

Abstract
The intercalated disc (ICD) is a unique structure to the heart and plays vital roles in communication and signaling among cardiomyocytes. ICDs are formed and matured during postnatal development through a profound redistribution of the intercellular junctions, as well as recruitment and assembly of more than 200 proteins at the termini of cardiomyocytes. The molecular mechanism underlying this process is not completely understood. The mouse orthologs (mXinα and mXinβ) of human cardiomyopathy-associated (CMYA)/Xin actin-binding repeat-containing protein (XIRP) genes (CMYA1/XIRP1 and CMYA3/XIRP2, respectively) encode proteins localized to ICDs. Ablation of mXinα results in adult late-onset cardiomyopathy with conduction defects and up-regulation of mXinβ. ICD structural defects are found in adult but not juvenile mXinα-null hearts. On the other hand, loss of mXinβ leads to ICD defects at postnatal day 16.5, a developmental stage when the heart is forming ICDs, suggesting mXinβ is required for ICD formation. Using quantitative Western blot, we showed in this study that mXinβ but not mXinα was uniquely up-regulated during the redistribution of intercellular junction from the lateral membrane of cardiomyocytes to their termini. In the absence of mXinβ, the intercellular junctions failed to be restricted to the termini of the cells, and the onset of such defect correlated with the peak expression of mXinβ. Immunofluorescence staining and subcellular fractionation showed that mXinβ preferentially associated with the forming ICDs, further suggesting that mXinβ functioned locally to promote ICD maturation. In contrast, the spatiotemporal expression profile of mXinα and the lack of more severe ICD defects in mXinα−/−;mXinβ−/− double knockout hearts than in mXinβ−/− hearts suggested that mXinα was not essential for the postnatal formation of ICDs. A two-step model for the development of ICD is proposed where mXinβ is essential for the redistribution of intercellular junction components from the lateral puncta to the cell termini.
Keywords: Xin repeat-containing family of proteins, Intercalated disc, Development, Maturation, N-cadherin, mXinα−/−;, mXinβ−/− double knockout
Introduction
The integration of the contraction and relaxation of billions of individual cardiomyocytes is essential for the heart to function. To carry out such integration, individual cardiomyocytes must be excited at the right moment so that they have coordinated contractions in each heartbeat, which requires electrical coupling between cardiomyocytes. The contractile forces generated by individual cardiomyocytes in turn must be transmitted to the correct neighbors so that the tiny forces from each cardiomyocyte is added up for the heart to perform mechanical work, which requires mechanical coupling between cardiomyocytes. These essential electrical and mechanical couplings are carried out by a cardiac specific structure, the intercalated discs (ICDs). The functions of ICDs have been classically attributed to three types of intercellular junctions (Forbes and Sperelakis, 1985). The gap junctions that are made of connexin permit ions to flow between cardiomyocytes for electrical coupling, the adherens junctions that are organized by N-cadherin confer continuity for the myofibrils between cardiomyocytes and are central for transmitting contractile forces, and the desmosomes that are organized by the desmosomal cadherins couple the sarcolemma to the intermediate filaments to maintain the mechanical integrity of the cardiomyocytes. In addition to these functions classically assigned to the ICDs, it is now increasingly realized that ICDs with more than 200 proteins (Estigoy et al., 2009) are specialized membrane domains of the cardiomyocytes and also function in chemical and mechanical signaling as well as ion transportation (Noorman et al., 2009).
Given the important roles of ICDs, it is not surprising that mutations in genes encoding ICD components can cause severe heart diseases, such as the arrhythmogenic right ventricular cardiomyopathy (Delmar and McKenna, 2010). Conversely, various heart diseases not directly related to mutations of ICD components lead to alterations in the ICDs, and such alterations are likely an important hallmark of the pathology of these diseases (Barker et al., 2002; Noorman et al., 2009; Wang and Gerdes, 1999). The importance of the ICDs in the hearts is further supported by the severe cardiac defects of a number of animal models (Ferreira-Cornwell et al., 2002; Kostetskii et al., 2005; Li et al., 2006; Li and Radice, 2010; Wang et al., 2010). An important theme of these cardiac diseases and defects, either originated from mutations of ICD components or from non-ICD related reasons, is that the molecular integrity of the ICDs are disrupted. The manifested phenotypes include altered morphology, abnormal expression levels and localizations of protein components, as well as changed protein–protein interaction profiles of the ICDs. Thus, the precise structure and the organization of the ICDs are vital for their specific functions, and such structure and organization are prone to be disturbed by pathological factors.
The components of the complex ICDs are assembled mainly during postnatal development as ICDs mature. Several studies have shown that the maturation of ICDs is characterized by drastic reorganization of the distribution of intercellular junctions (Angst et al., 1997; Hirschy et al., 2006; Peters et al., 1994). In embryos, N-cadherin and associated proteins such as β-catenin and mXinα are localized to almost the entire surface of cardiomyocytes in a rather diffused pattern (Sinn et al., 2002). Gap junction and desmosomal proteins are also distributed on the entire surface of cardiomyocytes where cell–cell contacts exist but show more spotted localization than the components of adherens junctions (Coppen et al., 2003; Pieperhoff and Franke, 2007). During postnatal development, the intercellular junctions undergo reorganization by which all three types of intercellular junctions are eventually localized to the ends of cardiomyocytes. Unique to the ICDs of adult mammalian hearts, the adherens junctions also intermix with desmosomes at the molecular level to form a newly identified structure, area composita (Pieperhoff and Franke, 2007, 2008). The molecular mechanisms for the maturation of ICDs are largely unknown. However, since all the classic components of the intercellular junctions are already expressed in the embryonic heart, the postnatal reorganization of the intercellular junctions for the maturation of ICDs must be dictated by additional factors that are expressed/activated during the postnatal life.
One such factor might be the intercalated protein mXinβ, a member of the Xin repeat-containing family of proteins, that is specifically localized to the ICDs in the adult cardiomyocytes (Lin et al., 2005). In mice, the Xin repeat-containing gene family has two members, the mXinα and mXinβ, which encode the mXinα alternatively splicing variants (mXinα and mXinα-a) and mXinβ alternatively splicing variants (mXinβ and mXinβ-a), respectively (Gustafson-Wagner et al., 2007; Wang et al., 2010). The Xin repeats are conserved protein motifs that interact with actin filaments (Choi et al., 2007; Pacholsky et al., 2004). Within its Xin repeat region, the mXinα variants have a conserved β-catenin-interacting domain (Choi et al., 2007). This β-catenin-interacting domain and its counterpart in mXinβ may be responsible for recruiting both the mXin proteins to the adherens junctions, where the mXin proteins may directly couple the N-cadherin–catenin complex to the underlying actin cytoskeleton (Grosskurth et al., 2008; Wang et al., 2012).
Evidence from our previous study indicates that mXinβ may play important roles in the postnatal formation and maturation of ICDs. We observed an up-regulation of mXinβ protein in the heart from postnatal day 0.5 (P0.5) to P13.5. We also found that in the mXinβ−/− hearts at P16.5, the intercellular junctions are punctate, and mature ICD-like structures are sparse (Wang et al., 2010). However, several gaps in our knowledge prevent us from drawing a firm conclusion about mXinβ’s roles in ICD formation. First, although previous studies showed that ICDs are formed postnatally, descriptions of the reorganization of intercellular junctions between P0.5 and P16.5 are not detailed enough for us to correlate this process with the expression profile of mXinβ. Second, the spatiotemporal expression profile was not fully characterized for mXinβ; we do not know the expression profile of mXinβ after P13.5 and whether mXinβ is localized to the adherens junctions throughout their reorganization. Third, the nature of the defects of ICDs in mXinβ−/− hearts was not addressed in our previous study; we do not know whether the ICDs are not formed at all in the mXinβ−/− hearts or are formed but then fail to be maintained. In this study, we asked what specific roles mXinβ plays in the formation of ICDs and answered this question by investigating the quantitative expression profiles of mXinβ and the core protein of adherens junctions, N-cadherin; we also provided a detailed description of the time course of reorganization of the intercellular junctions during ICD formation in wild-type and mXinβ−/− hearts. Our results suggest a direct involvement of mXinβ in the postnatal reorganization of intercellular junctions.
In addition to studying mXinβ’s roles in the maturation of ICDs, we examined mXinα variants’ roles in this process because mXinα variants share many conserved regions with mXinβ but seems incapable of compensating for the loss of mXinβ for the formation of ICDs (Wang et al., 2010). This is in contrast with the compensatory roles of mXinβ for the loss of mXinα we demonstrated previously (Gustafson-Wagner et al., 2007). We asked whether the inability of mXinα variants to compensate for the loss of mXinβ is due to insufficient expression during ICD formation or lack of essential protein function required for this process.
Materials and methods
Generation of mXinα−/−;mXinβ−/− double knockout (DKO) mice
All animal procedures were performed with the approval of the University of Iowa Animal Care and Use Committee and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals approved by the National Institutes of Health. Generation and initial characterization of mXinα−/− (Gustafson-Wagner et al., 2007) or mXinβ−/− (Wang et al., 2010) single knockout (SKO) mice have been previously reported. Both mouse lines were backcrossed to C57BL/6J for at least eight generations and the heterozygotes were maintained in C57BL/6J background. The mXinβ-null mice die before weaning, whereas the mXinβ+/− and the mXinα-null mice are viable and fertile. Therefore, we crossed mXinβ heterozygotes to mXinα-null mice to obtain double heterozygotes, which were then used for the experiments to generate the mXinα−/−;mXinβ−/− DKO mice. Gross morphology, histology and molecular characterization of DKO mice were carried out as previously described for mXinα−/− or mXinβ−/− SKO mice (Gustafson-Wagner et al., 2007; Wang et al., 2010).
Antibodies
Primary antibodies used for both immunofluorescence microscopy and Western blot analysis included rabbit polyclonal antibody (pAb) U1013 (against both mXinα and mXinβ) (Sinn et al., 2002), pAb U1697 (mXinα-specific) (Gustafson-Wagner et al., 2007), and pAb U1040 (mXinβ-specific) (Wang et al., 2010), mouse monoclonal antibody (mAb) 3B9 anti-N-cadherin (Invitrogen, Life Technologies, Grand Island, NY), rat mAb MNCD2 anti-mouse N-cadherin (Developmental Studies Hybridoma Bank, University of Iowa), rabbit pAb anti-connexin 43 (Cx43) (Zymed Laboratories Inc., Invitrogen), rabbit pAb AHP320 anti-desmoplakin (AbD Serotec, Raleigh, NC), mouse mAb 5H10 anti-β-catenin (Zymed Laboratories Inc., Invitrogen), mouse mAb 15D2 anti-p120-catenin (Zymed Laboratories Inc., Invitrogen), and mouse mAb 6C5 anti-GAPDH (RDI Research Diagnostics, Inc., Flanders, NJ). Secondary antibodies included Dylight 488 conjugated goat anti-mouse IgG (Thermo Scientific, Waltham, MA) and Cy5 conjugated goat anti-rabbit IgG (Chemicon International Inc., Temecula, CA) used for immunostaining as well as IRDye 800CW conjugated goat anti-rat IgG (Li-Cor Biosciences, Lincoln, NE), IRDye 800 conjugated goat anti-mouse IgG (Rockland, Gilbertsville, PA) and IRDye 800 conjugated goat anti-rabbit IgG (Rockland) used for Western blot.
Quantitative Western blot
To generate recombinant protein as standard for quantification of mXinβ, we subcloned a cDNA fragment of mXinβ encoding the entire region (aa#1–1293) recognized by anti-Xin antibody U1013 into pGEX4-T2 vector (GE Healthcare, Piscataway, NJ). The GST-fused mXinβ fragment predicted to have molecular weight of 177.1 kD was named GST-mXinβ5′. The GST-mXinβ5′ was expressed in BL21(DE3) pLysS bacteria and affinity purified with Glutathione Sepharose 4B beads (GE Healthcare). To determine the concentration of purified GST-mXinβ5′, we subjected the purified protein to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) alongside with serially diluted Bovine Serum Albumin (BSA) protein standards. The gel was stained by Coomassie Brilliant Blue for 2 h, de-stained, and imaged by EC3 imaging system (Ultra-Violet Products, Ltd., Upland, CA). The intensities of the protein bands were quantified with NIH ImageJ (<http://rsbweb.nih.gov/ij/>) and the concentration of the GST-mXinβ5′ was determined against the intensity–concentration standard curves established with BSA protein standards.
For quantification of mXinβ, heart samples and GST-mXinβ5′ standards were loaded into 6% SDS-polyacrylamide (SDS-PAGE) gels, and Western blot analyses were performed with U1013 antibody against mXin (examples shown in the left column of Fig. 1). For each postnatal stage, samples (each contains 0.125 mg of tissue) from three different hearts were loaded into each gel, and each heart sample was loaded into two lanes as duplicates. In order to generate standard curves, 12.5 ng, 6.25 ng, 3.13 ng, 1.56 ng and 0.88 ng or 50 ng, 25 ng, 12.5 ng, 6.25 ng, and 3.13 ng of GST-mXinβ5′ were loaded into separate lanes in each gel alongside the heart samples. Following electrophoresis, the proteins were transferred overnight at 20 V to nitrocellulose membranes (EMD Millipore, Billerica, MA). Then, Western blot experiments were carried out following the Western Blot Analysis protocol from Li-Cor Biosciences, using U1013 as the primary antibody. The blots were imaged by an Odyssey Imager (Li-Cor Biosciences) and the images were analyzed by ImageJ. One standard curve was generated for each blot by plotting the amount of GST-mXinβ5′ in each lane against the signal intensities of the corresponding protein bands in Western blot. Based on the standard curve, the amount of endogenous mXinβ in each heart sample was calculated (example for P3.5 samples shown in the left plot of Fig. 1).
Fig. 1.

Representative Western blots and plots for determining the amounts of mXinβ, mXinα variants (mXinα and mXinα-a) and N-cadherin in developing postnatal hearts. Western blot analyses of three different samples (I, II and III) in duplicates each at various developmental stages were carried out alongside with two-fold serial dilutions of purified recombinant proteins as standard curve for calculating the amounts of mXinβ (the left column), mXinα variants (the middle column) and N-cadherin (the right column). The red and purple gradient symbols on the top of the gel represent a two-fold serial dilutions of 12.5 ng and 50 ng GST-mXinβ5′, respectively. The blue and green gradient symbols on the top of the gel represent a two-fold serial dilutions of 2.5 ng GST-mXinα5′ and 5 ng 75 kD N-cadherin band, respectively. Examples of standard curve plots for determining the protein amounts at P3.5 samples were shown.
To quantify mXinα variants, we used purified GST-mXinα5′ (86.7 kD) as standard (the middle column and plot of Fig. 1). All procedures were the same as the above description for the quantification of mXinβ except that each of the samples loaded for Western blot contains contents from 0.0125 mg of heart tissue. The amounts of GST-mXinα5′ used for standard were 2.5 ng, 1.25 ng, 0.625 ng, 0.313 ng and 0.156 ng. U1013 was used for Western blot detection.
To quantify N-cadherin, a purified recombinant human N-cadherin extracellular region (Sino Biological Inc., Beijing, PRC) that was expressed in Chinese Hamster Ovary cells as secreted protein was used to generate standard curves. This commercial product contains both the pro- and mature form of the N-cadherin’s extracellular region and runs as two bands (90 kD and 75 kD) in SDS-PAGE under reducing condition. We quantified the mature 75 kD form (calculated molecular weight, 67.5 kD) with BSA standard as described above. For quantitative Western blot, the loading of the heart samples was identical to the ones in the mXinα quantification experiments. To generate standard curve, 5.00 ng, 2.50 ng, 1.25 ng, 0.625 ng and 0.313 ng of the N-cadherin recombinant protein were used. The primary antibody used for the Western blot was the Rat mAb MNCD2 (the right column and plot of Fig. 1).
Immunostaining
Immunostaining were carried out on 4-μm frozen sections of hearts from P3.5 to P60.5. The sections were fixed in 3.7% formaldehyde in phosphate buffered saline (PBS) for 10 min at room temperature, rinsed with PBS and permeabilized with cold acetone (−20 °C) for 5 min. After blocking the sections with the blocking reagent from the Vector Laboratories’s Mouse on Mouse kit (Vector Laboratories Inc., Burlingame, CA) for 1 h at room temperature and followed by blocking with 5% normal goat serum for 30 min, the sections were incubated with primary antibodies diluted with the Pierce Immunostain Enhancer (Thermo Scientific) for 30 min at 37 °C. The sections were then washed with PBS and incubated with secondary antibodies diluted in the Pierce Immunostain Enhancer for 10 min at 37 °C. After wash, the sections were sealed in antifading reagent (Gelvatol) and covered by coverslips. Sections were imaged with Leica TCS SPE confocal microscope with an ACS APO 40 × N.A. 1.15 oil objective.
Quantification of confocal images
Quantification was carried out with ImageJ. Briefly, confocal images (from each single optic section) of longitudinally sectioned cardiomyocytes were first randomly selected. The total integrated immunofluorescence signals (pixel number × intensity) were measured by ImageJ after subtracting background using its default threshold function. Then the ICD/ICD-like signals (defined as signal clusters that have one dimension as wide as the width of the cardiomyocyte’s terminus where the signals reside) were masked by black pixels in the images and the integrated immunofluorescence signals (non-ICD signals) were measured again. The ICD-signal was then calculated by subtracting the non-ICD signals from the total signals.
For quantification and statistical test of the ICD/total signal ratio, we used at least 5 confocal images of each heart and for each stage, at least two control hearts and two mutant hearts were used. The ICD/total signal ratio of each image was treated as a sample when carrying out Student’s t-test.
ImageJ’s colocalization plugin was initially used to analyze the spatial correlation between N-cadherin and desmoplakin/Cx43 from the confocal images of double-labeled samples. A strong overlap (colocalization) between N-cadherin and desmoplakin signals was obtained with a Person’s correlation coefficient, Rr=0.80. On the other hand, a reasonable but not very strong correlation between N-cadherin and Cx43 was observed with a Pearson’s correlation coefficient, Rr=0.24. To further examine the association between Cx43 and N-cadherin signals, we used the find maxima function of ImageJ to determine the x- and y-coordinates of each signal spot in confocal images and output the coordinates into Microsoft Excel. To find the closest N-cadherin spot for a specific Cx43 spot, the minimal distance from all N-cadherin spots in the same images were determined for the specific Cx43 spot with a custom Excel macro, RED/GREEN DOT PROCESSOR.
Subcellular fractionation
Subcellular fractionation experiments were carried out following a modified protocol that was used in isolating ICDs from large quantity of adult heart tissues (Colaco and Evans, 1981). Briefly, one or two hearts were homogenized in ice-cold homogenization buffer (10 mM imidazole buffer, pH 7.0, containing complete protease inhibitor cocktail from Roche) with a Multigen 7 homogenizer (Pro Scientific, Inc., Oxford, CT) for 2.0 min at full-speed and followed by Dounce treatment with pestle B (loose fitting) for 30 times. The homogenate was layered on top of a sucrose step gradient made with the homogenization buffer in the Beckman Ultra-Clear Tubes (14×89 mm). The steps of the gradient from the bottom to the top of the tubes were: 51.5% (w/w), 46.5%, 41.5%, 36.0%, 30.0% and 23.0%. After centrifugation for 20 h at 28,000 RPM with an SW41 rotor, 20 consecutive 0.6-ml fractions were collected from the bottom of the tube by puncturing the tube with a syringe needle. The fractions were analyzed by Western blot following a standard protocol.
Results
The adherens junction proteins, mXinβ, mXinα and N-cadherin, have unique temporal expression profiles during postnatal development
To determine the roles of mXinβ in the formation of ICDs, we first examined mXinβ’s temporal expression profile in the mouse heart during postnatal stages from the initiation to the completion of the maturation process of ICDs (P0.5–P60.5). The expression profile of mXinβ was established by quantitative Western blot experiments, which allowed us to determine the molar amount of mXinβ expressed in the heart. This facilitated comparisons of the expression of mXinβ among different developmental stages and to that of N-cadherin and mXinα. Using this approach, we measured the amounts of mXinβ, mXinα and N-cadherin expressed in the heart at multiple time points from P0.5 to P60.5, with three hearts for each time point. Fig. 2A shows that during this period, mXinβ’s expression in the heart fluctuates between 0.19±0.03 pmoles/mg heart at lowest expression level (P20.5) and 0.59±0.08 pmoles/mg heart at highest expression level (P13.5). Since mXinβ is expressed specifically in the cardiomyocytes, which make up the majority of the volume of the heart (Legato, 1979), the concentration of mXinβ in the cardiomyocytes is estimated to be about 0.2–0.6 μM. However, because mXinβ is not uniformly localized in the cardiomyocytes, the local concentration of mXinβ at the ICDs could be much higher. By plotting the expression level of mXinβ as concentration (Fig. 2A) and as amount per heart (Fig. 2B) against the age of the mice, our data revealed a three-phase expression profile of mXinβ between P0.5 and P60.5: a rapid up-regulation between P0.5 and P13.5, followed by a sharp down-regulation between P13.5 and P20.5 and then an up-regulation from P20.5 to P60.5. Since heart weight (HW) varies by body weight (BW) even at the same age, the expression levels of mXinβ were corrected with their respective normalized heart weights (i.e., HW/BW ratios). A similar three-phase expression profile of mXinβ was also evident (Fig. 3A). After corrected with the heart weights (data not shown), fold changes of mXinβ expression relative to that at P0.5 revealed a sharp 13.2-fold increase at P13.5, a graduate returning to only 4.6-fold increase at P20.5, and then continuous increase to 19.3-fold at P60.5 (Fig. 4A). Neither mXinα variants nor N-cadherin followed this type of fold change pattern of expression (Fig. 4A). Thus, between P0.5 and P60.5, the expression of mXinβ is dynamically regulated. The drastic up-regulation of mXinβ both in concentration and total amount between P0.5 and P13.5 suggests that mXinβ may play important roles during this period. In addition, a continuous increase of mXinβ expression after P30.5 further suggests that mXinβ may be required for the maintenance and integrity of adult ICDs.
Fig. 2.

Temporal expression profiles of mXinβ, mXinα variants and N-cadherin in developing postnatal hearts established by quantitative Western blot. The amounts of the proteins per mg of total heart extracts (A, C and E) and in the entire heart (B, D and F) are plotted against age to establish their developmental expression profiles. mXinβ (A and B); mXinα variants (C and D); N-cadherin (E and F). Each point represents the mean from three independent heart samples. Error bars represent standard errors.
Fig. 3.

Temporal expression profiles of mXinβ, mXinαvariants and N-cadherin after normalized with HW/BW ratio in developing postnatal hearts established by quantitative Western blot. mXinβ (A); mXinα variants (B); N-cadherin (C). Each point represents the mean from three independent heart samples. Error bars represent standard errors.
Fig. 4.

(A) Comparison of fold change plots for mXinβ, mXinα variants and N-cadherin expression. Temporal expression profiles shown in Fig. 2B, D and F were re-calculated as fold changes relative to that at P0.5 and plotted against age. (B) Comparison of the expressions of mXin proteins with that of N-cadherin. Data are compiled from the same quantitative Western blots as in Fig. 2B, D and F.
Since the young mXinα-null animals have normal ICDs (Chan et al., 2011; Gustafson-Wagner et al., 2007), we asked whether the mXinα variants would have a different expression profile compared to mXinβ during the period of ICD formation. We carried out quantitative Western blot experiments to determine the expression profiles of both mXinα variants with the same heart samples used for the mXinβ experiments. The data showed that each mXinα variant was expressed at about five times the level of mXinβ at P0.5, and the difference between mXinα variants and mXinβ became smaller as the heart matured (compare Fig. 2A and B with C and D). The concentrations of mXinα variants in the cardiomyocytes were estimated to be between 0.4 μM and 1.3 μM. The expression profiles of mXinα variants between P0.5 and P60.5 were distinct from the three-phase expression of mXinβ. Specifically, from P0.5 to P13.5, total amounts of mXinα and mXinα-a were expressed at relatively constant levels per mg of heart extract (Fig. 2C). Similar to mXinβ from P13.5 to P20.5, the amounts of mXinα and mXinα-a expressed per mg of heart tissue decreased by slightly more than half. After P20.5, the mXinα variants initially up-regulated and reached a peak at p30.5 but then down-regulated again, in contrast with the continuous up-regulation of mXinβ from P20.5 to P60.5 (Fig. 2C). The profiles of the total amounts of mXinα variants in each heart were accordingly very different from that of mXinβ (Fig. 2D). Interestingly, although mXinα and mXinα-a were expressed at almost the same levels at P0.5 (1.14±0.01 pmole/mg of heart tissue and 1.11±0.03 pmole/mg of heart tissue, respectively), their expression diverged at P3.5. mXinα-a was expressed at lower levels than mXinα from P3.5 onward, suggesting the mXinα variants may play different roles in prenatal and postnatal life. In summary, mXinα variants have distinct profiles of expression compared to mXinβ. In particular, concentrations of mXinα variants per mg of heart extract remain relatively constant between P0.5 and P13.5, similar to the expression profile of N-cadherin (Fig. 2E). These suggest that mXinα variants may play certain constitutive roles during this period but they are unlikely to be directly involved in initiating specific developmental changes, such as ICD formation, that start within this period.
The dynamic expression profiles of mXinβ and mXinα variants promoted us to ask whether their expression profiles reflect the change of the expression of the core adherens junction protein, N-cadherin. Thus, we quantified the expression profile of N-cadherin with the above heart samples by quantitative Western blot experiments. Plotting the expression of N-cadherin per mg of heart tissue against age showed that N-cadherin level followed a slowly reducing trend from P0.5 to P60.5 with minor fluctuations during the process (Fig. 2E). From P0.5 to P13.5, N-cadherin expression per mg heart reduced slightly by 8.9% (from 2.49±0.09 pmole to 2.26±0.09 pmole/mg of heart extract). By P60.5, N-cadherin was expressed at 1.50±0.14 pmole/mg of heart extract, a 39.6% reduction from P0.5. When the total N-cadherin expressed in each heart or after normalized with HW/BW ratio was plotted against age, it was clear that expression of N-cadherin had two phases: it rapidly increased from P0.5 to P15.5, and then remained almost unchanged from P15.5 to P60.5 (Figs. 2F and 3C). The expression profile of N-cadherin seemed to reflect the postnatal growth of the heart. Plotting the heart weights against age showed that postnatal growth of the heart also had two phases: a rapid growth phase from P0.5 to P15.5 and a slow growth phase from P15.5 to P60.5 (data not shown). The transition of the growth phases of the heart at P15.5 coincided with the time point when N-cadherin expressed in each heart reached the plateau. It is remarkable that after P15.5, total amount of N-cadherin per heart remained constant despite the slow albeit continuous growth of the heart. Thus, these data show that the expression profiles of mXinβ and mXinα variants do not simply follow that of N-cadherin; the unique rapid up-regulation of mXinβ between P0.5 and P13.5 likely reflects important roles that mXinβ plays during this period.
Total amount of mXin proteins is similar to that of N-cadherin except at P20.5
We also directly compared the total amount of mXin proteins with that of N-cadherin (Fig. 4B), because all the mXin proteins might associate with N-cadherin through β-catenin, thus a quantitative relationship might exist. Interestingly, between P0.5 and P60.5, the total amount of mXin proteins is similar to that of N-cadherin in a majority of the time points we studied. The correlation between the total levels of the mXin proteins and N-cadherin suggests that N-cadherin may influence the expression of mXin and/or mXin proteins may influence the level of N-cadherin. The comparison also revealed that mXinβ was only expressed at a small fraction of N-cadherin. At P0.5, mXinβ was only expressed at 8.7% of the level of N-cadherin; this number increased to 26.6% at P13.5, when mXinβ expression reached a peak. Such quantitative relationship indicates that even if a majority of mXinβ associates with N-cadherin/β-catenin complexes, only a minor population of N-cadherin/β-catenin complexes could have mXinβ as their partners. Because mXinβ is required for the development of normal ICDs by P16.5, we asked if mXinβ is specifically associated with the populations of N-cadherin that are being incorporated into the maturing ICDs.
mXinβ but not mXinα variants preferentially associates with a subpopulation of N-cadherin at the forming ICDs
To determine if mXinβ is specifically associated with the population of N-cadherin that is being incorporated into the forming ICDs, we did double-label immunofluorescence staining of mXinβ and N-cadherin on frozen sections of postnatal hearts. Representative confocal images of P7.5 and P24.5 sections are shown (Fig. 5). We found that at P7.5, N-cadherin was distributed extensively on the surface of cardiomyocytes (Fig. 5A and C). On the lateral surface of the many cardiomyocytes, N-cadherin staining is characterized by almost continuous signal interspaced with strongly stained puncta. Larger N-cadherin clusters can be found on the termini of the cardiomyocytes, where the ICDs are being formed (Fig. 5A, arrows). Interestingly, at this stage, mXinβ was sparse and associated with the bright N-cadherin puncta that were found mainly at the longitudinal termini of the cardiomyocytes (Fig. 5B and C). A similar phenomenon was found in both P3.5 and P13.5 hearts (data not shown). Thus, during ICD formation only a subpopulation of N-cadherin-containing complexes were associated with detectable levels of mXinβ, and importantly, the mXinβ–N-cadherin co-localization was primarily found at the forming ICDs located at termini of cardiomyocytes. At both P21.5 (data not shown) and P24.5, the laterally localized N-cadherin largely disappeared and almost all N-cadherin signals were found to be highly co-localized with mXinβ at the termini of the cardiomyocytes (white color in Fig. 5F). Thus, during ICD formation, mXinβ is preferentially associated with a subpopulation of N-cadherin at the forming ICDs located at the termini of cardiomyocytes. It should be noted that the relative amounts of mXinβ estimated by immunofluorescence at P7.5 versus P24.5 seems to contradict to that shown in Fig. 2A (concentration of mXinβ) determined by quantitative Western blot. However, consistent results could be obtained if the data after normalized with either heart weight (Fig. 2B) or HW/BW ratio (Fig. 3A) were plotted. Furthermore, the indirect-label immunofluorescence could not be in general used to accurately quantify/compare the amount of mXinβ from different sections/samples, because of non-uniform expression nature of mXinβ throughout the heart (Gustafson-Wagner et al., 2007; Wang et al., 2012).
Fig. 5.
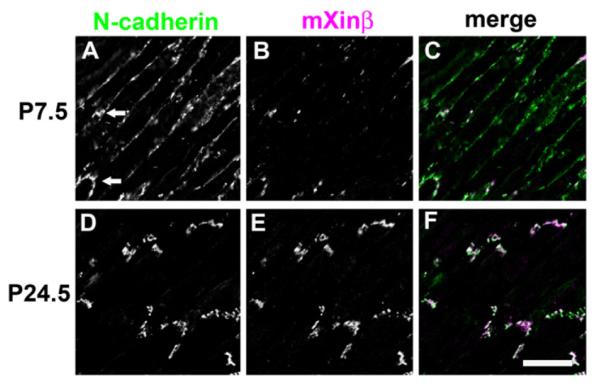
Characterization of the co-localization between mXinβ and N-cadherin during postnatal heart development. Confocal images of double-immunofluorescence labeled frozen sections of wild-type hearts for N-cadherin (A and D) and mXinβ (B and E). (A–C) P7.5 and (D–F) P24.5. Merged images are also shown (C and F). Bar=20 μm.
On the other hand, double labeling of N-cadherin and mXinα/mXinα-a showed that at P7.5 (Fig. 6A–C) a majority of the N-cadherin signal overlapped with the mXinα/mXinα-a signal (white color in Fig. 6C); only a few lateral surface areas showed weakly stained N-cadherin but no mXinα/mXinα-a. Importantly, mXinα/mXinα-a seems to have no preference for the forming ICDs at the termini of the cardiomyocytes. At both P21.5 (data not shown) and P24.5, N-cadherin was highly co-localized with mXinα/mXinα-a in the ICDs, but a fraction of non-colocalized mXinα variants remained evident (Fig. 6D–F). Although the varied microscopic localization of mXinβ versus mXinα variants seems to be dependent on the amount of expression of each of these isoforms in the cardiomyocytes, this is not completely true. These microscopic results together with the fold change plots (Fig. 4A) and the subcellular fractionations to be described below (Fig. 7) clearly suggest that only mXinβ, but not mXinα/mXinα-a preferentially associates with the subpopulation of N-cadherin at the termini of cardiomyocytes where the ICDs are being formed, further supporting mXinβ’s specific roles in the formation of ICDs.
Fig. 6.
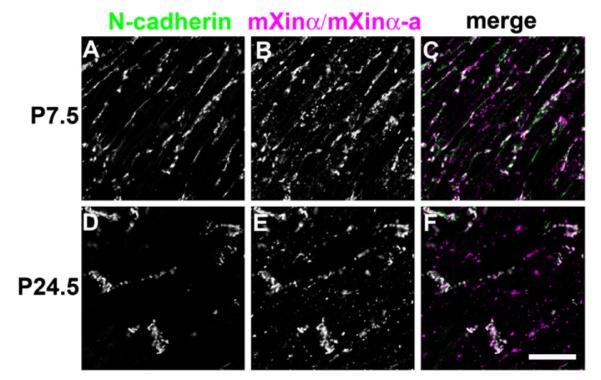
Characterization of the co-localization between mXinα variants and N-cadherin during postnatal heart development. Confocal images of double-immunofluorescence labeled frozen sections of wild-type hearts for N-cadherin (A and D) and mXinα variants (B and E). (A–C) P7.5 and (D–F) P24.5. Merged images are also shown (C and F). Bar=20 μm.
Fig. 7.

Subcellular fractionation provided evidence for the preferential association of mXinβ with the maturing/matured ICDs. (A) Representative profiles (from P39.5 hearts) of the distributions of N-cadherin and mXinβ in the sucrose gradient. The N-cadherin is clearly present in three distinct parts of the gradient, designated as peaks I, II, and III. mXinβ is concentrated in peak I. (B) Percentage of N-cadherin and mXinβ distributed in each peak at different developmental stages. A developmental increase of N-cadherin in the peak I indicates that peak I contains the mature ICDs, consistent with the previous identification of this peak fraction by electron microscopy (Kargacin et al., 2006). After P18.5, a larger proportion of mXinβ than N-cadherin is associated with peak I.
mXinβ preferentially associates with a subcellular fraction containing the forming ICDs
We provided an additional line of evidence to support the preferential association of mXinβ with the subpopulation of N-cadherin that is incorporated into the mature ICDs by subcellular fractionation of developing postnatal hearts. Total homogenates of hearts from P12.5, P18.5, P24.5, P39.5 and P90.5 were fractionated by sucrose buoyant density gradient centrifugation. When the subcellular fractions were analyzed by Western blot, most N-cadherin and mXinβ were present in three peaks of the gradient (I, II and III) that had different densities (representative P39.5 profile shown in Fig. 7A). We found an increasing proportion of N-cadherin in the peak I as the age of the mice increases from P12.5 to P90.5 (Fig. 7B), which strongly suggests that the peak I may contain the subcellular fraction of mature ICDs. Previous studies by electron microscopy also indicated that mature ICDs are enriched in this peak I (Colaco and Evans, 1982). Importantly, we found that a higher proportion of mXinβ than that of N-cadherin was present in the peak I of the gradient at stages examined from P18.5 to P90.5 (Fig. 7B), which further supported the observed preferential association of mXinβ with N-cadherin at the forming ICDs of cardiomyocytes.
ICD defects in mXinβ−/− hearts first appear at the time when mXinβ is normally expressed at its peak level in the wild-type hearts
Previously, we have observed that ICD components such as N-cadherin failed to be localized to the termini of cardiomyocytes in P16.5 mXinβ−/− hearts (Wang et al., 2010). To determine the timing of the first appearance of such defects and its relationship to the temporal expression profile of mXinβ in wild-type hearts, we examined the distribution pattern of N-cadherin during the formation of ICDs (Fig. 8). Confocal images of frozen sections labeled for N-cadherin showed that in the wild-type hearts at P3.5 and P7.5 (Fig. 8A and C), lateral puncta of N-cadherin signal were numerous and large clusters of N-cadherin at the termini were few. Terminal localization of N-cadherin clearly increased after P7.5 in that at P13.5, the lateral puncta diminished, while large clusters that demarcated ICDs at the termini became frequent (Fig. 8E). After P13.5, lateral N-cadherin puncta continued to disappear while the terminal N-cadherin signals were accentuated (Fig. 8G and I). In the mXinβ−/− hearts, N-cadherin staining pattern was initially indistinguishable from that of the wild-type hearts at P3.5 and P7.5 (Fig. 8B and D), but in the mutant hearts at later stages, lateral puncta of N-cadherin remained numerous and ICD-like structures were much less frequent (Fig. 8F, H and J). As previously reported (Wang et al., 2010), most (>70%) of mXinβ-null mice died within 2 weeks of age. Also, all mXinβ-null hearts examined showed various degree of mis-organized myocardium with no myofilament disarray. The P24.5 knockout mice used in this study may represent those with less degree of mis-organization in myocardium; however, all of these mXinβ-null mice at P24.5 showed severe growth retardation and reduced activity.
Fig. 8.

Time courses of ICD maturation in the postnatal wild-type and mXinβ−/− hearts characterized by N-cadherin localization. Confocal images of frozen sections labeled for N-cadherin in wild-type (A, C, E, G and I) and mXinβ−/− (B, D, F, H and J) hearts. Ages of the mice are shown at the left side of the images. Bar=15 μm.
To confirm the apparent onset of the defects in the distribution N-cadherin after P7.5, we quantified the incorporation of N-cadherin into the termini of cardiomyocytes from P3.5 to P60.5 in the confocal images (Fig. 9A). The ratios of fluorescence signals located at the termini of the cardiomyocytes versus the total signals in the entire cardiomyocytes were calculated from confocal images of frozen sections of each developmental stage. In the wild-type hearts, the terminal distribution of N-cadherin continuously increased during the developmental stages studied until P60.5 (Fig. 9A, blue bars), consistent with previously reported time course of ICD maturation (Angst et al., 1997). In contrast, the localization of N-cadherin to the termini proceeded slowly in the mXinβ-null hearts (Fig. 9A, orange bars). By P13.5, statistically significant differences of the terminal localization of N-cadherin between wild-type and mXinβ−/− hearts were observed. These observations suggest that the redistributions of intercellular junctions proceed quickly between P3.5 and P13.5, and mXinβ plays indispensable roles during this period. Without mXinβ, N-cadherin failed to be restricted to the termini of cardiomyocytes by the time when mXinβ was expressed at its peak level in the wild-type hearts. Based on previous observations of N-cadherin distribution in embryonic hearts (Hirschy et al., 2006; Pieperhoff and Franke, 2007; Sinn et al., 2002) and the current study on the postnatal process, we propose a bi-phasic process of ICD formation in mice. Firstly, N-cadherin molecules uniformly distributed at periphery of embryonic cardiomyocytes aggregate/cluster to form the lateral spots/puncta during the first week of postnatal life. Secondly, the lateral spots/puncta redistribute to the cell’s longitudinal termini to form ICD from the second week postnatally to at least P24.5 (Fig. 9B). In the mXinβ-null hearts, this second phase is blunted, leading to the failure of ICD formation.
Fig. 9.

Comparison of the proportions of ICD-localized N-cadherins during ICD maturation in wild-type and mXinβ−/− hearts. (A) Quantification of the ratios of terminally localized N-cadherin signals in wild-type and mXinβ−/− hearts. *p<0.05 significant difference between age-matched wild type and mXinβ-null hearts. (B) Bi-phasic process of ICD maturation. N-cadherin localization is schematically diagramed in the cardiomyocytes of developing mouse hearts. During the embryonic stages, N-cadherin locates uniformly along the periphery of cardiomyocytes, particularly at the cell–cell contacts (Sinn et al., 2002). From newborn to one week of age, N-cadherin aggregates into spots/puncta mainly localized along the lateral side of cardiomyocytes (the first step of ICD formation). The redistribution of these lateral N-cadherin puncta into terminal ICD localization initiates between P7.5 and P13.5 (the second step of ICD formation), coinciding with a surged expression of mXinβ. At P24.5 and older, the majority of N-cadherin forms mature ICD at cell’s termini. The mXinβ-null heart appears to have a normal first step of ICD formation but fails to redistribute N-cadherin from the lateral puncta to the terminal ICD (defective in the second step of ICD maturation). Cardiomyocytes’ surfaces drawn with gray lines and the N-cadherin immunofluorescence signals were shown in red.
Desmosomes and gap junctions also fail to be restricted to the termini of cardiomyocytes in mXinβ−/− hearts
During the establishment and maturation of intercellular junctions in various tissues, the adherens junctions have a leading role in determining the distribution of desmosomes and gap junctions (Green et al., 2010; Hertig et al., 1996a, 1996b). Thus, we asked if mXinβ−/− hearts have corresponding defects in the distributions of desmosomes and gap junctions. Quantification of the incorporation of desmoplakin (a desmosome marker) and Cx43 (a gap junction marker) in confocal images of developing hearts was carried out (Fig. 10). The terminal distribution of desmoplakin showed a trend (Fig. 10A) similar to that of N-cadherin between P3.5 and P24.5 in the wild-type hearts (Fig. 9A). Accordingly, a significantly lower proportion of desmoplakin in the mXinβ−/− cardiomyocytes’ termini was observed at P13.5 (Fig. 10A). On the other hand, the terminal distribution of Cx43 increased abruptly between P13.5 and P15.5 in the wild-type hearts, consistent with the reported lag of gap junction’s re-distribution during ICD maturation (Angst et al., 1997; Hirschy et al., 2006), and the defect of Cx43 localization in the mXinβ−/− hearts became significant at P15.5 (Fig. 10B). These data suggest that mXinβ−/− hearts have a general defect in the developmental reorganization of all three types of intercellular junctions, resulting in failure of forming ICDs.
Fig. 10.

Characterization of the distributions of desmosome and gap junctions in the postnatal wild-type and mXinβ−/− hearts. Quantification of the ratios of terminally localized desmoplakin (A) and Cx43 (B) signals in wild-type and mXinβ−/− hearts. *p<0.05 significant difference between age-matched wild type and mXinβ-null hearts.
Intercellular junction components retain their close spatial relationship in mXinβ−/− hearts despite being mis-localized
To determine if the failure to restrict intercellular junctions to the termini of cardiomyocytes in the mXinβ−/− hearts could be a result of disrupted association between intercellular junctions which is normally found in the mature ICDs, we used double-label immunofluorescence staining to examine the relationship between N-cadherin and desmoplakin (Fig. 11), as well as N-cadherin and Cx43 (Fig. 12) in the mXinβ−/− hearts. The results showed that co-localization between N-cadherin and desmoplakin was indistinguishable between wild-type and mXinβ−/− hearts at both P7.5 and P24.5 (Fig. 11) even though at P24.5, both proteins were grossly mis-localized at the cellular level in the mXinβ−/− hearts. Interestingly, it was noted that the mXinβ−/− hearts at P24.5, large clusters of N-cadherin/desmoplakin signals can occasionally be found at the termini of some cardiomyocytes, and these clusters resembled the ICDs of the wild-type hearts (Fig. 11L, arrows). Despite the clear defects in restricting the intercellular junctions to the termini of cardiomyocytes at P24.5, the mutant hearts at this stage also reduced the diffused N-cadherin signals on the lateral surfaces of the cardiomyocytes and assembled discrete clusters of N-cadherin that co-localized with desmoplakin (Fig. 11). Many of these lateral N-cadherin/desmoplakin clusters were elongated and adopted a perpendicular orientation relative to the longitudinal axis of the cardiomyocytes (Fig. 11L, yellow stars). This was in contrast with the N-cadherin spots in the P7.5 hearts, which were smaller and elongate along the longitudinal axis of cardiomyocytes on the lateral surfaces (Fig. 11A–F).
Fig. 11.

Confocal images of double labeled frozen sections demonstrating the preserved co-localization between N-cadherin and desmoplakin in the mXinβ−/− hearts. (A, D, G, and J) N-cadherin (green); (B, E, H, and K) desmoplakin (magenta); and (C, F, I, and L) merged images. Age and genotypes are labeled at the left side of the images. Bar=20 μm.
Fig. 12.

Confocal images of double labeled frozen sections demonstrating the preserved association between N-cadherin and Cx43 in the mXinβ−/− hearts. (A, D, G, and J) N-cadherin (green); (B, E, H, and K) Cx43 (magenta); and (C, F, I, and L) merged images. Age and genotypes are labeled at the left side of the images. Bar=20 μm.
Similarly, Cx43 also showed preserved association with N-cadherin both before and after the localization of intercellular junctions was grossly disturbed in the mutant hearts (Fig. 12). Close observation showed that in the mXinβ−/− hearts at P24.5, the numerous Cx43 spots at the lateral surface of the cardiomyocytes almost always had at least one N-cadherin spot nearby (Fig. 12L and Fig. 13). Since N-cadherin and Cx43 signals did not overlap, it was thus not possible to do co-localization test. We then measured the distance from each Cx43 spot to its closest N-cadherin spot (examples of center positions of the immunofluorescence signal spots identified by ImageJ were shown in Fig. 13A’ and B’). No statistically significant difference was found between such distances in wild-type and mXinβ−/− hearts at both P7.5 and P24.5 (Fig. 13C, Rank sum test, n>2000, p>0.05), suggesting that in the mXinβ−/− hearts, N-cadherin and Cx43 retained normal spatial relationship. Thus, the mXinβ−/− hearts seemed to have numerous miniature ICD-like structures containing all three types of intercellular junction components that were ectopically formed at the lateral surface of cardiomyocytes. The above results are consistent with our observations with electron microscopy, which showed that the ultrastructure of ICDs seemed to be largely preserved in P15.5 mXinβ−/− hearts and that the adherens junctions, gap junctions and desmosomes were all found in close proximity (Wang et al., 2010). Although we did not know whether the distance between mXinβ and Cx43 changed during ICD development, our data showed that there was no significant change at the distance between N-cadherin and nearest Cx43 during development (Fig. 13C).
Fig. 13.

Quantification of the distances between Cx43 and N-cadherin immunofluorescence signal spots. Frozen sections from P24.5 wild-type (A) and mXinβ−/− (B) hearts double-labeled for N-cadherin (green) and Cx43 (magenta) and the positions of immunofluorescence spots located by the find maxima function of ImageJ software from the confocal images (A’ and B’). Bar=5 μm. (C) Box plots of the distances between each Cx43 immunofluorescence spot to its closest N-cadherin spot in P7.5 and P24.5 wild-type and mXinβ−/− heart sections. No significant differences were found between the wild-type and mXinβ−/− hearts at same age by Rank Sum tests. In addition, there was no significant change in the distance between N-cadherin and Cx43 during development. N.S.: no significant difference.
mXinα variants are not essential for the formation of ICDs
While the mXinα and mXinα-a are the prevalent mXin proteins in the P0.5 and P30.5 (Figs. 1–4), the unique ICD formation defect in mXinβ−/− hearts, and the absence of apparent ICD defects in the mXinα−/− hearts from young animals strongly suggest that mXinα variants are not essential for ICD formation. To further test this idea, we generated mXinα−/−;mXinβ−/− DKO animals and studied their ICDs by immunostaining (Fig. 14) and Western blot (Fig. 15). The loss of both mXinα and mXinβ expression was confirmed by immunostaining (Fig. 14E) and Western blot (Fig. 15) with an antibody recognizing both mXinα and mXinβ (U1013). We found that the loss of both mXinα and mXinβ led to ICD defects indistinguishable from the defects caused by the loss of mXinβ alone (compare Fig. 14 to Figs. 11 and 12). In the P19.5 DKO hearts, the N-cadherin, desmoplakin and Cx43 were scattered as discrete spots on the surface of cardiomyocytes (Fig. 14G–L). Similar to the mXinβ−/− hearts, the ICD-like structures in the DKO hearts showed apparently normal co-localization between N-cadherin and desmoplakin, as well as normal association between N-cadherin and Cx43. Furthermore, Western blot analysis showed no difference in the expression of N-cadherin, desmoplakin and Cx43 in P13.5 wild-type, mXinα−/−, mXinβ−/− and DKO hearts (Fig. 15). Thus, mXinα variants are not essential for the formation of ICDs, and further loss of mXinα variants in the mXinβ−/− hearts does not contribute to more severe defects in the formation of ICDs.
Fig. 14.

Confocal images of double-labeled frozen sections from P19.5 wild-type (A–C) and mXinα−/−:mXinβ−/− double knockout (DKO) (D–L) hearts. (A, D, G, and J) N-cadherin (green); (B and E) total mXin (magenta); (H) desmoplakin (magenta); (K) Cx43 (magenta). (C, F, I, and L) are merged images from their corresponding left panels. Bar=20 μm.
Fig. 15.

Western blot detection of representative intercellular junction proteins in wild-type (lanes 1–3), mXinα−/− SKO (lanes 4–6), mXinβ−/− SKO (lanes 7–9) and mXinα−/− ;mXinβ−/− DKO (lanes 10–12) hearts. Total protein extracts were prepared from three individual hearts of each genotype mice at P12–14, separated by gradient SDS-PAGE and immunoblotted with specific antibodies against various intercellular junction proteins. The band (indicated by *) occasionally detected in the mXinβ blot of mXinβ−/− SKO sample but not DKO sample had a different mobility from mXinβ band. This band may represent the aggregate of mXinα-a. There was no statistically significant difference among wild-type, SKO and DKO hearts in the expressions of N-cadherin, β-catenin, p120-catenin, desmoplakin, and Cx43.
Discussion
mXinβ plays essential roles in the maturation of ICDs
We provided several lines of evidence to establish mXinβ’s critical roles in the postnatal reorganization of intercellular junctions that leads to the restricted localization of ICD components to the termini of cardiomyocytes. The first line of evidence is the strong correlation between the timing of reorganization of intercellular junctions, which leads to the maturation of ICDs, and mXinβ’s unique temporal expression profile. In this study, we provided a detailed description of the process of ICD maturation from P3.5 to P60.5. Our findings agree with previous reports that in mammals, ICD maturation is a postnatal process, during which the three types of intercellular junctions found in adult ICDs redistribute from the entire surface of cardiomyocytes to the longitudinal termini of these cells (Angst et al., 1997; Hirschy et al., 2006; Peters et al., 1994). Consistent with previous studies, we showed that maturation of ICDs takes more than a month in rodents. We also confirmed that the time courses of incorporating adherens junctions (N-cadherin as the marker) and desmosomes (desmoplakin as the marker) to the longitudinal termini of cardiomyocytes are similar. Meanwhile, the incorporation of gap junctions (Cx43 as the marker) lags behind the two types of adhering junctions. More importantly, we provided novel insights for the time course of ICD maturation by showing that the maturation of ICDs progressed most rapidly between P3.5 and P13.5. Indeed, the percentage of terminally localized N-cadherin increased by 3.3 fold in 10 days (from P3.5 to P13.5), whereas the next comparable degree of increase (2.3 fold) happened in 47 days (from P13.5 to P60.5) (Fig. 9A). Significantly, the sharp increase of the terminal localization of N-cadherin correlated very well with the very rapid increase of mXinβ concentration between P0.5 and P13.5, as established by quantitative Western blot, suggesting that mXinβ may have important roles in this phase of postnatal ICD maturation.
The second line of evidence supporting mXinβ’s role in ICD maturation is that during this process, mXinβ was preferentially targeted to the maturing ICDs at the termini of cardiomyocytes where it co-localized with N-cadherin (Fig. 5). Such preferential localization was unique to mXinβ because the mXinα variants were found to be co-localized with N-cadherin both at the termini and the lateral surface of the cardiomyocytes (Fig. 6). Although we did observe a small amount of mXinβ signals at the lateral surface that were co-localized with bright N-cadherin puncta, the majority of N-cadherin puncta at the lateral surface did not have detectable levels of mXinβ. The preferential association of mXinβ with a subpopulation of N-cadherin-containing complexes was further supported by subcellular fractionation experiments (Fig. 7), which revealed that mXinβ preferentially associated with the mature ICD-containing fraction during development. This preferential association of mXinβ with the maturing ICDs suggests that mXinβ may be directly involved in the initiation of ICD maturation. The facts that mXinβ messages are previously found to locate to the ICD of adult hearts (Gustafson-Wagner et al., 2007; Wang et al., 2012), and mXinβ possesses a conserved β-catenin-binding domain (Choi et al., 2007; Grosskurth et al., 2008) are in line of the mechanism by which mXinβ plays initiating role in the ICD maturation.
The third line of evidence supporting a direct involvement of mXinβ in localizing the intercellular junctions to the termini of cardiomyocytes is that loss of mXinβ led to the failure to restrict all three types of intercellular junctions to the termini of cardiomyocytes, and the onset of such defects correlated strongly with the peak expression of mXinβ in wild-type hearts (Figs. 8–10). In this study, we showed that at P13.5, significantly lower percentages of N-cadherin and desmoplakin were localized to cell termini in the mXinβ−/− hearts than in the wild-type hearts. On the other hand, a significant difference for Cx43 localization was not observed until P15.5, consistent with the well-known delay in the incorporation of Cx43 to the termini of cardiomyocytes. Thus, mXinβ is rapidly up-regulated from P0.5 and reaches its peak expression at P13.5 while loss of mXinβ during this period leads to failure of restricting the intercellular junction from the lateral puncta to the termini of cardiomyocytes. Taken together, the correlation between mXinβ’s temporal expression pattern with the time course of ICD maturation, the preferential targeting of mXinβ to the maturing ICDs, and the timing of the onset of the defects in ICD maturation when mXinβ is lost, all point to a direct involvement of mXinβ in the reorganization process of intercellular junctions leading to ICD maturation.
Defects of the mXinβ-null hearts provide novel insights for ICD formation in healthy and diseased hearts
Besides providing evidence for the important roles of mXinβ in ICD maturation, our results further imply that both the formation of area composita by the amalgamation of adhering junctions and the localization of gap junctions to the vicinity of adhering junctions are independent of the overall distribution of intercellular junctions at the cellular scale. During heart development, the amalgamation of the adhering junctions (adherens junction and desmosomes) is a prolonged process that initiates in the embryo and continues even at 3 weeks of age (Borrmann et al., 2006), overlapping with the redistribution of the adhering junctions at the cellular scale. Now, we have shown that despite the disruption of the overall distribution of intercellular junctions at the cellular scale in the mXinβ−/− hearts, the amalgamation between adhering junctions seemed to be unaffected (Fig. 11). Similarly, the association between the adherens junctions and the gap junctions remained unchanged in the mXinβ−/− hearts despite the extensive mis-localization of both types of junctions (Figs. 12 and 13). Therefore, mXinβ does not appear to be involved in these types of associations among intercellular junction components. In fact, accumulated lines of evidence suggest that linker molecules such as plakoglobin (γ-catenin), plakophilin 2, p0071, p120-catenin, and zonulaoccludens (ZO)-1 may play cross-linking roles between different intercellular junctions in the heart (review in Wang et al. (2012)).
Multiple ICDs are a pathological structure frequently found in the hypertrophied myocardium, including human and canine (Laks et al., 1970; Maron and Ferrans, 1973). They are defined as two or more ICDs lying in tandem along the longitudinal axis of a cardiomyocytes and are separated by less than 10 sarcomeres (Laks et al., 1970). These abnormally arranged ICDs are formed between one cardiomyocyte and the protruding processes from a neighboring cardiomyocyte with bizarre irregular shape (Maron and Ferrans, 1973). Maron and co-workers postulated that these cellular processes are established by sidewise addition of sarcomeres between two lateral intercellular junctions. Lateral intercellular junctions are prominent between cardiomyocytes in embryonic and neonatal hearts, as shown in this and previous studies on ICD development, but greatly reduce during the maturation of the ICDs in an mXinβ-dependent fashion. Thus, it is possible that dys-regulation of mXinβ or its related cellular pathways may play important roles in forming the pathological structure, multiple ICDs. The miniature ICD-like structures arranged in tandem in the p24.5 mXinβ−/− hearts further supports this possibility (Figs. 11 and 12).
Besides demonstrating the indispensable roles of mXinβ in postnatal maturation of ICDs, our results also indicate that mXinα variants are not essential for the formation of ICDs, and this is likely due to their lack of specific protein functions for this process. This notion is supported by the fact that although mXinα variants were more highly expressed than mXinβ during the course of ICD development (Figs. 2–4), loss of mXinα did not affect the formation of ICDs (Gustafson-Wagner et al., 2007). More importantly, in the mXinβ−/− background, additional loss of mXinα did not contribute to severer defects in ICD formation (Fig. 14). On the other hand, despite its lack of important roles in ICD formation, mXinα is required for the maintenance of ICDs in adult heart, evidenced by the ICD structure defects observed in adult mXinα-null hearts (Gustafson-Wagner et al., 2007; Otten et al., 2010). Interestingly, we have observed an increase in mXinβ expression after P20.5 in the wild-type hearts, and in the P60.5 hearts, mXinβ accounted for about 1/3 of the total mXin proteins. This phenomenon indicates that mXinβ may also play important roles in the ICDs of the adult hearts. Generation and characterization of mouse line carrying cardiac-restricted mXinβ conditional knockout allele will definitely answer this important question.
Molecular mechanisms of ICD formation and mXinβ’s function in this process
As diagramed in Fig. 9B, the components of adherens junctions undergo two phases of redistribution in order for ICDs to form (i.e., bi-phasic process). The first phase happens during late embryonic development and early postnatal stages, in which the diffusely distributed adherens junction components such as N-cadherins aggregate to form discrete spots/puncta. Similar to the formation of junctional complex in the epithelial cells, this process might be mediated by cadherin clustering. The extracellular domains of cadherins on opposing cell surfaces interact spontaneously to initiate weak contact/adhesion (cadherin engagement), while the intracellular domains of cadherins bound by β-catenin and p120-catenin regulate cadherins’ stability and turnover, resulting in a strong adhesion (Hong et al., 2010; Kusumi et al., 1999; Troyanovsky et al., 2007; Yap et al., 1998). The α-catenin is known to bind β-catenin and bundle actin filaments (Pokutta and Weis, 2000). However, it has been shown that α-catenin does not directly link the cadherin/catenin complex to the underlying actin cytoskeleton. Instead, it can act as a molecular switch in modulating actin cytoskeleton dynamic (Drees et al., 2005; Yamada et al., 2005). In order to understand the mechanism of cadherin clustering during ICD maturation, several questions need to be addressed. The first question is what molecules can directly link the cadherin/catenin complex to the underlying actin cytoskeleton. It has been shown that in epithelial cells, EPLIN (epithelial protein lost in neoplasm) via its ability to interact with both α-catenin and actin filaments can directly link the cadherin/catenin complex to the underlying cytoskeleton in an α-catenin-dependent manner (Abe and Takeichi, 2008). The depletion of EPLIN by siRNA treatment leads to failure of forming adhesion belts (Abe and Takeichi, 2008). We have previously shown that mXinα can also bind β-catenin and bundle actin filaments through β-catenin- and actin-interacting domains(Choi et al., 2007), and that mXinβ likely does so by the presence of both domains (Grosskurth et al., 2008). Thus, the mXin proteins might be the cardiac counterpart of the EPLIN. The next question is how the engaged cadherin/catenin complexes further cluster into discrete puncta/aggregates on cell surface. In the epithelial cells, it has been shown that the engaged cadherins induce the transient and cyclic activations of both phosphatidylinositol 3-kinase and Rac1, which would enhance membrane and actin cytoskeleton dynamics (Perez et al., 2008). The enhanced dynamics together with the intrinsic property of cadherin’s extracellular domains to form a stable dimers at contact sites (Hong et al., 2010; Troyanovsky et al., 2007) would greatly facilitate the lateral clustering of cadherin to form aggregates (a diffusion-mediated trapping mechanism). It remains to be demonstrated whether such a diffusion-mediated trapping mechanism also operates in the late embryonic and early postnatal cardiomyocytes. The third question is what factors control the size of aggregates. In addition to what has been known that β-catenin and p120-catenin can tightly regulate the stability and turnover of cadherin/catenin clusters, ATP-dependent mechanisms by which cadherin molecules are actively removed from each cluster have been identified in the epithelial cells were shown to be required for maintaining the cluster size (Hong et al., 2010). Therefore, cadherin cluster size is maintained by a balance between spontaneous assembly and active disassembly of cadherin/catenin complexes. Since mXinβ-null hearts showed no apparent defects in the formation of cadherin clusters (Figs. 8–10), it should not be involved in this first process of redistribution. Despite of the failure to form mature ICD, mXinβ-null hearts still preserve the co-localization between N-cadherin and desmoplakin as well as the close association between N-cadherin and Cx43.
In the second phase, the clusters of adherens junction redistribute to the termini of cardiomyocytes; specifically, the contacts at the cell termini expand to form mature ICDs while the lateral clusters reduce both in number and in signal intensity. mXinβ-null hearts showed severe defects in this process, which resulted in the failure to restrict clusters of the adherens junctions as well as desmosomes and gap junctions to the termini of the cells. In addition to initiating and recruiting adherens junctions to ICDs by mXinβ as shown in the present study, the second phase of ICD maturation would also require ICD membrane expansion and ICD structure maintenance/stabilization. Although the molecular mechanisms underlying these processes remain unclear, studies on the spot adherens junctions in Drosophila embryonic epithelial cells have revealed two types of cadherin-mediated intercellular contacts: a mobile and α-catenin-dependent contact associated with a dynamic actin network as well as a stable and α-catenin-independent contact associated with a much more stable actin patch (Cavey and Lecuit, 2009; Cavey et al., 2008). The existence of this stable contact leads Cavey and colleagues to suggest that an unidentified X protein helps linking the cadherin/catenin clusters to actin patch to form stable contact. In the maturation and maintenance of ICDs in the heart, the role of this unidentified X protein may be served by the mXinα and mXinβ proteins. Supporting this idea, we have shown that adult hearts without mXinα proteins exhibit progressive ICD structural defects and cardiomyopathy, suggesting mXinα via its ability to interact with β-catenin and actin filaments plays important roles in maintaining ICD integrity (Gustafson-Wagner et al., 2007). In mXinα-null hearts, mXinβ is found to up-regulate its messages and proteins (Gustafson-Wagner et al., 2007). mXinβ with conserved binding domains for β-catenin and actin filaments (Grosskurth et al., 2008) may also stabilize adherens junctions but preferentially at the termini of cardiomyocytes by linking the adherens junctions to the actin cytoskeleton. Through its localized translation (Gustafson-Wagner et al., 2007; Wang et al., 2012), mXinβ could increase the stability of N-cadherin clusters locally, leading to increased accumulation of N-cadherin at the cell termini at the expense of lateral N-cadherin clusters, thus promoting the maturation of ICDs. Without mXinβ, the N-cadherin clusters at the lateral side and the termini of cardiomyocytes might be equally stable/unstable, causing the failure of N-cadherin accumulation in the termini.
Previously, we observed that in the mXinβ-null hearts, Rac1 activity is significantly down-regulated (Wang et al., 2010). Since Rac1 plays important roles in adherens junction formation and expansion, the reduction of Rac1 could lead to defects in expanding the adherens junctions of ICDs at the cell termini. Consistent with this possibility, transmission electron microscopic observation showed that the membranes of the maturing ICDs in the mXinβ-null hearts are smoother than those in the wild-type hearts, suggesting a depressed Rac1-mediated membrane ruffling activity in the mutant hearts (Wang et al., 2010). Current findings that mXinβ is preferentially localized to the maturing ICDs further suggests that mXinβ may function locally to promote the expansion of adherens junctions through Rac1.
Through the above mechanisms, mXinβ may directly regulate the redistribution of adherens junctions during ICD maturation. The redistribution of adherens junctions may in turn facilitate the redistribution of both desmosomes and gap junctions. It has been shown that classic cadherin-mediated adherens junctions form prior to desmosomes at newly established intercellular contacts, and adherens junctions are the prerequisites for the formation and correct location of the desmosomes and gap junctions (Green et al., 2010). In the heart, induced deletion of N-cadherin in adult cardiomyocytes leads to dissolution of the entire ICDs (Kostetskii et al., 2005), further supporting the central role of adherens junctions in maintaining the desmosomes and gap junctions. Thus, by regulating the localization of adherens junctions, mXinβ could control of the organization of the desmosomes and gap junctions indirectly.
Conclusions
Based on previous and present studies, we have proposed a biphasic process of ICD maturation. We have shown that mXinβ is required for the second step of the ICD maturation (i.e., the reorganization of intercellular junctions to establish mature ICDs) in postnatal cardiomyocytes. mXinβ likely carries out this role by nucleating the formation of maturing ICDs at the termini of cardiomyocytes and preventing the retention of intercellular contacts and formation of ectopic ICDs at the lateral surfaces. One important question that remains to be addressed is what roles of mXinβplay in the maintenance of ICDs in the adult heart.
Preserved co-localization between N-cadherin and desmoplakin as well as preserved association between N-cadherin and Cx43 detected in both P7.5 and P24.5 mXinβ-null hearts suggest that the formation of lateral aggregates/puncta (i.e., the first step of the ICD maturation) from the diffused localization is independent of mXinβ. Although the molecular mechanisms for these co-localization and association remain unclear, many known junctional proteins, such as γ-catenin, PKP2 and ZO-1, that can shuttle and/or link among different junctions of ICD could, in theory, play critical roles in this first step of ICD maturation. By characterizing mXinα−/−;mXinβ−/− DKO mice, we have further provided evidence to show that mXinα are not essential for the formation of ICDs, although mXinα has been previously shown to play a role in maintaining the ICD integrity in the adult hearts (Gustafson-Wagner et al., 2007; Otten et al., 2010).
Acknowledgments
We thank Drs. Peter Rubenstein, Diane Slusarski, Chris Stipp and Chung-Fang Wu for critical reading and comments of the manuscript. This work was supported by the National Institutes of Health Grant HL107383.
Role of the funding sources None.
References
- Abe K, Takeichi M. EPLIN mediates linkage of the cadherin–catenin complex to F-actin and stabilizes the circumferential actin belt. Proc. Natl. Acad. Sci. U. S. A. 2008;105:13–19. doi: 10.1073/pnas.0710504105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angst BD, Khan LU, Severs NJ, Whitely K, Rothery S, Thompson RP, Magee AI, Gourdie RG. Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ. Res. 1997;80:88–94. doi: 10.1161/01.res.80.1.88. [DOI] [PubMed] [Google Scholar]
- Barker RJ, Price RL, Gourdie RG. Increased association of ZO-1 with connexin43 during remodeling of cardiac gap junctions. Circ. Res. 2002;90:317–324. doi: 10.1161/hh0302.104471. [DOI] [PubMed] [Google Scholar]
- Borrmann CM, Grund C, Kuhn C, Hofmann I, Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adhaerens molecules in the intercalated disk. Eur. J. Cell Biol. 2006;85:469–485. doi: 10.1016/j.ejcb.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Cavey M, Lecuit T. Molecular bases of cell–cell junctions stability and dynamics. Cold Spring Harb. Perspect. Biol. 2009;1:a002998. doi: 10.1101/cshperspect.a002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavey M, Rauzi M, Lenne PF, Lecuit T. A two-tiered mechanism for stabilization and immobilization of E-cadherin. Nature. 2008;453:751–756. doi: 10.1038/nature06953. [DOI] [PubMed] [Google Scholar]
- Chan FC, Cheng CP, Wu KH, Chen YC, Hsu CH, Gustafson-Wagner EA, Lin JL, Wang Q, Lin JJ, Lin CI. Intercalated disc-associated protein, mXin-alpha, influences surface expression of ITO currents in ventricular myocytes. Front. Biosci. 2011;3:1425–1442. doi: 10.2741/e344. (Elite Ed) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Gustafson-Wagner EA, Wang Q, Harlan SM, Sinn HW, Lin JL, Lin JJ. The intercalated disc protein, mXinα, is capable of interacting with β-catenin and bundling actin filaments. J. Biol. Chem. 2007;282:36024–36036. doi: 10.1074/jbc.M707639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaco CA, Evans WH. A biochemical dissection of the cardiac intercalated disk: isolation of subcellular fractions containing fascia adherentes and gap junctions. J. Cell Sci. 1981;52:313–325. doi: 10.1242/jcs.52.1.313. [DOI] [PubMed] [Google Scholar]
- Colaco CA, Evans WH. Partial purification of an intercalated disc-containing cardiac plasma membrane fraction. Biochim. Biophys. Acta. 1982;684:40–46. doi: 10.1016/0005-2736(82)90046-3. [DOI] [PubMed] [Google Scholar]
- Coppen SR, Kaba RA, Halliday D, Dupont E, Skepper JN, Elneil S, Severs NJ. Comparison of connexin expression patterns in the developing mouse heart and human foetal heart. Mol. Cell. Biochem. 2003;242:121–127. [PubMed] [Google Scholar]
- Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ. Res. 2010;107:700–714. doi: 10.1161/CIRCRESAHA.110.223412. [DOI] [PubMed] [Google Scholar]
- Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actinfilament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estigoy CB, Ponten F, Odeberg J, Herbert B, Guilhuas M, Charleston M, Ho JWK, Cameron D, dos Remedies CG. Intercalated discs: multiple proteins perform multiple functions in non-failing and failing human hearts. Biophys. Rev. 2009;1:43–49. doi: 10.1007/s12551-008-0007-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira-Cornwell MC, Luo Y, Narula N, Lenox JM, Lieberman M, Radice GL. Remodeling the intercalated disc leads to cardiomyopathy in mice misexpressing cadherins in the heart. J. Cell Sci. 2002;115:1623–1634. doi: 10.1242/jcs.115.8.1623. [DOI] [PubMed] [Google Scholar]
- Forbes MS, Sperelakis N. Intercalated discs of mammalian heart: a review of structure and function. Tissue Cell. 1985;17:605–648. doi: 10.1016/0040-8166(85)90001-1. [DOI] [PubMed] [Google Scholar]
- Green KJ, Getsios S, Troyanovsky S, Godsel LM. Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb. Perspect. Biol. 2010;2:a000125. doi: 10.1101/cshperspect.a000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskurth SE, Bhattacharya D, Wang Q, Lin JJ. Emergence of Xin demarcates a key innovation in heart evolution. PLoS One. 2008;3:e2857. doi: 10.1371/journal.pone.0002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson-Wagner EA, Sinn HW, Chen YL, Wang DZ, Reiter RS, Lin JL, Yang B, Williamson RA, Chen J, Lin CI, Lin JJ. Loss of mXinα, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H2680–H2692. doi: 10.1152/ajpheart.00806.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertig CM, Butz S, Koch S, Eppenberger-Eberhardt M, Kemler R, Eppenberger HM. N-cadherin in adult rat cardiomyocytes in culture. II. Spatiotemporal appearance of proteins involved in cell–cell contact and communication. Formation of two distinct N-cadherin/catenin complexes. J. Cell Sci. 1996a;109:11–20. doi: 10.1242/jcs.109.1.11. [DOI] [PubMed] [Google Scholar]
- Hertig CM, Eppenberger-Eberhardt M, Koch S, Eppenberger HM. N-cadherin in adult rat cardiomyocytes in culture. I. Functional role of N-cadherin and impairment of cell–cell contact by a truncated N-cadherin mutant. J. Cell Sci. 1996b;109:1–10. doi: 10.1242/jcs.109.1.1. [DOI] [PubMed] [Google Scholar]
- Hirschy A, Schatzmann F, Ehler E, Perriard JC. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev. Biol. 2006;289:430–441. doi: 10.1016/j.ydbio.2005.10.046. [DOI] [PubMed] [Google Scholar]
- Hong S, Troyanovsky RB, Troyanovsky SM. Spontaneous assembly and active disassembly balance adherens junction homeostasis. Proc. Natl. Acad. Sci. U. S. A. 2010;107:3528–3533. doi: 10.1073/pnas.0911027107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kargacin GJ, Hunt D, Emmett T, Rokolya A, McMartin GA, Wirch E, Walsh MP, Ikebe M, Kargacin ME. Localization of telokin at the intercalated discs of cardiac myocytes. Arch. Biochem. Biophys. 2006;456:151–160. doi: 10.1016/j.abb.2006.06.018. [DOI] [PubMed] [Google Scholar]
- Kostetskii I, Li J, Xiong Y, Zhou R, Ferrari VA, Patel VV, Molkentin JD, Radice GL. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ. Res. 2005;96:346–354. doi: 10.1161/01.RES.0000156274.72390.2c. [DOI] [PubMed] [Google Scholar]
- Kusumi A, Suzuki K, Koyasako K. Mobility and cytoskeletal interactions of cell adhesion receptors. Curr. Opin. Cell Biol. 1999;11:582–590. doi: 10.1016/s0955-0674(99)00020-4. [DOI] [PubMed] [Google Scholar]
- Laks MM, Morady F, Adomian GE, Swan HJ. Presence of widened and multiple intercalated discs in the hypertrophied canine heart. Circ. Res. 1970;27:391–402. doi: 10.1161/01.res.27.3.391. [DOI] [PubMed] [Google Scholar]
- Legato MJ. Cellular mechanisms of normal growth in the mammalian heart. I. Qualitative and quantitative features of ventricular architecture in the dog from birth to five months of age. Circ. Res. 1979;44:250–262. doi: 10.1161/01.res.44.2.250. [DOI] [PubMed] [Google Scholar]
- Li J, Patel VV, Radice GL. Dysregulation of cell adhesion proteins and cardiac arrhythmogenesis. Clin. Med. Res. 2006;4:42–52. doi: 10.3121/cmr.4.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Radice GL. A new perspective on intercalated disc organization: implications for heart disease. Dermatol. Res. Pract. 2010;2010:207835. doi: 10.1155/2010/207835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JJ-C, Gustafson-Wagner EA, Sinn HW, Choi S, Jaacks SM, Wang DZ, Evans S, Lin JL-C. Structure, expression, and function of a novel intercalated disc protein, Xin. J. Med. Sci. 2005;25:215–222. doi: 10.1901/jaba.2005.25-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron BJ, Ferrans VJ. Significance of multiple intercalated discs in hypertrophied human myocardium. Am. J. Pathol. 1973;73:81–96. [PMC free article] [PubMed] [Google Scholar]
- Noorman M, van der Heyden MA, van Veen TA, Cox MG, Hauer RN, de Bakker JM, van Rijen HV. Cardiac cell–cell junctions in health and disease: electrical versus mechanical coupling. J. Mol. Cell. Cardiol. 2009;47:23–31. doi: 10.1016/j.yjmcc.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Otten J, van der Ven PF, Vakeel P, Eulitz S, Kirfel G, Brandau O, Boesl M, Schrickel JW, Linhart M, Hayess K, Naya FJ, Milting H, Meyer R, Furst DO. Complete loss of murine Xin results in a mild cardiac phenotype with altered distribution of intercalated discs. Cardiovasc. Res. 2010;85:739–750. doi: 10.1093/cvr/cvp345. [DOI] [PubMed] [Google Scholar]
- Pacholsky D, Vakeel P, Himmel M, Lowe T, Stradal T, Rottner K, Furst DO, van der Ven PFM. Xin repeats define a novel actin-binding motif. J. Cell Sci. 2004;117:5257–5268. doi: 10.1242/jcs.01406. [DOI] [PubMed] [Google Scholar]
- Perez TD, Tamada M, Sheetz MP, Nelson WJ. Immediate-early signaling induced by E-cadherin engagement and adhesion. J. Biol. Chem. 2008;283:5014–5022. doi: 10.1074/jbc.M705209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters NS, Severs NJ, Rothery SM, Lincoln C, Yacoub MH, Green CR. Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium. Circulation. 1994;90:713–725. doi: 10.1161/01.cir.90.2.713. [DOI] [PubMed] [Google Scholar]
- Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates—IV: coalescence and amalgamation of desmosomal and adhaerens junction components—late processes in mammalian heart development. Eur. J. Cell Biol. 2007;86:377–391. doi: 10.1016/j.ejcb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Pieperhoff S, Franke WW. The area composita of adhering junctions connecting heart muscle cells of vertebrates. VI. Different precursor structures in non-mammalian species. Eur. J. Cell Biol. 2008;87:413–430. doi: 10.1016/j.ejcb.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Pokutta S, Weis WI. Structure of the dimerization and beta-catenin-binding region of alpha-catenin. Mol. Cell. 2000;5:533–543. doi: 10.1016/s1097-2765(00)80447-5. [DOI] [PubMed] [Google Scholar]
- Sinn HW, Balsamo J, Lilien J, Lin JJ. Localization of the novel Xin protein to the adherens junction complex in cardiac and skeletal muscle during development. Dev. Dyn. 2002;225:1–13. doi: 10.1002/dvdy.10131. [DOI] [PubMed] [Google Scholar]
- Troyanovsky RB, Laur O, Troyanovsky SM. Stable and unstable cadherin dimers: mechanisms of formation and roles in cell adhesion. Mol. Biol. Cell. 2007;18:4343–4352. doi: 10.1091/mbc.E07-01-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Lin JL, Reinking BE, Feng HZ, Chan FC, Lin CI, Jin JP, Gustafson-Wagner EA, Scholz TD, Yang B, Lin JJ. Essential roles of an intercalated disc protein, mXinβ, in postnatal heart growth and survival. Circ. Res. 2010;16:1468–1478. doi: 10.1161/CIRCRESAHA.109.212787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Lin JL, Wu KH, Wang DZ, Reiter RS, Sinn HW, Lin CI, Lin CJ. Xin proteins and intercalated disc maturation, signaling and diseases. Front. Biosci. 2012;17:2566–2593. doi: 10.2741/4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gerdes AM. Chronic pressure overload cardiac hypertrophy and failure in guinea pigs: III. Intercalated disc remodeling. J. Mol. Cell. Cardiol. 1999;31:333–343. doi: 10.1006/jmcc.1998.0886. [DOI] [PubMed] [Google Scholar]
- Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin–catenin–actin complex. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap AS, Niessen CM, Gumbiner BM. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J. Cell Biol. 1998;141:779–789. doi: 10.1083/jcb.141.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]