

Abstract
Retrovirus-based vectors are commonly used as delivery vehicles to correct genetic diseases because of their ability to integrate new sequences stably. However, adverse events in which vector integration activates proto-oncogenes, leading to clonal expansion and leukemogenesis hamper their application. The host cell-encoded lens epithelium-derived growth factor (LEDGF/p75) binds lentiviral integrase and targets integration to active transcription units. We demonstrated earlier that replacing the LEDGF/p75 chromatin interaction domain with an alternative DNA-binding protein could retarget integration. Here, we show that transient expression of the chimeric protein using mRNA electroporation efficiently redirects lentiviral vector (LV) integration in wild-type (WT) cells. We then employed this technology in a model for X-linked chronic granulomatous disease (X-CGD) using myelomonocytic PLB-985 gp91−/− cells. Following electroporation with mRNA encoding the LEDGF-chimera, the cells were treated with a therapeutic lentivector encoding gp91phox. Integration site analysis revealed retargeted integration away from genes and towards heterochromatin-binding protein 1β (CBX1)-binding sites, in regions enriched in marks associated with gene silencing. Nevertheless, gp91phox expression was stable for at least 6 months after electroporation and NADPH-oxidase activity was restored to normal levels as determined by superoxide production. Together, these data provide proof-of-principle that transient expression of engineered LEDGF-chimera can retarget lentivector integration and rescues the disease phenotype in a cell model, opening perspectives for safer gene therapy.
Keywords: gene therapy, LEDGF/p75, lentiviral vector, retargeted integration
Introduction
Over the past two decades, gene therapy using integrating viral vectors has made incredible progress for the correction of monogenic disorders. However, gene transfer has also been associated with some cases of vector-induced leukemia.1,2,3 Initial clinical trials using Moloney murine leukemia virus-based retroviral vectors were prone to insertional mutagenesis because of their integration site preferences close to gene promoters and near known proto-oncogenes leading to enhancer-mediated expression. Insertional mutagenesis, or genotoxicity, is a major drawback for the use of retroviral vectors for gene therapy.
The first case was reported in a French gene therapy trial for X-linked severe combined immunodeficiency where Moloney murine leukemia virus-based retroviral vectors were used to express IL2Rγ in CD34+ bone marrow progenitor cells.4 In total, 5 out of 20 treated patients developed T-cell leukemia due to integration in the proximity of proto-oncogenes, like LIM domain only 2 (LMO2).1,4 In addition to this malignant integration event, other acquired mutations played an important role in leukemia development including gain-of-function mutation in NOTCH1, deletion of the tumor suppressor gene locus cyclin-dependent kinase 2A (CDKN2A), and translocation of the TCR-β (T-cell receptor) region to the STIL-TAL1 (STIL, stem cell leukemia gene/TAL-1 interrupting locus; TAL, T-cell acute lymphocytic leukemia) locus.5 Unfortunately, genotoxicity was not confined to X-linked severe combined immunodeficiency. Recent reports of insertional mutagenesis leading to myelodysplastic syndrome in a trial for X-linked chronic granulomatous disease (X-CGD)2 and a case of leukemia in a trial for Wiskott–Aldrich syndrome,3 underscored that this type of toxicity can be found in other disease settings, raising concerns regarding the safety of the current therapies. The cause of the adverse event in case of X-CGD was pinpointed to retroviral-mediated transcriptional activation of ecotropic viral integration site 1 (EVI1),2 and the same genes as found in X-linked severe combined immunodeficiency- and X-CGD–related genotoxicity, were found to be targeted in a gene therapy trial treating Wiskott–Aldrich syndrome.
In that sense, lentivirus-based viral vectors were generally believed to be safer because of their favored integration into the body of transcription units, and because HIV infection is not associated with insertional activation of proto-oncogenes. However, lentiviral vectors (LVs) can still lead to insertional mutagenesis. A clonal expansion of erythroid cells was reported in a β-thalassaemia trial after lentiviral integration in the high mobility group adenine/thymine (AT)-hook 2 gene (HMGA2).6 Although the clonal expansion is still benign, it emphasizes that with these therapeutic benefits also come significant clinical risks and underscores the need to develop strategies to improve safety of LVs.
Retroviral integration in the host genome is not a random process, but is specific for the retrovirus used: γ-retroviral vectors (like Moloney murine leukemia virus) preferentially integrate in promoter–proximal regions, thereby affecting promoter activity,7 whereas LVs tend to integrate in active transcription units, disfavoring promoter regions.8 Understanding the mechanisms that dictate integration site selection will increase our insight in retrovirology and help the development of gene therapy. For lentivirus-based vectors, the integration bias is triggered by the cellular cofactor lens epithelium-derived growth factor (LEDGF/p75).9 LEDGF/p75, an epigenetic reader recognizing H3K36me3 marks on the chromatin through its N-terminal PWWP domain,10,11 binds downstream of the transcription start site of active transcription units.12 The ubiquitously expressed protein consists of an ensemble of chromatin-binding motifs including the PWWP domain,11,13 AT-hook–like motifs, and three charged regions (CR1-3)14 (Figure 1a) at its N-terminal end. Through a protein-binding domain at the C-terminal end, called integrase (IN)-binding domain (aa 347-429),15 LEDGF/p75 interacts with lentiviral integrases15,16 and binds to several cellular proteins including JPO2,17,18 PogZ,19 Cdc-7/ASK,20 and MLL/menin.21
Figure 1.

Schematic representation of the LEDGF/p75 domain structure and of the CBX1-LEDGF325-530 fusion. (a) LEDGF/p75 carries a conserved PWWP-domain and several charged regions (CR) at its N-terminus. Together with the nuclear localization signal (NLS) and the AT hook-like domains (AT), these elements form the DNA-binding domain of LEDGF/p75. The C-terminal IBD domain is responsible for the interaction with HIV-IN. D366 is essential for the interaction with HIV-IN (indicated by an arrowhead): mutation of this residue to Asn (D366N) results in loss of interaction. (b) The CBX1-LEDGF325-530 and CBX1-LEDGF325-530 D366N fusions are depicted. All protein elements are drawn to scale. Numbers indicate amino acids. CBX1, heterochromatin protein 1β IBD, integrase-binding domain; IN, integrase; LEDGF/p75, lens epithelium-derived growth factor; PWWP, Pro-Trp-Trp-Pro domain.
Artificial fusion proteins in which the LEDGF/p75 chromatin-binding domain is replaced with alternative chromatin interaction domains retarget integration towards regions bound by the respective chromatin-binding domain.22,23,24 We previously engineered an artificial LEDGF/p75-based tether by fusing the C-terminal IN-binding fragment of LEDGF/p75 (LEDGF325-530) to heterochromatin-binding protein 1β (CBX1), resulting in CBX1-LEDGF325-530 (Figure 1b).22 CBX1 binds di- and trimethylated H3K925 and locates to centromeric heterochromatin and transcriptionally silent regions, generally disfavored for lentiviral integration. Expression of CBX1-LEDGF325-530 in LEDGF/p75-depleted HeLaP4 cells (referred to as LEDGF KD cells) retargeted LV integration towards regions enriched in CBX1-binding sites.22 Although, this study provided proof-of-principle for functional retargeting of LV integration, the system required two supplemental viral vector transductions: one vector to deplete endogenous LEDGF/p75, and a second to express the retargeting CBX1-LEDGF325-530 tether.
In an effort to develop safer gene therapy, we have evaluated mRNA electroporation to transiently overexpress CBX1-LEDGF325-530, both in LEDGF KD and in wild-type (WT) cells, and demonstrated retargeting of LV integration. In addition, we validated this technology in a cellular model for X-CGD. X-CGD is an immunodeficiency disorder, where a mutation in the gene-encoding gp91phox affects phagocytic neutrophils by limiting the generation of reactive oxygen species which is essential for their microbicidal activity. The X-CGD model was generated by disrupting the gp91phox gene using homologous recombination in myelomonocytic PLB-985 cells (X-CGD PLB-985).26 After electroporation of X-CGD PLB-985 cells with CBX1-LEDGF325-530 mRNA, we transduced the cells with a therapeutic lentivector encoding a functional copy of gp91. Integration site analysis demonstrated retargeting and rescue of the disease phenotype was indicated by restoration of superoxide production. Analysis of single clones derived from these cells confirmed functional rescue. Together, these findings demonstrate that integration of gene therapy vectors can be retargeted towards gene–poor regions, where the genotoxicity may be reduced, without hampering functional rescue of the disease phenotype.
Results
mRNA electroporation results in efficient CBX1- LEDGF325-530 expression
We previously reported that lentiviral integration can be retargeted.22 Next to the therapeutic viral vector, this technology required two additional viral vector transductions: one for stable knockdown of the natural tether, LEDGF/p75, and a second for stable overexpression of the designed tether, e.g., CBX1 fused to the C-terminus of LEDGF/p75 (referred to as CBX1-LEDGF325-530).
In a subsequent step towards safer gene therapy, we evaluated mRNA electroporation to transiently express CBX1-LEDGF325-530. mRNA was produced from a modified pST1 plasmid template27 that promotes mRNA stability and efficient translation. In parallel, the mRNA for full-length LEDGF/p75 and the interaction-deficient CBX1-LEDGF325530D366N mutant were generated as controls (Figure 1b). Residue D366 in LEDGF/p75 is pivotal for the interaction with IN, and substitution with Asn (D366N) abolishes the interaction.28 In addition, eGFP (enhanced green fluorescent protein) mRNA was produced to spike the electroporation mixture of the different constructs to assess the electroporation efficiency.
First, the different mRNA constructs were electroporated in LEDGF KD HeLaP4 cells (referred to as LEDGF KD). Efficient mRNA electroporation was confirmed by eGFP expression in 100% of the cells as observed by fluorescence-activated cell sorting analysis (data not shown). Expression of the different proteins was evaluated by western blot analysis (Supplementary Figure S1). LEDGF/p75 was absent in the LEDGF KD cell line,22 whereas mRNA electroporation resulted in protein bands migrating at the predicted molecular weight. In parallel, subcellular distribution was evaluated by immunocytochemistry (Figure 2). One day after mRNA electroporation, CBX1-LEDGF325-530, CBX1-LEDGF325-530D366N, and LEDGF/p75 were expressed (Figure 2).22 We also evaluated the turnover of the LEDGF325-530 hybrids by monitoring protein expression levels on 2, 3, and 4 days post-electroporation (Figure 2). All fusion proteins were abundantly present in the nuclei of the cells at 24 and 48 hours after electroporation. Expression levels decreased only from day 3 onwards, and were nearly completely absent at day 4 post-electroporation, corroborating the transient nature of expression after mRNA electroporation.
Figure 2.

Translation of electroporated mRNA in LEDGF/p75 KD HeLaP4 cells. Confocal imaging and immunocytochemistry were used to evaluate peak protein expression for the indicated constructs following mRNA electroporation of LEDGF-depleted HeLaP4 cells. Images were taken at 1, 2, 3, and 4 days after electroporation. Left panels demonstrate expression of CBX1-LEDGF325-530, CBX1-LEDGF325-530D366N, and LEDGF/p75 as detected with A300-848a antibody. Right panels demonstrate DAPI-stained nuclei. CBX1, heterochromatin protein 1β DAPI, 4′,6-diamidino-2-phenylindole; LEDGF/p75, lens epithelium-derived growth factor.
Transient expression of CBX1-LEDGF325-530 rescues LV integration in LEDGF KD cells and retargets integration away from genes
After demonstrating efficient and transient translation of the electroporated mRNA, we investigated whether lentivector transduction was supported. We transduced LEDGF KD HeLaP4 cells 24 hours after mRNA electroporation with a lentiviral reporter vector (LV_eGFP-T2A-fLuc).29 Integrated proviral vector copies were quantified by quantitative PCR (qPCR) (Supplementary Figure S2a). Electroporation of CBX1-LEDGF325-530 and LEDGF/p75 mRNA significantly rescued lentivector integration compared with mock-electroporated cells (1.7- and 1.8-fold more integrated copies, P = 0.0006 and P = 0.0199, respectively), whereas the interaction-deficient CBX1-LEDGF325-530D366N did not rescue integration.
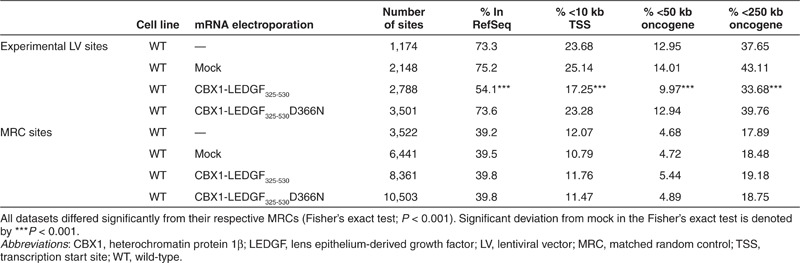
We next asked whether transient CBX1-LEDGF325-530 expression retargeted integration to genomic sites bound by CBX1. Integration sites were amplified and analyzed as described previously.22 Random control sites were generated computationally, and matched to experimental sites with respect to the distance to the nearest MseI cleavage site (matched random control (MRC)). In the analysis that follows, the distribution of experimental LV integration sites is normalized to that of the MRC sites, as a control for recovery bias due to cleavage by restriction enzymes. First, we evaluated LV integration frequencies following transient CBX1-LEDGF325-530 expression in RefSeq transcription units (Supplementary Table S1). In line with previous reports,22,30,31 integration in LEDGF KD cells occurred significantly less in transcription units (57.3% integration in RefSeq genes compared with 73.3% in WT cells (P < 0.001)). Mock electroporation (no mRNA) did not alter integration in RefSeq genes (58.3% compared with 57.3% in KD cells, P = 0.6452). Electroporation with the interaction-deficient CBX1-LEDGF325-530D366N mRNA altered integration in RefSeq genes only slightly (62.6% compared with 58.3% in mock, P = 0.0261). Upon electroporation of LEDGF/p75 mRNA, integration in RefSeq genes was rescued to WT levels (78.7% for LEDGF/p75). In line with previous data,22 upon CBX1-LEDGF325-530 mRNA electroporation, integration was significantly disfavored in transcription units (39.9%, P = 0.8946 compared with MRC), consistent with the distribution pattern of CBX1 in heterochromatic regions, which are generally gene–poor. In addition, we evaluated integration site distribution relative to histone modifications, employing high-resolution maps for the genome-wide distribution of 39 histone modifications determined in human CD4+ T-cells.32,33 In accordance, integration was enriched near histone modifications bound by CBX1 (Supplementary Figure S2b; H3K9me2 and H3K9me3), and near markers associated with transcriptionally silent regions or heterochromatin (H4K20me3 and H3K79me3), whereas the D366N control did not (Supplementary Figure S2b; compare CBX1-LEDGF325-530 and CBX1-LEDGF325-530D366N with LEDGF KD).
CBX1-LEDGF325-530 mRNA electroporation retargets integration to more favorable, intergenic regions in WT cells;
In a next step, we employed the mRNA electroporation strategy to retarget vector integration in WT HeLaP4 cells, expressing endogenous LEDGF/p75, using CBX1-LEDGF325-530 and CBX1-LEDGF325-530D366N mRNA. Fusion proteins were expressed (western blot analysis; Figure 3a) and LV integration frequency was comparable to mock (integrated proviral copies; Figure 3b). Analysis of integration site distribution revealed high frequency of integration in transcription units in WT cells and WT cells that were mock-electroporated (Table 1; 73.3 and 75.2% in RefSeq, respectively). Electroporation of WT cells with CBX1-LEDGF325-530 mRNA resulted in significantly less integration in transcription units (54.1%, P < 0.001, compared with WT or mock), whereas expression of CBX1-LEDGF325-530D366N control did not affect distribution compared with WT (73.6%, P = 0.8869). In addition to integration in RefSeq genes, we evaluated integration near genomic features that would indicate an increased likelihood of deregulation of the neighboring genes, such as transcription start sites and oncogenes. Integration within 10 kb of the transcription start site of protein-coding genes and near the 5′-end of known proto-oncogenes or tumor suppressor genes (within 50 or 250 kb distance) occurred significantly less frequently following CBX1-LEDGF325-530 mRNA electroporation (Table 1; P < 0.001 when compared with WT or mock), whereas the CBX1-LEDGF325-530D366N mRNA control condition was indistinguishable from WT. More detailed analysis showed that integration associated with histone modifications bound by CBX1 (H4K20me3, H3K9me3, and H3K9me2; Figure 3c). In WT or mock-electroporated cells, LV integration frequency negatively associated with H3K9me2 and H3K9me3 histone modifications (yellow tiles) and positively correlated with histone modifications generally associated with active transcription, such as all acetylations and some histone methylations (blue tiles, Figure 3c). Transient CBX1-LEDGF325-530 expression during vector transduction reversed most of these correlations (***P < 0.001, Wald statistics referred to χ2 distribution compared with mock; Figure 3c), underscoring redistribution of integration sites, shifting more towards random (less intense yellow and blue tiles). The pattern in CBX1-LEDGF325-530D366N control cells was not different from WT cells. Similar data were obtained in SupT1 cells (data not shown). These results demonstrate that retargeting of viral vector integration employing transient CBX1-LEDGF325-530 expression in WT cells is feasible.
Figure 3.

Transient expression of CBX1-LEDGF325-530 retargets LV integration into CBX1-rich heterochromatin regions. (a) Western blot analysis showing protein expression of CBX1-LEDGF325-530(D366N) and endogenous LEDGF/p75 (mock) compared with WT HeLaP4 cells 48 hours after mRNA electroporation. Loading was controlled by α-tubulin. (b) Following mRNA electroporation, WT HeLaP4 cells were transduced with LV_eGFP-T2A-fLuc. Integrated vector copies were measured by quantitative PCR. (c) Correlations were made between integration sites and the density of a panel of 39 histone modifications. Associations of integration and histone methylation/acetylation were quantified using ROC areas, comparing the association of integration site data sets with the frequency in corresponding MRC sets. A ROC area scale is shown along the bottom of the panel. Tile color indicates whether a specific feature is favored (blue, enrichment relative to random) or disfavored (yellow, negative correlation compared with random) for integration (10 kb window) in the respective data sets relative to their MRCs. P values showing significance of departures from mock-treated WT cells (overlaid with dashes) are shown with asterisks (***P < 0.001, Wald statistics referred to χ2 distribution compared with mock). CBX1, heterochromatin protein 1β LEDGF/p75, lens epithelium-derived growth factor; LV, lentiviral vector; MRC, matched random control; ROC, receiver operating characteristic; WT, wild-type.
Table 1. Integration frequency near mapped genomic features in the genome in HeLaP4 cells.
Retargeted integration of a therapeutic vector (LV_gp91) following mRNA electroporation rescues a model for X-CGD
Subsequently, we validated our technology in human myelomonocytic X-CGD PLB-985 cells (further referred to as X-CGD PLB),26 a cell model of X-CGD with a disrupted CYBB gene, the gene encoding the gp91phox subunit of the phagocyte NADPH-oxidase complex. WT PLB-985 cells were compared as a control (referred to as PLB WT). X-CGD PLB cells were electroporated with CBX1-LEDGF325-530 and CBX1-LEDGF325-530D366N mRNA, respectively, and transduced the next day with a LV encoding codon-optimized gp91phox (LV_gp91),34 or a control vector expressing eGFP (LV_eGFP) (Figure 4a). Western blot analysis for the respective LEDGF fusions showed bands at the expected molecular weight (data not shown). Transduction efficiency was determined by flow cytometry, monitoring gp91 expression (Figure 4b). Transduction of X-CGD PLB cells electroporated with CBX1-LEDGF325-530 or D366N control mRNA resulted in equal gp91-positive cells as PLB WT (91.8 and 91.1% gp91+ cells, respectively, compared with 91.4% in PLB WT; Figure 4b) and comparable gp91 expression levels (1.4 × 103 and 1.5 × 103 arbitrary units as measured by mean fluorescence intensity, respectively). In addition, comparable integrated vector copies were detected by qPCR in X-CGD PLB cells electroporated with CBX1-LEDGF325-530 or D366N control mRNA (3.6 and 3.15 vector copy numbers (VCN), respectively). Next, LV integration sites were determined in the respective cell lines for LV_gp91 and LV_eGFP (Table 2). In line with our previous results (Table 1), the vector integration in mock-treated X-CGD PLB cells preferentially occurred in RefSeq genes (Table 2, 80.65%) and was similar to CBX1-LEDGF325-530D366N mRNA-electroporated cells (79.87 and 78.8% for LV_eGFP and LV_gp91, respectively), whereas integration shifted out of RefSeq transcription units in cells electroporated with CBX1-LEDGF325-530 mRNA (Table 2; 59.36 and 56.22% for LV_eGFP and LV_gp91, respectively). Although integration shifted towards random and the reduction was significant (***P < 0.001, for LV_eGFP and LV_gp91 comparing CBX1-LEDGF325-530 mRNA electroporation to mock), integration events were still favored in transcription units (***P < 0.001 compared with MRC). Although CBX1-LEDGF325-530 mRNA electroporation did not alter the integration frequency within 10 kb of the transcription start site of protein-coding genes in X-CGD PLB-985 cells, integration occurred significantly less frequently within 50 or 250 kb distance of the 5′-end of known proto-oncogenes or tumor suppressor genes (Table 2; P < 0.001 when compared with mock). Again, electroporation with the CBX1-LEDGF325-530D366N mRNA control did not affect integration near these features compared with mock. Likewise, integration occurred more frequently near markers for silenced chromatin (H3K9me2, H3K9me3, and H4K20me3; ***P < 0.001, Wald statistics referred to χ2 distribution compared with mock) and less in regions rich in markers for active chromatin following CBX1-LEDGF325-530 mRNA electroporation (Figure 5; compare CBX1-LEDGF325-530 and CBX1-LEDGF325-530D366N with mock) corroborating redistribution of integration sites out of transcription units and more random.
Figure 4.

Vector constructs and expression efficiency after electroporation. After CBX1-LEDGF325-530(D366N) mRNA electroporation, X-CGD PLB-985 cells were transduced with LV_gp91 or LV_eGFP and subsequently differentiated into granulocytes. (a) Schematic representation of transfer plasmids used for vector production. The SFFV promoter drives gp91 or eGFP expression. All lentiviral vectors are based on HIV-1NL4.3. (b) Flow cytometry was used to determine expression levels of gp91 (7D5 antibody) in electroporated cells. CBX1, heterochromatin protein 1β eGFP, enhanced green fluorescent protein; gp91, glycoprotein 91; LEDGF/p75, lens epithelium-derived growth factor; LTR, long terminal repeat; LV, lentiviral vector; SFFV, spleen focus-forming virus; SIN, self-inactivating; X-CGD, X-linked chronic granulomatous disease; Ψ, packaging signal.
Table 2. Integration frequency near mapped genomic features in X-CGD PLB cells.
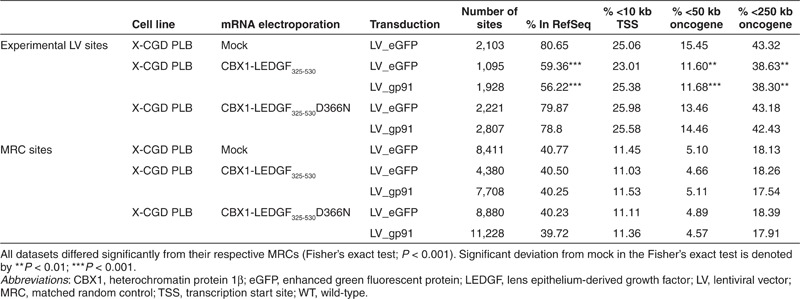
Figure 5.

Transient expression of CBX1-LEDGF325-530 retargets LV integration into CBX1-rich heterochromatin regions in X-CGD PLB cells. Correlations were made between integration sites and the density of a panel of 39 histone modifications (Figure 3c). P values showing significance of departures from mock-treated WT cells (overlaid with dashes) are shown with asterisks (***P < 0.001, Wald statistics referred to χ2 distribution compared with mock). CBX1, heterochromatin protein 1β eGFP, enhanced green fluorescent protein; gp91, glycoprotein 91; LEDGF/p75, lens epithelium-derived growth factor; LV, lentiviral vector.
Next, we verified whether the expression of the retargeted LV_gp91 was sufficient to reconstitute the superoxide production. Cells were treated with dimethyl sulfoxide to induce granulocytic differentiation. Differentiation was comparable for all cell lines as measured by flow cytometry (Supplementary Figure S3; upper quadrants, CD11b+). Subsequently, NADPH-oxidase activity was assessed using the cytochrome C assay. Contrary to LV_eGFP cells, cells transduced with LV_gp91 reached comparable superoxide levels as in PLB WT cells (about 2.8 nmol/min × 106 cells; Figure 6a), irrespective of the mRNA used for electroporation, indicating that even though the therapeutic vector was retargeted, NADPH-oxidase activity was restored to WT levels. In parallel, we evaluated the potential of the different gp91-reconstituted cells to reduce nitroblue tetrazolium (NBT) to NBT-formazan (blue dye). X-CGD PLB cells transduced with LV_gp91 reduced NBT as efficient as WT PLB cells, irrespective of retargeted integration (86% for both electroporated cells compared with 81% in PLB WT cells, Figure 6b), whereas no reduction was observed in cells transduced with LV_eGFP control vector. Together, these results indicate that even when vector integration is altered, retargeted out of transcription units, and towards regions enriched in heterochromatin-associated marks, equal integrated VCNs are obtained and transgene expression is sufficient to rescue a disease phenotype.
Figure 6.

gp91 expression rescues superoxide production after retargeting to heterochromatin. Superoxide production was measured in the transduced and electroporated X-CGD PLB cells. (a) Superoxide production was measured by the continuous cytochrome C reduction assay. (b) Percentage of NBT formazan-positive cells are depicted after reduction of NBT as measure for superoxide production. Mean ± SD of three measurements are shown. (c) Monoclonals were grown from the LV_gp91-transduced CBX1-LEDGF325-530(D366N)-electroporated X-CGD PLB cells. MFI of gp91 expression per VCN. The MFI determined from the different gp91-expressing monoclonals was divided by the VCN of the respective monoclonal. (d) Superoxide production was measured by the continuous cytochrome C reduction assay for all different monoclonals and normalized to VCNs. CBX1, heterochromatin protein 1β eGFP, enhanced green fluorescent protein; gp91, glycoprotein 91; LEDGF/p75, lens epithelium-derived growth factor; MFI, mean fluorescence intensity; NBT, nitroblue tetrazolium; VCN, vector copy number; WT, wild-type; X-CGD, X-linked chronic granulomatous disease.
Alternatively, the observed rescue of superoxide production can be explained by a few WT integrations mediated by endogenous LEDGF/p75. Single clones of LV_gp91 X-CGD PLB cells were grown by limiting dilution (10 clones derived from cells electroporated with CBX1-LEDGF325-530 mRNA and 9 clones from cells electroporated with CBX1-LEDGF325-530D366N mRNA). Individual clones were characterized by determining VCN, integration site distribution, gp91 expression levels, and production of reactive oxygen species. Integrated proviral copies were in the same range, with CBX1-LEDGF325-530 cells carrying 1–4 VCN and CBX1-LEDGF325-530D366N cells carrying 2–3 VCN (Supplementary Table S2). One CBX1-LEDGF325-530D366N clone did not carry an integrated provirus and thus was excluded from the analysis. gp91 expression levels were determined for cells electroporated either with CBX1-LEDGF325-530 or with CBX1-LEDGF325-530D366N mRNA and normalized for VCN (Figure 6c). Although gp91 expression in both groups was not significantly different (P = 0.0565; unpaired two-sided t-test), more variance of gene expression levels was observed in clones obtained after electroporation with CBX1-LEDGF325-530 mRNA when compared with CBX1-LEDGF325-530D366N. Individual clones were cultured in the presence of dimethyl sulfoxide to induce myeloid differentiation and to score their capacity to produce superoxide and to rescue the X-CGD phenotype using the cytochrome C assay (Figure 6d). LV_gp91-transduced cells derived from CBX1-LEDGF325-530 and CBX1-LEDGF325-530D366N mRNA-electroporated X-CGD PLB cells produced equal levels of superoxide (P = 0.3674), even though reactive oxygen species production levels differed more in the cells generated using CBX1-LEDGF325-530 mRNA. In addition, we determined the integration sites of the LV_gp91 vectors in the single clones, resulting in 18 and 14 unique sites for CBX1-LEDGF325-530 and CBX1-LEDGF325-530D366N mRNA, respectively (Supplementary Table S3). Analysis of integration site distribution underscored retargeted integration in clones electroporated with CBX1-LEDGF325-530 mRNA: 66.67% integration in RefSeq genes for clones grown following CBX1-LEDGF325-530 mRNA electroporation compared with 78.57% for CBX1-LEDGF325-530D366N clones. Also, integration in the CBX1-LEDGF325-530 clones occurred more frequently near H4K20me3, H3K9me3, and H3K9me2 histone modifications that are recognized by CBX1 (data not shown).
These results indicate that even when integration is retargeted and takes place in heterochromatin, transgene expression is sufficient to rescue a disease phenotype regardless of integration site placement and expression levels.
Discussion
Clinical trials to treat several blood cell diseases have demonstrated that long-term gene correction is feasible and in some patients may provide equal clinical benefit with less risk as compared with standard treatments. Alongside, the therapeutic benefits has also come with the recognition of associated clinical risks. Initial clinical trials using γ-retroviral vectors were prone to insertional mutagenesis, in part due to their integration site preferences close to gene promoters and near proto-oncogenes leading to enhancer-mediated expression. Accordingly, LVs were generally believed to be safer because of their favored integration into the body of active transcription units. However, LV integration can lead to insertional mutagenesis and clonal expansion, as reported in the β-thalassaemia trial,6 which underscores the need to develop strategies that improve the safety of viral vectors.
The increasingly sophisticated understanding of the mechanisms by which the therapeutic vectors caused leukemia, provides new avenues to develop vectors that maintain or even increase the efficacy seen in the first-generation retroviral vectors but that potentially offer a safer alternative. Several approaches have been pursued to overcome the issues accompanying the current therapies: insulator elements have been introduced to act as both enhancer blockers and boundaries against potential silencing of integrating viral vectors;35 genetic regions have been selected that might function as safe harbors for integration;36 alternative integration systems with a more random integration profile have been developed, such as foamy virus or α-retroviral vectors;37,38 progress has been made in transposon-based gene transfer;39 specific promoters have been selected to support cell-specific expression6,40 and to limit unwanted transgene expression in off-target cells;41 and alternative tethers have been generated that redirect integration towards defined genetic regions.22,23,24
We previously reported retargeting of lentiviral integration in LEDGF/p75 KD HeLaP4 cells stably overexpressing CBX1-LEDGF325-530. That system required two extra vectors: one to create the LEDGF/p75 KD and another to express CBX1-LEDGF325-530. With the cellular function of LEDGF/p75 remaining largely unknown, modification of LEDGF/p75 expression levels (stable knockdown or overexpression of artificial chimera) is likely to interfere with the natural function of LEDGF/p75. Indeed, akin to its effect on HIV-1 IN, LEDGF/p75 orchestrates the chromatin association of several proteins or protein complexes, such as JPO2,18 pogZ,19 MLL/menin,21 and Cdc7-ASK.20 Overexpression of the IN-binding C-terminal end of the LEDGF/p75 protein might affect these interactions and thus their downstream pathways. In a next step towards safer gene therapy, we therefore opted for transient overexpression of CBX1-LEDGF325-530 using mRNA electroporation and proved that transient expression is sufficient to redirect integration towards regions enriched in CBX1-binding sites, both in LEDGF/p75 KD (Supplementary Table S1 and Supplementary Figure S2) and in WT (Table 1 and Figure 3) cells.
Moreover, we reported retargeting of LV_gp91 in CBX1-LEDGF325-530 mRNA-electroporated X-CGD PLB-985 cells, a cell model for X-CGD (Table 2 and Figure 5) and demonstrated functional rescue of the disease phenotype after retargeting to CBX1-binding sites (Figure 6). Although a clear shift in integration out of RefSeq genes was detected (80.65 and 56.22% for mock and CBX1-LEDGF325-530 mRNA-electroporated cells, respectively), the frequency in the presence of the engineered was still different from random (40.24% for MRC). This can be explained by residual integration targeted by endogenous LEDGF/p75. To overcome this, CBX1-LEDGF325-530 mRNA electroporation may be combined with electroporation of small interfering RNA targeting the endogenous LEDGF/p75 mRNA.
One question that remained was whether the functional rescue observed in the cytochrome C and the NBT assay (Figure 6a,b) originated from integrations in hetero- or euchromatin, and whether the integration event was mediated by endogenous LEDGF/p75 or CBX1-LEDGF325-530. To address this issue, we generated single clones and evaluated phenotypic rescue. Our results demonstrated that integrated VCNs and gp91 expression levels were comparable for X-CGD PLB-985 cells electroporated with CBX1-LEDGF325-530 and CBX1-LEDGF325-530 D366N mRNA (Figures 4 and 6c). The fact that these measurements were performed on cells that were cultured for at least 6 months underscores that, even with retargeted integration (out of transcription units and towards regions enriched in heterochromatin-associated marks), expression levels are sufficient to rescue the disease phenotype and are not more silenced over time than with WT integration. However, the increased variation in gene expression per VCN in single clones obtained after CBX1-LEDGF325-530 mRNA electroporation may reflect the impact of integration near heterochromatin markers on vector gene expression, also referred to as position effect variegation. Indeed, the spleen focus-forming virus promoter is reported to be prone to silencing on the long-term.2 Although this does not lead to significantly lower reactive oxygen species production in our setting (gp91 is not rate-limiting at high expression levels),26 this effect may become more relevant when weaker promoters are used. One strategy to overcome this issue is the inclusion of a ubiquitous acting chromatin-opening element into the LV, an element that has been shown to reduce position effect variegation and to boost expression levels.34 Alternatively, insulator sequences could be included in the long terminal repeat to achieve a similar effect.42
Electroporation-mediated mRNA transfection is an efficient approach for gene expression that does not result in permanent genetic modification of cells. Successful gene transfer by mRNA electroporation into primary T lymphocytes has been demonstrated for T-cell receptor43 and chimeric antigen receptors.44 Likewise, mRNA-mediated gene delivery into human CD34+ progenitor cells promoted efficient protein expression, allowing transitory manipulation of the stem cell characteristics.45 In a next step towards safer vectors for gene therapy, targeting integration to a specific site within the genome, preferably unique and safe, will be advantageous. Two candidate loci are the CCR5 and adeno-associated virus (AAV) integration site 1 loci. The CCR5 locus is not essential in humans, because individuals carrying a homozygous null mutation develop normally and are healthy; AAV integration site 1 is a common integration site of the human non-pathogenic AAV.46 Therefore, fusions of LEDGF325-530 linked to artificial zinc fingers or transcription activator-like effectors that specifically bind the CCR5 or AAV integration site 1 locus could be generated to direct integration towards these safe harbors.
Our findings open possibilities to engineer viral vectors that incorporate LEDGF hybrids to target integration into safe-landing sites, thereby reducing the risk of insertional mutagenesis. Hare and colleagues have reported47 a set of amino acid substitutions in HIV-IN that abolish LEDGF/p75 binding, together with mutations in the LEDGF/p75 protein that restore binding. Gene delivery vectors could thus use an altered IN/LEDGF pair to direct integration, even in the presence of WT LEDGF/p75. To date, the altered IN does not show WT integration activity, but this may be improved with further engineering.47
In conclusion, we demonstrate that integration can be retargeted in the presence of endogenous LEDGF/p75 following transient expression of CBX1-LEDGF325-530, and that expression after retargeting remains efficient and can rescue the X-CGD disease phenotype in a cell culture model for the disease.
Materials and Methods
Cell culture. SupT1 cells (NIH AIDS Research and Reference Reagent Program, http://www.aidsreagent.org/) and X-CGD PLB-985 cells, derived from the PLB-985 myelomonocytic leukemia cell line by disrupting the gp91phox gene by homologous recombination,26 were grown in RMPI (Gibco, Invitrogen, Merelbeke, Belgium) supplemented with 10% heat-inactivated fetal calf serum (Sigma-Aldrich, Bornem, Belgium) and 50 µg/ml gentamicin (Gibco). HEK 293T cells (ATCC, LGC Standards S.a.r.I., Molsheim Cedex, France) were grown in Dulbecco's modified Eagle's medium with Glutamax (Gibco) supplemented with 8% fetal calf serum and 50 µg/ml gentamicin. HeLaP4 cells, a kind gift from Pierre Charneau, Institut Pasteur, Paris, France, were grown in Dulbecco's modified Eagle's medium with Glutamax supplemented with 5% fetal calf serum, 50 µg/ml gentamicin, and 0.5 mg/ml geneticin (Gibco). All cells were grown at 37 °C in a humidified atmosphere containing 5% CO2. Stable monoclonal LEDGF/p75 KD HeLaP4 cells were generated previously.22
Plasmids and in vitro RNA synthesis. All primers used are listed in Supplementary Table S4. All enzymes used were obtained from Fermentas (St Leon-Rot, Germany).
Three plasmids were generated for in vitro RNA synthesis using pST1_eGFP27 as backbone plasmid: pST1_CBX1-LEDGF325-530BC, pST1_CBX1-LEDGF325-530BC-D366N, and pST1_LEDGF/p75BC. BC (backcomplemented) indicates that the mRNA encoded by these constructs is resistant to the miRNA-based hairpin expressed in the LEDGF KD cells. The CBX1-LEDGF325-530BC and CBX1-LEDGF325-530BC-D366N cassettes were PCR-amplified with the primers CBX1_s NheI and LEDGF325-530_as SalI, using the templates pLNC-CBX1-LEDGF325-530BC-IRES-BsdR22 and pLNC-CBX1-LEDGF325-530BC-D366N-IRES-BsdR,22 respectively. These fragments were digested with NheI and SalI, and subsequently cloned into pST1_eGFP that was digested with SpeI and XhoI to remove eGFP. The pST1_LEDGF/p75BC construct was generated similarly by PCR amplification with the primers LEDGF_s NheI and LEDGF325-530_as SalI, and cloning it into the digested pST1_eGFP backbone.
The mMESSAGE mMACHINE T7 Kit (Applied Biosystems, Gent, Belgium) was used to generate 5′-capped and 3′-polyA containing mRNA transcripts, starting from 5 µg linearized plasmid containing a T7 promoter upstream of the target open-reading frame. Following linearization of pST1_eGFP, pST1_CBX1-LEDGF325-530BC, pST1_CBX1-LEDGF325-530BC-D366N, and pST1_LEDGF/p75BC with LguI, mRNA was synthesized according to the manufacturer's protocol, resulting in four mRNA preparations, encoding eGFP, CBX1-LEDGF325-530, CBX1-LEDGF325-530 D366N, and LEDGF/p75, respectively.
mRNA electroporation. In a first step, protein expression levels following eGFP mRNA electroporation were scored, comparing square wave and exponential decay pulse in different cell lines (HeLa, SupT1, PLB-985). Four million cells in Optimem without phenol red (Gibco) were mixed with mRNA encoding eGFP (5 µg). This cell suspension was then transferred to a Gene Pulser/Micropulser Electroporation Cuvette with a 0.4 cm gap (BioRad, Nazareth, Belgium) and subjected to a square wave pulse with 500 V and pulse length 5 ms or an exponential decay pulse starting at 450 V by a capacitance of 150 µF, using the Gene Pulser Xcell electroporation system (BioRad). In all cell lines tested, the square wave pulse resulted in higher expression levels than exponential decay pulse (data not shown). Hence, all mRNA electroporations in this study were performed using square wave pulse.
Four million cells in Optimem without phenol red (Gibco) were mixed with mRNA encoding eGFP (1 µg) and CBX1-LEDGF325-530 (5 µg) or CBX1-LEDGF325-530D366N (5 µg) or LEDGF/p75 (5 µg) before electroporation with the square wave pulse. After electroporation, cells were seeded in the appropriate growth medium.
Plasmids and LV production. LV production was performed as described earlier.29 Briefly, vesicular stomatitis virus glycoprotein pseudotyped HIV-based particles were produced by poly(ethylenimine) transfection using pCHMWS_eGFP-T2A-fLuc,29 SgpW,34 and SEW48 as a transfer plasmid for LV_eGFP-T2A-fLuc, LV_gp91, and LV_eGFP, respectively.
Transduction. Electroporated HeLaP4 cells were seeded in a 96-well plate 1 day before transduction (20,000 cells/well). All electroporated SupT1 cells and X-CGD PLB-985 were seeded in a 24-well plate for 24 hours, then reseeded in a 96-well plate at 40,000 cells/well and immediately transduced. After 72 hours, 90% of cells were reseeded into two plates (fluorescence-activated cell sorting analysis and Luc assay). The remainder was cultured for qPCR or integration site analysis for at least 20 days to eliminate non-integrated DNA.
Expression analysis. Immunocytochemistry and western blot analysis were performed as previously described.22 Briefly, LEDGF325-530 fusions were detected using A300-848a antibody (1/500 dilution; Bethyl Laboratories, Montgomery, TX) and Alexa555-labeled goat anti-rabbit secondary antibodies (Life Technologies, Ghent, Belgium). All images were acquired using a laser scanning microscopy 510 META imaging unit (Carl Zeiss, Zaventem, Belgium). Alexa555 was excited at 543 nm (HeNe laser), and 4′,6-diamidino-2-phenylindole (DAPI) at 790 nm (MaiTai laser; Spectra Physics, Mountain View, CA). For western blot analysis, SDS (1%) protein extracts were separated by gel electrophoresis. LEDGF/p75 fusions were detected using A300-848a antibody (1:2,000; Bethyl Laboratories) and visualized by chemiluminescence (ECLplus; GE Healthcare, Diegem, Belgium). Equal loading was confirmed with α-tubulin antibody (1:4,000, T-4026; Sigma).
qPCR. Integrated proviral copies were quantified by real-time qPCR on an iQ5 Multicolor RT PCR detection system (BioRad) as reported earlier.22 Briefly, cells transduced with HIV-based LVs were cultured for at least 2 weeks to eliminate all non-integrated DNA. Genomic DNA was extracted using the GenElute Mammalian genomic DNA miniprep kit (Sigma). Samples corresponding to 100 ng of genomic DNA were used as input for qPCR. Integrated copies for HIV-based LVs were measured using a Gag-derived primer–probe set: RT-GAG-1, 5′-ATC AAG CAG CCA TGC AAA TGT T; RT-GAG-2, 5′-CTG AAG GGT ACT AGT AGT TCC TGC TAT GTC; RT-GAG probe, 5′-FAM-ACC ATC AAT GAG GAA GCT GCA GAA TGG GA-Tamra-3′. RNaseP was used as an endogenous housekeeping control (TaqMan RNaseP control reagent; Life Technologies). All samples were run in quadruplet and subjected to an initial 3 minutes denaturation at 95 °C followed by 50 cycles of 10 seconds at 95 °C and 30 seconds at 55 °C. Data were analyzed with iQ5 Optical System Software (BioRad). Relative numbers of integrated LV copies were calculated by normalizing the Gag-signal over RNaseP signal.
The mean VCN per cell was determined by real-time qPCR using a Roche LightCycler 480 (Roche, Mannheim, Germany) applying advanced quantification (LightCycler 480 Software 1.5.0; Roche) with a reference sample known to harbor one single vector integrant in serial dilution. Samples were measured in triplicates using 50–100 ng of genomic DNA mixed with Roche LC480 Probes Master Mix (Roche). Primers and probe specific for the codon-optimized gp91phox coding region and the human EpoR as endogenous standard were used to determine the amount of viral sequences per genome in a duplex reaction. A serial dilution of genomic DNA from a known PLB-985 clone harboring a single lentiviral provirus was used for quantification of VCNs. Primers and probe for hEpo-R: forward 5′-ATG CCA GAC TAG ACC CAG AC and reverse 5′-GGA AAG GAA CTA ACA AAG GGA C, probe sequence 5′-tct tgg gga ctt tca cct gat ttt cct tct ac. Primers and probe for codon-optimized gp91: forward 5′-CCA GCA GCA CCA AGA CCA T and reverse 5′-CCG ATG AAA AAG ATC ACG AAC AG, probe sequence 5′-ACC AGA ACA CCT CGA AGT AGC TCC GCC (designed by PrimerDesign, Southampton, UK).
Luciferase activity assay. Cells were lysed with 70 µl of lysis buffer (50 mmol/l Tris, pH 7.5; 200 mmol/l NaCl; 0.2% NP40; 10% glycerol). The lysate was assayed according to the manufacturer's protocol (ONE-Glo; Promega, Leiden, The Netherlands). Luciferase activity was normalized for total protein (BCA; Pierce, Aalst, Belgium).
Integration site amplification. Integration sites were amplified as previously described.22 Genomic DNA was digested using MseI, and linkers were ligated. Provirus–host junctions were amplified by nested PCR using bar-coded primers. This enabled pooling of PCR products into one sequencing reaction. Products were gel-purified and sequenced on a 454 GS-FLX instrument (454 Life Sciences; Roche, Basel, Switzerland).
Bioinformatic analysis. For integration sites to be authentic, sequences needed a best unique hit when aligned to the human genome (hg18) using BLAT, the alignment began within 3 bp of the viral long terminal repeat end, and had >98% sequence identity. Statistical methods are detailed in Berry et al.49 Random control sites were generated computationally, and matched to experimental sites with respect to the distance to the nearest MseI cleavage site (MRC). In all analyses, the distribution of experimental LV integration sites is normalized to that of the MRC sites, as a control for recovery bias due to cleavage by restriction enzymes. Integration site counts were compared with MRCs by a Fisher's exact test (where stated), or by multiple regression models for integration intensity and a c-logit test for significance.49 Analysis was carried out using R (http://www.r-project.org). Frequency of integration sites near oncogenes or within tumor suppressor genes was calculated using a list of genes that were collected to generate a comprehensive list of cancer-related genes (http://www.bushmanlab.org/links/genelists). Epigenetic heat maps show the relationship between integration sites and the density of 39 histone modifications are summarized using receiver operating characteristic area. Histone modification data determined in primary T cells from Barski et al.32 and Wang et al.33 were used. Tile color indicates whether a chosen feature is favored (blue) or disfavored (yellow) for integration in the respective data sets relative to their MRCs. Asterisks indicate significant P values when comparing all samples (columns) to mock-treated WT sample (overlaid with dashes). Detailed information on the statistics and the interpretation of the heat maps can be found at doi:10.1371/journal.ppat.1001313.s011.
CBX1-binding sites were analyzed using data from Vogel et al.25 The number of sequence tags from the ChIP-Solexa data sets in a defined window around each LV integration site or MRC, was calculated. For each DamID probe set available, probes were aligned onto the hg18 using BLAT, and their associated log2-binding ratios used to select the top 5% of sites. For each integration site or MRC, the average number of high-affinity probes within a defined window around the site was calculated.
Fluorescence-activated cell sorting analysis. gp91phox was detected using the 7D5-FITC antibody (MBL international no. D162-4). Granulocytic differentiation was detected using the CD11b-PE-Cy7 (clone M1/70) and dead cells were excluded using Dead Cell Stain eFluor780 (both from eBiosciences, Frankfurt, Germany). All data acquisitions were performed on a BD FACSCanto II flow cytometer (BD Biosciences, Heidelberg, Germany) and analyzed using the BD FACSDiva 6.0 software.
Cytochrome C and NBT assay. Cytochrome C assays were carried out according to Mayo and Curnutte50 using a Spectra MAX 340 reader (Molecular Devices, Sunnyvale, CA) and the SOFTmax Version 2.02 PRO software. The NBT assay was performed as described earlier.40 Briefly, electroporated X-CGD PLB-985 cells differentiated to neutrophils were used in 96-well plates. To the sample and control tubes, 200 µl of NBT reagent (1 µg/ml of NBT dye (Sigma) in 0.9% NaCl) were added in the presence or absence of 1 µg/ml PMA (phorbol 12-myristate 13-acetate) (Sigma). Cells were incubated at 37 °C for 30 minutes. Neutrophils containing the blue/black formazan deposits, indicative of superoxide production, were classed as NBT-positive and quantified using light microscopy with oil immersion.
SUPPLEMENTARY MATERIAL Figure S1. Translation of electroporated mRNA in LEDGF KD cells. Figure S2. Transient expression of CBX1-LEDGF325-530 in LEDGF KD cells rescues LV transduction and retargets LV integration into CBX1-rich heterochromatin regions. Figure S3. Differentiation status of electroporated X-CGD PLB cells. Table S1. Integration frequency near mapped genomic features in LEDGF KD HeLaP4 cells. Table S2. Characterization of single clones. Table S3. Vector integration sites in single X-CGD PLB clones. Table S4. Primers used for cloning.
Acknowledgments
We thank Paulien Van de Velde and Frances Male for excellent technical assistance. S.V. is funded by the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen). J. De R. had a Mathilde-Krim postdoctoral fellowship from amfAR. This work was supported by KU Leuven Research Council (grant OT/09/047); the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen) CellCoVir SBO grant (60813); the Flanders Research Foundation (FWO) grant (G.0530.08); European Commission THINC grant (HEALTH-F3-2008-201032) to Z.D. This work was supported in part by grants from the European Union (FP7 integrated project CELL-PID, HEALTH-2010-261387) and from the LOEWE Center for Cell and Gene Therapy Frankfurt. The Georg-Speyer-Haus is supported by the Bundesministerium für Gesundheit and the Hessisches Ministerium für Wissenschaft und Kunst. The authors declared no conflict of interest.
Supplementary material
Translation of electroporated mRNA in LEDGF KD cells.
Transient expression of CBX1-LEDGF325-530 in LEDGF KD cells rescues LV transduction and retargets LV integration into CBX1-rich heterochromatin regions.
Differentiation status of electroporated X-CGD PLB cells.
Integration frequency near mapped genomic features in LEDGF KD HeLaP4 cells.
Characterization of single clones.
Vector integration sites in single X-CGD PLB clones.
Primers used for cloning.
References
- Hacein-Bey-Abina, S, Garrigue, A, Wang, GP, Soulier, J, Lim, A, Morillon, Eet al. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest 118: 3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, S, Ott, MG, Schultze-Strasser, S, Jauch, A, Burwinkel, B, Kinner, Aet al. (2010). Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med 16: 198–204. [DOI] [PubMed] [Google Scholar]
- Boztug, K, Schmidt, M, Schwarzer, A, Banerjee, PP, Díez, IA, Dewey, RAet al. (2010). Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med 363: 1918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina, S, Von Kalle, C, Schmidt, M, McCormack, MP, Wulffraat, N, Leboulch, Pet al. (2003). LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302: 415–419. [DOI] [PubMed] [Google Scholar]
- Howe, SJ, Mansour, MR, Schwarzwaelder, K, Bartholomae, C, Hubank, M, Kempski, Het al. (2008). Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest 118: 3143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo, M, Payen, E, Negre, O, Wang, G, Hehir, K, Fusil, Fet al. (2010). Transfusion independence and HMGA2 activation after gene therapy of human ß-thalassaemia. Nature 467: 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell, RS, Beitzel, BF, Schroder, AR, Shinn, P, Chen, H, Berry, CCet al. (2004). Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol 2: E234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, AR, Shinn, P, Chen, H, Berry, C, Ecker, JR and Bushman, F (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110: 521–529. [DOI] [PubMed] [Google Scholar]
- Ciuffi, A, Llano, M, Poeschla, E, Hoffmann, C, Leipzig, J, Shinn, Pet al. (2005). A role for LEDGF/p75 in targeting HIV DNA integration. Nat Med 11: 1287–1289. [DOI] [PubMed] [Google Scholar]
- Pradeepa, MM, Sutherland, HG, Ule, J, Grimes, GR and Bickmore, WA (2012). Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet 8: e1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijsbers, R, Vets, S, De Rijck, J, Ocwieja, KE, Ronen, K, Malani, Net al. (2011). Role of the PWWP domain of lens epithelium-derived growth factor (LEDGF)/p75 cofactor in lentiviral integration targeting. J Biol Chem 286: 41812–41825. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- De Rijck, J, Bartholomeeusen, K, Ceulemans, H, Debyser, Z and Gijsbers, R (2010). High-resolution profiling of the LEDGF/p75 chromatin interaction in the ENCODE region. Nucleic Acids Res 38: 6135–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix, J, Gijsbers, R, De Rijck, J, Voet, A, Hotta, J, McNeely, Met al. (2011). The transcriptional co-activator LEDGF/p75 displays a dynamic scan-and-lock mechanism for chromatin tethering. Nucleic Acids Res 39: 1310–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turlure, F, Maertens, G, Rahman, S, Cherepanov, P and Engelman, A (2006). A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res 34: 1653–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov, P, Devroe, E, Silver, PA and Engelman, A (2004). Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J Biol Chem 279: 48883–48892. [DOI] [PubMed] [Google Scholar]
- Busschots, K, Vercammen, J, Emiliani, S, Benarous, R, Engelborghs, Y, Christ, Fet al. (2005). The interaction of LEDGF/p75 with integrase is lentivirus-specific and promotes DNA binding. J Biol Chem 280: 17841–17847. [DOI] [PubMed] [Google Scholar]
- Maertens, GN, Cherepanov, P and Engelman, A (2006). Transcriptional co-activator p75 binds and tethers the Myc-interacting protein JPO2 to chromatin. J Cell Sci 119(Pt 12): 2563–2571. [DOI] [PubMed] [Google Scholar]
- Bartholomeeusen, K, De Rijck, J, Busschots, K, Desender, L, Gijsbers, R, Emiliani, Set al. (2007). Differential interaction of HIV-1 integrase and JPO2 with the C terminus of LEDGF/p75. J Mol Biol 372: 407–421. [DOI] [PubMed] [Google Scholar]
- Bartholomeeusen, K, Christ, F, Hendrix, J, Rain, JC, Emiliani, S, Benarous, Ret al. (2009). Lens epithelium-derived growth factor/p75 interacts with the transposase-derived DDE domain of PogZ. J Biol Chem 284: 11467–11477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, S, Jenkins, V, Dar, MJ, Engelman, A and Cherepanov, P (2010). Transcriptional co-activator LEDGF interacts with Cdc7-activator of S-phase kinase (ASK) and stimulates its enzymatic activity. J Biol Chem 285: 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, A and Cleary, ML (2008). Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 14: 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijsbers, R, Ronen, K, Vets, S, Malani, N, De Rijck, J, McNeely, Met al. (2010). LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol Ther 18: 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris, AL, Wu, X, Hughes, CM, Stewart, C, Smith, SJ, Milne, TAet al. (2010). Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc Natl Acad Sci USA 107: 3135–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvers, RM, Smith, JA, Schowalter, M, Litwin, S, Liang, Z, Geary, Ket al. (2010). Modification of integration site preferences of an HIV-1-based vector by expression of a novel synthetic protein. Hum Gene Ther 21: 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, MJ, Guelen, L, de Wit, E, Peric-Hupkes, D, Lodén, M, Talhout, Wet al. (2006). Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res 16: 1493–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen, L, King, AA, Xiao, Y, Chanock, SJ, Orkin, SH and Dinauer, MC (1993). Gene targeting of X chromosome-linked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox. Proc Natl Acad Sci USA 90: 9832–9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtkamp, S, Kreiter, S, Selmi, A, Simon, P, Koslowski, M, Huber, Cet al. (2006). Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108: 4009–4017. [DOI] [PubMed] [Google Scholar]
- Cherepanov, P, Sun, ZY, Rahman, S, Maertens, G, Wagner, G and Engelman, A (2005). Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75. Nat Struct Mol Biol 12: 526–532. [DOI] [PubMed] [Google Scholar]
- Ibrahimi, A, Vande Velde, G, Reumers, V, Toelen, J, Thiry, I, Vandeputte, Cet al. (2009). Highly efficient multicistronic lentiviral vectors with peptide 2A sequences. Hum Gene Ther 20: 845–860. [DOI] [PubMed] [Google Scholar]
- Marshall, HM, Ronen, K, Berry, C, Llano, M, Sutherland, H, Saenz, Det al. (2007). Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting. PLoS ONE 2: e1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrijvers, R, De Rijck, J, Demeulemeester, J, Adachi, N, Vets, S, Ronen, Ket al. (2012). LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog 8: e1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski, A, Cuddapah, S, Cui, K, Roh, TY, Schones, DE, Wang, Zet al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837. [DOI] [PubMed] [Google Scholar]
- Wang, Z, Zang, C, Rosenfeld, JA, Schones, DE, Barski, A, Cuddapah, Set al. (2008). Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40: 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel, C, Müller-Kuller, U, Schultze-Strasser, S, Stein, S, Chen-Wichmann, L, Krattenmacher, Aet al. (2012). Physiological regulation of transgene expression by a lentiviral vector containing the A2UCOE linked to a myeloid promoter. Gene Ther 19: 1018–1029. [DOI] [PubMed] [Google Scholar]
- Montini, E, Cesana, D, Schmidt, M, Sanvito, F, Bartholomae, CC, Ranzani, Met al. (2009). The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest 119: 964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo, A, Cesana, D, Genovese, P, Di Stefano, B, Provasi, E, Colombo, DFet al. (2011). Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat Methods 8: 861–869. [DOI] [PubMed] [Google Scholar]
- Suerth, JD, Maetzig, T, Brugman, MH, Heinz, N, Appelt, JU, Kaufmann, KBet al. (2012). Alpharetroviral self-inactivating vectors: long-term transgene expression in murine hematopoietic cells and low genotoxicity. Mol Ther 20: 1022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlwein, O and McClure, MO (2010). Progress and prospects: foamy virus vectors enter a new age. Gene Ther 17: 1423–1429. [DOI] [PubMed] [Google Scholar]
- Mátés, L, Chuah, MK, Belay, E, Jerchow, B, Manoj, N, Acosta-Sanchez, Aet al. (2009). Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet 41: 753–761. [DOI] [PubMed] [Google Scholar]
- Santilli, G, Almarza, E, Brendel, C, Choi, U, Beilin, C, Blundell, MPet al. (2011). Biochemical correction of X-CGD by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells. Mol Ther 19: 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentner, B, Visigalli, I, Hiramatsu, H, Lechman, E, Ungari, S, Giustacchini, Aet al. (2010). Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci Transl Med 2: 58ra84. [DOI] [PubMed] [Google Scholar]
- Hanawa, H, Yamamoto, M, Zhao, H, Shimada, T and Persons, DA (2009). Optimized lentiviral vector design improves titer and transgene expression of vectors containing the chicken beta-globin locus HS4 insulator element. Mol Ther 17: 667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y, Zheng, Z, Cohen, CJ, Gattinoni, L, Palmer, DC, Restifo, NPet al. (2006). High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther 13: 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, DM, Zhao, Y, Liu, X, Jiang, S, Carpenito, C, Kalos, Met al. (2011). Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther 22: 1575–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiehe, JM, Ponsaerts, P, Rojewski, MT, Homann, JM, Greiner, J, Kronawitter, Det al. (2007). mRNA-mediated gene delivery into human progenitor cells promotes highly efficient protein expression. J Cell Mol Med 11: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samulski, RJ, Zhu, X, Xiao, X, Brook, JD, Housman, DE, Epstein, Net al. (1991). Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J 10: 3941–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare, S, Shun, MC, Gupta, SS, Valkov, E, Engelman, A and Cherepanov, P (2009). A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75. PLoS Pathog 5: e1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaison, C, Parsley, K, Brouns, G, Scherr, M, Battmer, K, Kinnon, Cet al. (2002). High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency [correction of imunodeficiency] virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum Gene Ther 13: 803–813. [DOI] [PubMed] [Google Scholar]
- Berry, C, Hannenhalli, S, Leipzig, J and Bushman, FD (2006). Selection of target sites for mobile DNA integration in the human genome. PLoS Comput Biol 2: e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo, LA and Curnutte, JT (1990). Kinetic microplate assay for superoxide production by neutrophils and other phagocytic cells. Meth Enzymol 186: 567–575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Translation of electroporated mRNA in LEDGF KD cells.
Transient expression of CBX1-LEDGF325-530 in LEDGF KD cells rescues LV transduction and retargets LV integration into CBX1-rich heterochromatin regions.
Differentiation status of electroporated X-CGD PLB cells.
Integration frequency near mapped genomic features in LEDGF KD HeLaP4 cells.
Characterization of single clones.
Vector integration sites in single X-CGD PLB clones.
Primers used for cloning.