20(4):490–500. doi: 10.1038/sj.onc.1204112. [DOI] [PubMed] [Google Scholar]
- Reiss Krzysztof KK, Luis Del Valle. In: JC virus association with brain tumors: The role of T-antigen and Insulin-like growth factor I in DNA repair fidelity. Khalili Kamel JK-T, editor. Hoboken, New Jersey: John Wiley & Sons and Blackwell Publishing; 2010. [Google Scholar]
- Ricciardiello L, Baglioni M, Giovannini C, Pariali M, Cenacchi G, Ripalti A, Landini MP, Sawa H, Nagashima K, Frisque RJ, Goel A, Boland CR, Tognon M, Roda E, Bazzoli F. Induction of chromosomal instability in colonic cells by the human polyomavirus JC virus. Cancer Res. 2003;63(21):7256–7262. [PubMed] [Google Scholar]
- Rossi A, Caracciolo V, Russo G, Reiss K, Giordano A. Medulloblastoma: from molecular pathology to therapy. Clin Cancer Res. 2008;14(4):971–976. doi: 10.1158/1078-0432.CCR-07-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz-Robles MT, Sullivan CS, Pipas JM. Transforming functions of Simian Virus 40. Oncogene. 2001;20(54):7899–7907. doi: 10.1038/sj.onc.1204936. [DOI] [PubMed] [Google Scholar]
- Schmitz-Peiffer C, Whitehead JP. IRS-1 regulation in health and disease. IUBMB Life. 2003;55(7):367–374. doi: 10.1080/1521654031000138569. [DOI] [PubMed] [Google Scholar]
- Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18(6):598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Shuda M, Feng H, Kwun HJ, Rosen ST, Gjoerup O, Moore PS, Chang Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(42):16272–16277. doi: 10.1073/pnas.0806526105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisci D, Morelli C, Cascio S, Lanzino M, Garofalo C, Reiss K, Garcia M, Russo A, Ando S, Surmacz E. The estrogen receptor alpha:insulin receptor substrate 1 complex in breast cancer: structure-function relationships. Ann Oncol. 2007a;18(Suppl 6):vi81–85. doi: 10.1093/annonc/mdm232. [DOI] [PubMed] [Google Scholar]
- Sisci D, Morelli C, Garofalo C, Romeo F, Morabito L, Casaburi F, Middea E, Cascio S, Brunelli E, Ando S, Surmacz E. Expression of nuclear insulin receptor substrate 1 in breast cancer. J Clin Pathol. 2007b;60(6):633–641. doi: 10.1136/jcp.2006.039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Pipas JM. T antigens of simian virus 40: molecular chaperones for viral replication and tumorigenesis. Microbiol Mol Biol Rev. 2002;66(2):179–202. doi: 10.1128/MMBR.66.2.179-202.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Tu X, Prisco M, Wu A, Casiburi I, Baserga R. Insulin-like growth factor I receptor signaling and nuclear translocation of insulin receptor substrates 1 and 2. Mol Endocrinol. 2003;17(3):472–486. doi: 10.1210/me.2002-0276. [DOI] [PubMed] [Google Scholar]
- Sun XJ, Miralpeix M, Myers MG, Jr, Glasheen EM, Backer JM, Kahn CR, White MF. Expression and function of IRS-1 in insulin signal transmission. J Biol Chem. 1992;267(31):22662–22672. [PubMed] [Google Scholar]
- Sun XJ, Pons S, Asano T, Myers MG, Jr, Glasheen E, White MF. The Fyn tyrosine kinase binds Irs-1 and forms a distinct signaling complex during insulin stimulation. J Biol Chem. 1996;271(18):10583–10587. doi: 10.1074/jbc.271.18.10583. [DOI] [PubMed] [Google Scholar]
- Surmacz E, Bartucci M. Role of estrogen receptor alpha in modulating IGF-I receptor signaling and function in breast cancer. J Exp Clin Cancer Res. 2004;23(3):385–394. [PubMed] [Google Scholar]
- Thacker J. The role of homologous recombination processes in the repair of severe forms of DNA damage in mammalian cells. Biochimie. 1999;81(1–2):77–85. doi: 10.1016/s0300-9084(99)80041-8. [DOI] [PubMed] [Google Scholar]
- Trojanek J, Croul S, Ho T, Wang JY, Darbinyan A, Nowicki M, Del Valle L, Skorski T, Khalili K, Reiss K. T-antigen of the human polyomavirus JC attenuates faithful DNA repair by forcing nuclear interaction between IRS-1 and Rad51. Journal of cellular physiology. 2006a;206(1):35–46. doi: 10.1002/jcp.20425. [DOI] [PubMed] [Google Scholar]
- Trojanek J, Ho T, Croul S, Wang JY, Chintapalli J, Koptyra M, Giordano A, Khalili K, Reiss K. IRS-1-Rad51 nuclear interaction sensitizes JCV T-antigen positive medulloblastoma cells to genotoxic treatment. International journal of cancer. 2006b;119(3):539–548. doi: 10.1002/ijc.21828. [DOI] [PubMed] [Google Scholar]
- Trojanek J, Ho T, Del Valle L, Nowicki M, Wang JY, Lassak A, Peruzzi F, Khalili K, Skorski T, Reiss K. Role of the insulin-like growth factor I/insulin receptor substrate 1 axis in Rad51 trafficking and DNA repair by homologous recombination. Mol Cell Biol. 2003;23(21):7510–7524. doi: 10.1128/MCB.23.21.7510-7524.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu X, Batta P, Innocent N, Prisco M, Casaburi I, Belletti B, Baserga R. Nuclear translocation of insulin receptor substrate-1 by oncogenes and Igf-I. Effect on ribosomal RNA synthesis. J Biol Chem. 2002;277(46):44357–44365. doi: 10.1074/jbc.M208001200. [DOI] [PubMed] [Google Scholar]
- Urbanska K, Pannizzo P, Lassak A, Gualco E, Surmacz E, Croul S, Del Valle L, Khalili K, Reiss K. Estrogen receptor beta-mediated nuclear interaction between IRS-1 and Rad51 inhibits homologous recombination directed DNA repair in medulloblastoma. Journal of cellular physiology. 2009;219(2):392–401. doi: 10.1002/jcp.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentinis B, Navarro M, Zanocco-Marani T, Edmonds P, McCormick J, Morrione A, Sacchi A, Romano G, Reiss K, Baserga R. Insulin receptor substrate-1, p70S6K, and cell size in transformation and differentiation of hemopoietic cells. J Biol Chem. 2000;275(33):25451–25459. doi: 10.1074/jbc.M002271200. [DOI] [PubMed] [Google Scholar]
- Valverde AM, Mur C, Pons S, Alvarez AM, White MF, Kahn CR, Benito M. Association of insulin receptor substrate 1 (IRS-1) y895 with Grb-2 mediates the insulin signaling involved in IRS-1-deficient brown adipocyte mitogenesis. Mol Cell Biol. 2001;21(7):2269–2280. doi: 10.1128/MCB.21.7.2269-2280.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscidi RP, Shah KV. Cancer. A skin cancer virus? Science. 2008;319(5866):1049–1050. doi: 10.1126/science.1155048. [DOI] [PubMed] [Google Scholar]
- Vuori K, Ruoslahti E. Association of insulin receptor substrate-1 with integrins. Science. 1994;266(5190):1576–1578. doi: 10.1126/science.7527156. [DOI] [PubMed] [Google Scholar]
- Wang JY, Del Valle L, Peruzzi F, Trojanek J, Giordano A, Khalili K, Reiss K. Polyomaviruses and cancer--interplay between viral proteins and signal transduction pathways. J Exp Clin Cancer Res. 2004;23(3):373–383. [PubMed] [Google Scholar]
- Wang JY, Grabacka M, Marcinkiewicz C, Staniszewska I, Peruzzi F, Khalili K, Amini S, Reiss K. Involvement of alpha1beta1 integrin in insulin-like growth factor-1-mediated protection of PC12 neuronal processes from tumor necrosis factor-alpha-induced injury. J Neurosci Res. 2006;83(1):7–18. doi: 10.1002/jnr.20712. [DOI] [PubMed] [Google Scholar]
- Wang JY, Gualco E, Peruzzi F, Sawaya BE, Passiatore G, Marcinkiewicz C, Staniszewska I, Ferrante P, Amini S, Khalili K, Reiss K. Interaction between serine phosphorylated IRS-1 and beta1-integrin affects the stability of neuronal processes. J Neurosci Res. 2007;85(11):2360–2373. doi: 10.1002/jnr.21400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283(3):E413–422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- White MK, Gordon J, Reiss K, Del Valle L, Croul S, Giordano A, Darbinyan A, Khalili K. Human polyomaviruses and brain tumors. Brain Res Brain Res Rev. 2005;50(1):69–85. doi: 10.1016/j.brainresrev.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Wolf G, Trub T, Ottinger E, Groninga L, Lynch A, White MF, Miyazaki M, Lee J, Shoelson SE. PTB domains of IRS-1 and Shc have distinct but overlapping binding specificities. J Biol Chem. 1995;270(46):27407–27410. doi: 10.1074/jbc.270.46.27407. [DOI] [PubMed] [Google Scholar]
- Woods C, LeFeuvre C, Stewart N, Bacchetti S. Induction of genomic instability in SV40 transformed human cells: sufficiency of the N-terminal 147 amino acids of large T antigen and role of pRB and p53. Oncogene. 1994;9(10):2943–2950. [PubMed] [Google Scholar]
- Wu A, Chen J, Baserga R. Nuclear insulin receptor substrate-1 activates promoters of cell cycle progression genes. Oncogene. 2008;27(3):397–403. doi: 10.1038/sj.onc.1210636. [DOI] [PubMed] [Google Scholar]
- Wu A, Sciacca L, Baserga R. Nuclear translocation of insulin receptor substrate-1 by the insulin receptor in mouse embryo fibroblasts. Journal of cellular physiology. 2003;195(3):453–460. doi: 10.1002/jcp.10261. [DOI] [PubMed] [Google Scholar]
- Wu A, Tu X, Prisco M, Baserga R. Regulation of upstream binding factor 1 activity by insulin-like growth factor I receptor signaling. J Biol Chem. 2005;280(4):2863–2872. doi: 10.1074/jbc.M406138200. [DOI] [PubMed] [Google Scholar]
- Wu S, Zhou B, Xu L, Sun H. Interaction between nuclear insulin receptor substrate-2 and NF-kappaB in IGF-1 induces response in breast cancer cells. Oncol Rep. 2010;24(6):1541–1550. doi: 10.3892/or_00001016. [DOI] [PubMed] [Google Scholar]
- Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y, Heng H, Livingston D. SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev. 2004;18(11):1305–1316. doi: 10.1101/gad.1182804. [DOI] [PMC free article] [PubMed] [Google Scholar]