

Abstract
Human papillomavirus (HPV) infection is severely limited in its natural host, primary human keratinocytes. Our data show HPV infectivity in primary keratinocytes is over 100- and 1,000-fold lower than in established keratinocyte cell lines NIKS and HaCaT, respectively. Here, we show that the basal level of autophagy in primary human foreskin keratinocytes (HFKs) is higher than in immortalized keratinocytes, and that HPV16 virions significantly induce autophagy in HFKs. Interestingly, HPV16 infectivity is dramatically enhanced by knockdown of essential autophagy genes as well as biochemical inhibition of autophagy. The increase in HPV16 infectivity by autophagy inhibition is most significant in HFKs, showing an inverse correlation with basal HPV16 infectivity in HFK, NIKS, HaCaT, and 293FT cells. Further, inhibition of autophagy delays degradation of HPV16 capsid proteins during virus trafficking, indicating that host autophagy induced by HPV16 virions inhibits infection of primary keratinocytes through rapid degradation of viral capsid proteins.
Keywords: HPV, Papillomavirus, Keratinocyte, HFK, Trafficking, Entry, Autophagy, Autophagosome, 3-MA, 3-methyladenine
Introduction
Human papillomaviruses (HPVs) are small non-enveloped double-stranded DNA viruses that strictly infect mucosal and cutaneous epithelia. HPVs are causally associated with multiple human cancers including >99% of cervical cancers, ~50% of other anogenital cancers, and ~25% of head and neck cancers (zur Hausen, 1996, 1999; Gillison and Shah, 2001; Burd, 2003; Gillison and Lowy, 2004). HPV-associated cancers account for about half a million deaths worldwide every year, with cervical cancer being the third leading cause of cancer deaths in women (zur Hausen, 1996, 1999; Burd, 2003). Over 100 genotypes of HPVs have been identified and categorized by oncogenicity, with HPV16 being the most prevalent genotype found in HPV-associated cancers (Burd, 2003).
HPV genomic DNA is encased by a ~55 nm icosahedral capsid, which is comprised of the major capsid protein, L1, and the minor capsid protein, L2 (Modis et al., 2002). Virions initially bind to the basement membrane of the mucosal epithelium through interactions between L1 and heparan sulfate proteoglycans (HSPGs), triggering conformational changes in both L1 and L2 capsid proteins (Joyce et al., 1999; Giroglou et al., 2001; Selinka et al., 2003; Yang et al., 2003). Subsequent steps in infectious entry remain poorly understood, as more specific interactions necessary to confer the strict tropism to basal keratinocytes have not yet been defined.
Several conflicting results regarding the internalization and subsequent endocytic trafficking of HPV virions during entry have been reported. Previously, multiple studies showed that HPV16 internalization is blocked by biochemical inhibition of clathrin and dynamin-2, suggesting that HPV16 entry requires clathrin-dependent endocytosis (Bousarghin et al., 2003; Day et al., 2003; Abban et al., 2008; Laniosz et al., 2009). However, recent studies using genetic knockdowns and dominant negative mutants showed that HPV16 internalization is independent of clathrin, dynamin, caveolin, and lipid rafts (Spoden et al., 2008; Schelhaas et al., 2012). These studies also suggested that HPV16 entry requires tetraspanin-enriched microdomains and endocytosis similar to, but distinct from, classical macropinocytosis (Spoden et al., 2008; Schelhaas et al., 2012). These discrepancies may be attributed to the different cell culture models used in these studies. For example, some of the experiments that showed a clathrin-dependent mechanism of HPV16 entry were conducted in 293 and COS-7 cells (Bousarghin et al., 2003; Abban et al., 2008), whereas the more recent experiments showing a clathrin-independent mechanism of HPV16 entry were carried out in HeLa and HaCaT cells (Spoden et al., 2008; Schelhaas et al., 2012). Even among different keratinocytes, we have observed significant differences in HPV16 infectivity (Fig. 1A), indicating that the mechanisms of HPV16 entry may differ depending on the cell culture model used to study it.
Fig. 1. Primary keratinocytes show limited HPV16 infectivity and high levels of basal autophagy.
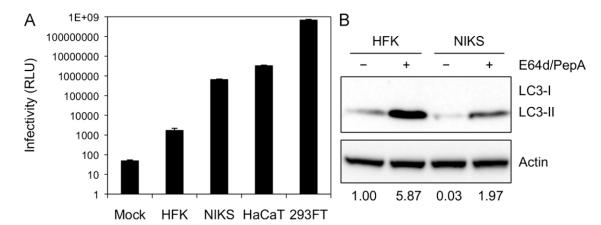
(A) The indicated cell lines were inoculated with 10,000 vge/cell of HPV16-LucF and incubated for 48 h. HPV16 infectivity and cell viability were measured by Bright-GloTM Luciferase Assay System (Promega)and CellTiter-Glo Luminescent Cell Vi ability Assay (Promega), respectively. Infectivity data normalized to cell viability are shown as average relative luminescence units (RLU) from quadruplicate samples. The data shown here are from one representative of three independent experiments. (B) HFK and NIKS cells were treated with the protease inhibitors E64d and Pepstatin A (10 μg/mL each) or DMSO for 6 h, and harvested for immunoblotting of LC3, using Actin as an internal control.The data shown here are from one representative of three independent experiments.
One point of consensus, however, is that all HPVs require low pH for infectious entry, as they must pass through acidified compartments (i.e. late endosomes, lysosomes), where it is thought that viral DNA uncoating and egress from the endosomal pathway occurs (Selinka et al., 2002; Day et al., 2003; Kämper et al., 2006; Abban et al., 2008; Smith et al., 2008; Spoden et al., 2008). Interestingly, a recent study showed that HPV16 virions colocalize with Rab5-containing compartments, but not with other markers of early endosomes, indicating trafficking to acidified vesicles via alternate Rab5-containing vesicles (Schelhaas et al., 2012). Rab5 plays critical roles in the biogenesis and coordination of early endosomes as well as autophagosomes, suggesting that virions may transit through autophagosomes during entry (Zerial et al., 1999a; Zerial et al., 1999b; Ravikumar et al., 2008). Autophagy has essential functions in cell homeostasis, as autophagosomes engulf bulk cytoplasm, including damaged organelles, and shuttle them to lysosomes for degradation and recycling of nutrients (reviewed in Levine and Kroemer, 2008). Recent studies demonstrated that autophagy also has significant roles in host defense including detection, degradation, and antigen presentation of several intracellular pathogens (reviewed in Levine and Kroemer, 2008; Kudchodkar and Levine, 2009; Mack and Münger, 2012). Interestingly, Rab5 is required for induction of autophagy by Hepatitis C Virus (Su et al., 2011).
Here, we show a role of host autophagy in HPV16 entry into its natural host, primary human keratinocytes. HPV16 infectivity in primary keratinocytes was significantly enhanced by inhibition of autophagy using 3-methyladenine (3-MA), as well as shRNA-mediated knockdown of essential autophagy genes, PIK3C3 and ATG7. Interestingly, autophagy inhibition by 3-MA increased HPV16 infectivity more dramatically in primary keratinocytes with high levels of basal autophagy compared to immortalized keratinocytes with low levels of autophagy, showing an inverse correlation with basal HPV16 infectivity. Furthermore, using the autophagosome marker, LC3, we found that HPV16 virions induce host autophagy several hours post inoculation. Degradation of HPV16 capsid proteins was also delayed by 3-MA mediated autophagy inhibition during entry. Our results suggest that host autophagy is induced by HPV16 virions as a host defense mechanism to inhibit infection by facilitating degradation of incoming virions.
Results
HPV16 infectivity is limited in natural host primary keratinocytes coincident with high levels of basal autophagy
As the natural host for HPV infection, primary human keratinocytes have been accepted as one of the most relevant in vitro cell culture models of HPV infection. However, HPV infection of these cells is known to be particularly difficult compared to immortalized and transformed keratinocytes (Meyers et al., 1997; Ozbun, 2002; Day et al., 2008). To directly compare the differences in HPV16 infectivity among cell culture models commonly used to study HPV entry and infection, we infected primary human foreskin keratinocytes (HFK), two spontaneously immortalized keratinocyte cell lines, NIKS and HaCaT, and the human embryonic kidney cell line, 293FT, with HPV16 pseudovirions containing a luciferase reporter gene (HPV16-LucF). At 48 h post infection (hpi), HPV infectivity was measured by relative luciferase activity (Pyeon et al., 2009). We found that, whileHPV infectivity is very limited in HFKs, it is well over 100-, 1,000-, and 100,000-folds greater in NIKS, HaCaT, and 293FT cells, respectively (Fig. 1A). Notably, HPV16 infectivity was significantly increased in NIKS cells compared to HFKs (~400-fold increase). Interestingly, of all cell culture models tested, HFK and NIKS cells are most similar to one another in that both are considered normal, near diploid keratinocytes that support normal differentiation programs.
The observed limited infectivity in HFKs could potentially be explained by the previously reported lack of furin and L2 cleavage of basement membrane-bound HPV16 virions (Day et al., 2008), as well as differences in cell proliferation between cell lines (Pyeon et al., 2009). We hypothesized that host autophagy could be an additional restriction factor against HPV infection that contributes to the low infectivity in primary keratinocytes. Autophagy is a host defense mechanism against infection, directly destroying intracellular pathogens and inducing innate immune responses (reviewed in Levine and Kroemer, 2008; Kudchodkar and Levine, 2009; Mack and Münger, 2012). To determine the relative levels of autophagy in HFK and NIKS cells, we detected protein levels of the established autophagosome marker, LC3 (Fig. 1B). LC3 is constitutively expressed in its soluble form, LC3-I, and upon autophagy induction is lipidated to form LC3-II (Kabeya et al., 2000, 2004). LC3-II stably associates with autophagosome membranes, but is also a substrate for autophagic protein degradation in autolysosomes following autophagosome-lysosome fusion (Tanida et al., 2005). As a standard procedure to prevent outflux of LC3-II in the autolysosome, we cultured cells with the protease inhibitors E64d and Pepstatin A. Immunoblotting of LC3 showed higher levels of autophagy in HFKs compared with NIKS cells (Fig. 1B). This finding implies that autophagy may restrict HPV16 infection in primary keratinocytes.
HPV16 virions induce host autophagy in primary keratinocytes
Many intracellular pathogens have been shown to induce autophagy as a host defense mechanism to sequester and deliver them to the lysosome for degradation (reviewed in Levine and Kroemer, 2008; Kudchodkar and Levine, 2009). To determine whether HPV16 virions induce autophagy in primary keratinocytes, GFP-LC3 expressing HFKs were established and infected with HPV16 virions containing the full-length HPV16 W12 genome (HPV16-W12) for 6 h (Flores et al., 1999). GFP puncta represent autophagosome membrane-bound LC3-II distinct from soluble LC3-I shown as diffuse GFP staining (Kabeya et al., 2000; Mizushima, 2004). The GFP-LC3 puncta were visualized using confocal microscopy (representative images shown in Fig. 2A) and the number of puncta in each cell were counted using NIH ImageJ in a blinded manner. To avoid potential artifacts from incubating the cells with excess virions, 10-fold fewer virions were used in these assays than for infectivity experiments. Interestingly, HFKs incubated with HPV16-W12 showed significantly increased numbers of GFP-LC3 puncta per cell (Fig. 2A and B). We further validated our finding by detecting endogenous LC3 in HFKs infected with HPV16-W12. At 6 hpi, we observed a significant increase in LC3-II, indicating that HPV16 virions induced autophagy in HFKs. This increase in LC3-II by HPV16-W12 was comparable in magnitude to the increase observed by starvation conditions, one of the most potent means for inducing autophagy (Fig. 2C). These results indicate that autophagy is induced by HPV16 virions during entry into primary keratinocytes.
Fig. 2. HPV16 virions induce host autophagy in primary keratinocytes.

(A) HFKs were transduced with GFP-LC3-containing lentiviruses for 48 h, inoculated with 1,000 vge/cell of HPV16-W12, and incubated for 6 h. Cells were fixed with 4% paraformaldehyde and mounted with ProLong® Gold Antifade Reagent with DAPI (Life Technologies). At least 25 cells were imaged for each condition. Representative images are shown. (B) Quantification of GFP-LC3 puncta obtained in (A). GFP-LC3 puncta were counted using NIH ImageJ image analysis software and plotted to show the number of GFP-LC3 puncta per cell. One-way analysis of variance between groups (ANOVA) statistical analysis to determine the p-value was performed using GraphPad Prism software (GraphPad Software, La Jolla, CA). Data shown are from one representative of two independent experiments. (C) HFKs were incubated with Krebs–Ringer Bicarbonate buffer for starvation condition, or 1,000 vge/cell of HPV16-W12 for 6 h. LC3 and Actin were detected by immunoblotting. The data shown here are from one representative of three independent experiments.
Inhibition of autophagy by 3-MA significantly enhances HPV16 infectivity of primary keratinocytes
To determine whether autophagy inhibits HPV16 infection in primary keratinocytes, we tested the effect of the established autophagy inhibitor 3-MA on HPV16 infectivity in HFKs. 3-MA is an inhibitor of class III phosphatidylinositol-3 kinase (PIK3C3), which plays an essential role in autophagosome formation (Seglen and Gordon, 1982; Blommaart et al., 1997; Petiot et al., 2000). We found that 3-MA treatment enhanced HPV16 infectivity in a dose-dependent manner up to almost 40-fold in HFKs (Fig. 3A). The increased HPV16 infectivity by 3-MA corresponded to decreased levels of autophagy, as measured by LC3 immunoblotting of cells treated with E64d and Pepstatin A (Fig. 3B). The magnitude of autophagy inhibition by 3-MA was consistent with previous reports (Seglen and Gordon, 1982; Fuertes et al., 2003; Kawai et al., 2006). To determine whether the effect of autophagy inhibition on HPV16 infectivity is unique to primary keratinocytes, we tested the effect of 3-MA on HPV16 infectivity in NIKS, HaCaT, and 293FT cells in addition to HFKs. Interestingly, the increase in HPV16 infectivity by 3-MA was most significant in HFKs, showing an inverse correlation with HPV16 infectivity of untreated cells presented in Fig. 1A (Fig. 3C). We also tested the effect of furin cleavage on HPV16-LucF infectivity of human keratinocytes by infecting the cells in the presence or absence of exogenous furin. Consistent with the previous report (Day et al., 2008), we observed an increase in infectivity by treatment with furin, which was most dramatic in HFKs (~8.5-fold increase) compared to NIKS and HaCaT cells (Fig. 3D). These results suggest that while the lack of furin cleavage of L2 significantly limits HPV16 infection of HFKs, high levels of host autophagy may be a critical restriction factor that further contributes to the low basal infectivity of primary keratinocytes.
Fig. 3. Inhibition of autophagy by 3-MA significantly enhances HPV16 infectivity in primary keratinocytes.

(A) Following pretreatment with the indicated concentrations of 3-MA or vehicle for 4 h, HFKs were inoculatedwith 10,000 vge/cell of HPV16-LucF and incubated for 48 h in the presence of 3-MA or vehicle. Infectivity and cell viability were measured as described above. Normalized infectivity data are presented as fold change in infectivity by 3-MA over vehicle control, and were averaged from quadruplicate samples. (B) HFKs were treated with 6 mM 3-MA or vehicle for 6 h with protease inhibitors E64d and Pepstatin A (10 μg/mL each) or DMSO, and harvested for immunoblotting of LC3 and Actin. (C) The indicated cell lines were treated with 6 mM 3-MA or vehicle, and inoculated with HPV16-LucF as in (A). Normalized infectivity data are shown as fold changein infectivity by 3-MA over vehicle control. (D) The indicated cell lines were treated with 8 U/mL furin or vehicle, and inoculated with HPV16-LucF as in (A). Normalized infectivity data are shown as fold change in infectivity by furin over vehicle control. The data shown here are from one representative of three independent experiments. Student’s t-test was performed to determine p-values between 3-MA treatments and vehicle control using StatPlus® (AnalystSoft, Inc.).
To confirm that 3-MA does not directly increase reporter gene expression, we examined the effect of 3-MA in 293FT cells transfected with the luciferase reporter plasmid (pLucF) compared to 293FT cells infected with HPV16-LucF. 293FT cells were used in this assay due to the low transfection efficiency of keratinocytes. 3-MA did not affect luciferase activity in the pLucF-transfected cells, whereas it significantly enhanced luciferase activity in HPV16-LucF-infected cells (Fig. S1). This result confirms that the enhancement of HPV16 infectivity by 3-MA is specific to virus infection and is not caused by directly altering reporter gene expression.
To determine whether the enhancement of HPV16 infectivity by 3-MA could be extended to low MOI infections, we tested the effect of 3-MA on HPV16 infectivity with decreasing viral genome equivalents (vge) of HPV16-LucF in HaCaT cells. HaCaT cells were used in this assay, as low MOI infections are easily detectable in HaCaT cells, allowing for more reliable infection assays. The result showed that 3-MA enhanced HPV16 infectivity by approximately 3 to 4 fold regardless of MOI (Fig. S2 and Fig. 3C). Furthermore, by approximating the percentage of infected cells using a GFP reporter, we estimate that the MOI of infection for HFKs using 10,000 vge/cell is approximately 0.02. These results indicate that autophagy inhibition by 3-MA enhances HPV16 infection regardless of the amount of virus used.
Knockdown of essential autophagy genes PIK3C3 and ATG7 enhances HPV16 infectivity of keratinocytes
To determine whether genetic knockdown of PIK3C3, the target of 3-MA, enhances HPV16 infectivity, we generated HFKs expressing shRNA against PIK3C3 (shR-PIK3C3) by lentiviral delivery, followed by puromycin selection for 4 days to avoid any potential artifacts due to coinfection of lentivirus and HPV. HPV16-LucF infection was slightly increased by PIK3C3 knockdown in HFKs (data not shown). However, this increase in HPV16 infectivity was not statistically significant, which may be attributed to the cellular toxicity of the shR-PIK3C3 expression in HFKs, indicated by their significantly altered morphology and decreased growth rate. Constitutive knockdown of PIK3C3 for more than one week was toxic for primary keratinocytes. Thus, as an alternative approach, we established NIKS cell lines that stably express shR-PIK3C3 by puromycin selection for >2 weeks. Using the stable NIKS cell lines, which showed no detectable toxicity, we found that HPV16 infectivity was significantly enhanced ~4-fold by PIK3C3 knock down (Fig. 4A and B).
Fig. 4. HPV16 infectivity is increased by knockdown of PIK3C3.
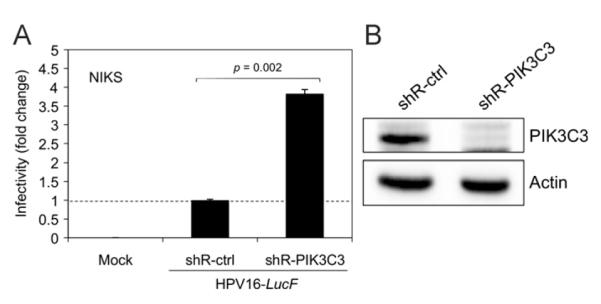
(A) NIKS cells were transduced with lentiviruses containing shRNAs against PIK3C3 (shR-PIK3C3) or a non-target control (shR-ctrl). At 48 h post transduction, the transduced cells were selected by culture in medium containing 1.5 μg/mL of puromycin for 2 weeks to establish stable semi-clonal cell lines. Puromycin-selected cells were inoculated with 10,000 vge/cell of HPV16-LucF and incubated for 48 h. Infectivity was measured and normalized to cell viability as described in Fig. 1. Normalized infectivity data are shown as fold change in infectivity by shR-PIK3C3 over shR-ctrl, and were averaged from quadruplicate samples. Statistical analysis was performed as described in Fig. 3A. (B) In parallel to the infection assay in (A), immunoblotting was performed with the puromycin-selected cells using anti-PIK3C3 (Life Technologies) and anti-Actin antibodies. The data shown here are from one representative of three independent experiments.
A recent report showed evidence of a Pik3c3-independent autophagy pathway in mouse sensory neurons, and that Atg7 is required for both Pik3c3-dependent and Pik3c3-independent autophagy (Zhou et al., 2010). To determine the effect of the essential and specific autophagy gene ATG7 on HPV16 infectivity in primary keratinocytes, we tested HPV16 infectivity in HFKs knocked down for expression of ATG7. Our results showed an ~5-fold enhancement of HPV16 infectivity by ATG7 knockdown (Fig. 5A and B), with minimal toxicity. Thus, our results consistently indicate that autophagy inhibits HPV16 infection in primary keratinocytes.
Fig. 5. HPV16 infectivity is increased by knockdown of the essential autophagy gene ATG7.
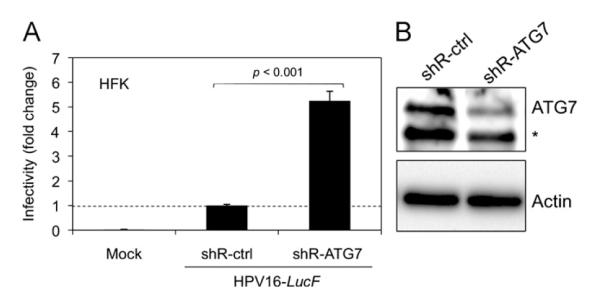
(A) HFKs were transduced with lentiviruses containing shRNAs against ATG7 (shR-ATG7) or shR-ctrl and puromycin-selected as in Fig. 3A. Infection assay and analysis of puromycin-selected cells was conducted as in Fig. 3A, and data are shown as fold change in infectivity by shR-ATG7 over shR-ctrl. Statistical analysis was performed as described in Fig. 3A. (B) In parallel to the infection assay in (A), immunoblotting was performed using anti-ATG7 antibody (ProSci) and Actin. The data shown here are from one representative of three independent experiments. *Non-specific band.
Inhibition of autophagy by 3-MA protects incoming virions from degradation during entry into primary keratinocytes
Next, we investigated which steps of virus entry are modulated by autophagy including virus attachment, internalization, and capsid degradation. To determine whether autophagy inhibition by 3-MA affects virus attachment to the host cell surface, HFKs were pretreated with 3-MA for 4 h, chilled at 4 °C and incubated with HPV16-W12 for 1 h at 4 °C to allow virus binding while preventing internalization. After washing off unbound virus particles, we detected the L1 major capsid protein by immunoblotting. Our results showed that 3-MA treatment did not affect the amount of virus attached to host cells (Fig. 6A). To examine whether autophagy inhibition by 3-MA affects virus internalization, HFKs treated as above were transferred to 37 °C to initiate internalization. At 1 and 2 hpi, the cells were treated with trypsin to eliminate bound but non-internalized virus particles and the L1 capsid protein was detected by immunoblotting. Again, 3-MA treatment of HFKs resulted in no difference in the amount of internalized virions (Fig. 6B).
Fig. 6. The autophagy inhibitor, 3-MA, delays degradation of HPV16 L1 capsid proteins during entry.

(A) Effect of 3-MA on virus binding. HFKs were pretreated with 6 mM 3-MA for 4 h. Cells were chilled to 4 °C to inhibit endocytosis, inoculated with 10,000 vge/cell of HPV16-W12 and incubated with or without 3-MA for 1 h at 4 °C. Unbound virions were washed out and cells were lysed directly to observe total bound virions, followed by immunoblotting of L1 capsid protein (CAMVIR-1, Abcam). (B) Effect of 3-MA on virus internalization. Virus internalization was initiated by shifting cells treated as in (A) to 37 °C. At the indicated hours post infection (hpi), cells were trypsinized prior to lysis to observe only internalized virions, followed by immunoblotting of L1 capsid protein. (C) Effect of 3-MA on capsid protein degradation during entry. Experiment was conducted as in (A) and (B) except 3-MA was added only when cells were shifted to 37 °C to initiate internalization (no pretreatment). The data shown here are from one representative of three independent experiments. Boxed bands are from a longer exposure. M, mock; N/A, not applicable. *Non-specific band.
To investigate the effect of autophagy inhibition by 3-MA on capsid protein degradation during entry into HFKs, the HFKs treated with 3-MA and inoculated with HPV16-W12 as described above were further incubated at 37 °C for 2, 4, 6, and 12 h. By immunoblotting with the anti-HPV16 L1 antibody (CAMVIR-1), we analyzed commonly detected degradation products of the L1 capsid protein during HPV entry (Chen and Okayama, 1987; Campos et al., 2012). Interestingly, 3-MA treatment delayed the rate of L1 degradation, which is most apparent by the decrease in the amount of the 22 kDa degradation product at 2 and 4 hpi as well as the faint 12 kDa degradation product at 6 and 12 hpi (Fig. 6C). These results indicate that host autophagy facilitates HPV16 capsid protein degradation during virus entry into primary keratinocytes.
Discussion
Our study revealed that host autophagy inhibits HPV16 infection in its natural host cell, primary human keratinocytes, as HPV16 infectivity was significantly enhanced by treatment with the autophagy inhibitor, 3-MA, as well as knockdown of essential autophagy genes PIK3C3 and ATG7. Interestingly, 3-MA treatment increased HPV16 infectivity most dramatically in primary keratinocytes, where basal infectivity is much lower and autophagy levels are higher than other established keratinocyte cell lines, such as NIKS and HaCaT. These data indicate that the low level of basal HPV16 infectivity in primary keratinocytes may be partly due to the high levels of host autophagy in these cells, in addition to other restriction factors such as the lack of furin and extracellular matrix. Furthermore, we showed that HPV16 virions efficiently induce host autophagy and that inhibition of autophagy by 3-MA delayed the degradation of L1 capsid proteins during entry. Taken together, these results suggest that HPV16 virions induce host autophagy as a host defense mechanism in primary keratinocytes, which inhibits HPV16 infection by rapidly degrading incoming virions.
Several previous publications have shown conflicting results regarding the mechanisms of HPV internalization and trafficking in host cells (Bousarghin et al., 2003; Day et al., 2003; Abban et al., 2008; Spoden et al., 2008; Laniosz et al., 2009; Schelhaas et al., 2012). One potential explanation is the use of different cell culture models to study HPV entry, many of which include immortalized and transformed cells not of keratinocyte origin such as 293 and COS-7 cell lines (Bousarghin et al., 2003; Abban et al., 2008). We found that autophagy inhibition by 3-MA increased HPV16 infectivity in normal mouse epithelial cells, but not in mouse fibroblasts (data not shown). This result indicates that those two mouse cell types may have different host mechanisms that support or restrict HPV16 entry. In human keratinocytes, the natural host for HPV16 infection, the increase in HPV16 infectivity by 3-MA vary among different cell culture models. These data highlight the importance of using appropriate cell culture models to understand physiologically relevant mechanisms of HPV infection. As no procedure exists to isolate authentic HPV virions, studies of HPV entry have solely relied on the use of in vitro-generated HPV virions either from transfected 293T cells or from organotypic raft cultures of keratinocytes. These in vitro-generated HPV virions may not perfectly resemble authentic virions isolated from HPV-infected tissues.
Mechanistically, autophagy inhibition by 3-MA in primary keratinocytes delayed HPV16 capsid protein degradation during entry (Fig. 6C). However, degradation of capsid proteins was not completely blocked by 3-MA. This could be due to the fact that 3-MA treatment did not completely inhibit autophagy, as we observed ~30% decrease in LC3-II turnover by 3-MA treatment (Fig. 3B), consistent with previous findings (Fuertes et al., 2003; Zhou et al., 2010). Another explanation for partial inhibition of capsid degradation by 3-MA is that there may be alternative routes for HPV16 trafficking in the host cytoplasm other than autophagosomes. We speculate that inhibition of autophagosome formation redirects HPV16 virions to these alternative trafficking routes, which could represent productive pathways for HPV16 infection. Further studies will be necessary to elucidate these alternative trafficking routes and assess their role in productive HPV16 infection.
Autophagy originally was regarded as a mechanism to maintain cell homeostasis by removing damaged organelles and recycling nutrients (Levine and Kroemer, 2008). It is now well accepted that autophagy also plays an essential role in host defense by efficiently degrading intracellular pathogens (Kudchodkar and Levine, 2009). For example, the function of the autophagy gene Atg5 is critical for protection against lethal Sindbis virus (SIN) infection of the mouse central nervous system (Orvedahl et al., 2010). The p62 adapter protein interacts with the SIN capsid protein to target the virus to autophagosomes for degradation of viral proteins (Orvedahl et al., 2010). While autophagy plays a critical role in host defense against several viruses, autophagosomes can also provide a beneficial environment for replication of other viruses such as influenza virus (Kudchodkar and Levine, 2009). Here, we report that autophagy is induced by HPV16-infection and that this cellular response functions as a host defense mechanism that limits the efficiency of HPV infection of the natural host cell for this virus, human keratinocytes.
As a housekeeping pathway, autophagy can be induced by many cellular stresses, such as nutrient deprivation and hypoxia (He and Klionsky, 2009). However, as a host defense mechanism, autophagy can be induced by pathogen-associated molecular patterns (PAMPs) and their cognate pattern recognition receptors (PRRs) such as toll-like receptors (Delgado et al., 2009; Delgado and Deretic, 2009). Autophagy induced by PRRs has been shown to directly eliminate intracellular pathogens, such as mycobacterium (Delgado et al., 2008). Based on our findings that autophagy is induced by HPV16 to inhibit infection, we speculate that HPV16 virions are recognized by certain PRRs in primary keratinocytes, which trigger activation of innate immune signaling leading to autophagy induction. Further studies will be necessary to understand the mechanism(s) of innate immune activation and autophagy induction by HPV16 in primary keratinocytes.
Materials and methods
Reagents
3-methyladenine (3-MA) was purchased from Fisher (Acros, New Jersey, US) and was dissolved in the appropriate cell culture medium. E64d and Pepstatin A were purchased from Enzo Life Sciences (Ann Arbor, MI) and were dissolved in DMSO. Furin was purchased from New England BioLabs (Ipswich, MA).
Cell lines
293FT cells were purchased from Invitrogen (Grand Island, NY) and maintained in Dulbecco’s modified eagle’s medium (DMEM) (Thermo Scientific/HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS) (HyClone). HaCaT and NIKS cells were obtained from Dr. Paul Lambert (University of Wisconsin-Madison) and were cultured in E-medium (3 parts DMEM, 1 part Ham’s F-12 nutrient mixture) supplemented with 5% FBS. NIKS cells were co-cultured with mitomycin C-treated NIH 3T3 cells in E-medium supplemented with 2.5% FBS, 0.4 μg/mL hydrocortisone, 8.4 ng/mL cholera toxin, 5 μg/mL insulin, 24 μg/mL adenine, and 10 ng/mL epidermal growth factor, as previously described(Allen-Hoffmann et al., 2000). Mitomycin C-treated NIH-3T3 cells were present throughout all experiments with NIKS cells, as no difference in infectivity or autophagy was observed between NIKS cells cultured alone and NIKS cells co-cultured with 3T3s (data not shown). HFKs, derived from 3 neonatal foreskin donors, were purchased from Invitrogen (Cascade Biologics, Portland, OR) and cultured in EpiLife medium with 60 μM calcium supplemented with human keratinocyte growth supplement (Invitrogen/Cascade Biologics) according to the manufacturer’s protocol.
Production of HPV16 virions and pseudovirions
HPV16 virions and pseudovirions were prepared as described previously (Buck et al., 2004, 2005; Pyeon et al., 2005). Briefly, 293FT cells were cotransfected with a plasmid expressing HPV16 capsid proteins (p16SheLL) and either the full-length W12 genome (Flores et al., 1999) for HPV16-W12 virion production or pLucF (Johnson et al., 2009) for HPV16-LucF pseudovirion production. Plasmid constructs p16SheLL and pLucF were kindly provided by Dr. John Schiller (National Cancer Institute).
Production of lentiviruses
Lentiviruses were prepared by cotransfecting packaging plasmids pVSV-g (Dr. Jerome Schaack, University of Colorado School of Medicine), pCMV-HIVdeltaR8.2 (Addgene, #12263), adapted from an established protocol (Chen and Okayama, 1987). The GFP-LC3 expression plasmid pCDH-GFP-LC3, was generated by subcloning the GFP-LC3 gene from the pEGFP-C3 vector backbone (Addgene plasmid #11546) into the pCDH-puro lentivirus expression vector (Dr. Jerome Schaack). shRNA expression plasmids, shR-PIK3C3 (TRCN0000196290), ATG7 (TRCN0000007587), and shR-ctrl plasmid (shc002), were obtained from Sigma-Aldrich (The RNAi Consortium, Cambridge, MA), through the Functional Genomics Facility at the University of Colorado Boulder.
Infectivity assays
To measure HPV infectivity, cells were inoculated with 10,000 vge/cell HPV16-LucF and luciferase activity was measured at 48 hpi, using the Bright-GloTM Luciferase Assay System (Promega, Madison, WI) according to the manufacturer’s instructions. A virus concentration of 10,000 vge/cell HPV16-LucF was used to allow for easy detection of infected cells across all cell lines tested. In a parallel experiment, relative cell viability was measured using CellTiter-Glo® Luminescent Cell Viability Assay (Promega) according to the manufacturer’s instructions. Luminescence was measured with a GloMax®-Multi+ Detection System with InstinctTM software (Promega).
Immunoblotting
Cell lysates were prepared using a 1% SDS/PBS lysis buffer containing cOmplete EDTA-free Mini Protease Inhibitor Cocktail (Roche) and boiling for 10 min to ensure complete dissociation of membrane bound LC3-II. QIAshredder homogenizer columns (Qiagen, Valencia, CA) were used to reduce viscosity. Total protein concentration was determined using the Pierce® BCA Protein Assay Kit (Thermo Scientific/Pierce Biotechnology, Rockford, IL) and 20–40 μg total protein was used for immunoblotting. Antibodies: mouse monoclonal anti-β-actin (1:5000, Cell Signaling Technology, Danvers, MA), rabbit polyclonal anti-LC3B (1:1000, Cell Signaling Technology), rabbit polyclonal anti-ATG7 (1:1000, ProSci, San Diego, CA), rabbit polyclonal anti-VPS34 to detect PIK3C3 (1:500, Life Technologies, Grand Island, NY), mouse monoclonal anti-L1 (1:1000, CAMVIR-1, Abcam, Cambridge, MA) and HRP-conjugated secondary antibodies (1:10,000, Jackson ImmunoResearch Laboratories, West Grove, PA). Proteins were visualized using SuperSignal® West Pico Chemiluminescent Substrate (Thermo Scientific/Pierce Biotechnology) and the ChemiDocTM XRS+System (Bio-Rad, Hercules, CA).
Confocal microscopy
HFKs were transduced with GFP-LC3 expressing lentiviruses for 48 h prior to indicated treatments. Cells were fixed with 4% paraformaldehyde and mounted with ProLong® Gold Antifade Reagent with DAPI (Life Technologies). Imaging analysis was performed on an Olympus FluoView® FV1000 confocal microscope at the Advanced Light Microscopy Core Facility at the University of Colorado School of Medicine.
Supplementary Material
Acknowledgments
We thank Jerry Schaack for providing pVSV-g and pCDH-puro and useful suggestions for lentivirus production, John Schiller for providing pLucF and p16SheLL, Jacob Gump and Andrew Thorburn for advice on autophagy assays, and Mark Karlok for preparing HPV16 virions and pseudovirions. We also thank Chris Buck and Patricia Day for technical assistance and members of our lab for useful support and suggestions.
Footnotes
Appendix A. Supporting information Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2012.12.004.
References
- Abban CY, Bradbury NA, Meneses PI. HPV16 and BPV1 infection can be blocked by the dynamin inhibitor dynasore. Am. J. Ther. 2008;15:304–311. doi: 10.1097/MJT.0b013e3181754134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen-Hoffmann BL, Schlosser SJ, Ivarie CA, Sattler CA, Meisner LF, O’Connor SL. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J. Invest. Dermatol. 2000;114:444–455. doi: 10.1046/j.1523-1747.2000.00869.x. [DOI] [PubMed] [Google Scholar]
- Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelárová H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- Bousarghin L, Touzé A, Sizaret P-Y, Coursaget P. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J. Virol. 2003;77:3846–3850. doi: 10.1128/JVI.77.6.3846-3850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck CB, Pastrana DV, Lowy DR, Schiller JT. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 2004;78:751–757. doi: 10.1128/JVI.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck CB, Thompson CD, Pang YYS, Lowy DR, Schiller JT. Maturation of papillomavirus capsids. J. Virol. 2005;79:2839–2846. doi: 10.1128/JVI.79.5.2839-2846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd EM. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 2003;16:1–17. doi: 10.1128/CMR.16.1.1-17.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos SK, Chapman JA, Deymier MJ, Bronnimann MP, Ozbun MA. Opposing effects of bacitracin on human papillomavirus type 16 infection: enhancement of binding and entry and inhibition of endosomal penetration. J. Virol. 2012;86:4169–4181. doi: 10.1128/JVI.05493-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day PM, Lowy DR, Schiller JT. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology. 2003;307:1–11. doi: 10.1016/s0042-6822(02)00143-5. [DOI] [PubMed] [Google Scholar]
- Day PM, Lowy DR, Schiller JT. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J. Virol. 2008;82:12565–12568. doi: 10.1128/JVI.01631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado M, Singh S, De Haro S, Master S, Ponpuak M, Dinkins C, Ornatowski W, Vergne I, Deretic V. Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 2009;227:189–202. doi: 10.1111/j.1600-065X.2008.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MA, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ. 2009;16:976–983. doi: 10.1038/cdd.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Allen-Hoffmann BL, Lee D, Sattler CA, Lambert PF. Establishment of the human papillomavirus type 16 (HPV-16) life cycle in an immortalized human foreskin keratinocyte cell line. Virology. 1999;262:344–354. doi: 10.1006/viro.1999.9868. [DOI] [PubMed] [Google Scholar]
- Fuertes G, Martín De Llano JJ, Villarroya A, Rivett AJ, Knecht E. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem. J. 2003;375:75–86. doi: 10.1042/BJ20030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillison ML, Lowy DR. A causal role for human papillomavirus in head and neck cancer. Lancet. 2004;363:1488–1489. doi: 10.1016/S0140-6736(04)16194-1. [DOI] [PubMed] [Google Scholar]
- Gillison ML, Shah KV. Human papillomavirus-associated head and neck squamous cell carcinoma: mounting evidence for an etiologic role for human papillomavirus in a subset of head and neck cancers. Curr. Opin. Oncol. 2001;13:183–188. doi: 10.1097/00001622-200105000-00009. [DOI] [PubMed] [Google Scholar]
- Giroglou T, Florin L, Schäfer F, Streeck RE, Sapp M. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 2001;75:1565–1570. doi: 10.1128/JVI.75.3.1565-1570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. Papillomavirus infections—a major cause of human cancers. Biochim. Biophys. Acta. 1996;1288:F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Viruses in human cancers. Eur. J. Cancer. 1999;35:1174–1181. doi: 10.1016/s0959-8049(99)00113-6. [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KM, Kines RC, Roberts JN, Lowy DR, Schiller JT, Day PM. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 2009;83:2067–2074. doi: 10.1128/JVI.02190-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce JG, Tung JS, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 1999;274:5810–5822. doi: 10.1074/jbc.274.9.5810. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell. Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- Kawai A, Takano S, Nakamura N, Ohkuma S. Quantitative monitoring of autophagic degradation. Biochem. Biophys. Res. Commun. 2006;351:71–77. doi: 10.1016/j.bbrc.2006.09.168. [DOI] [PubMed] [Google Scholar]
- Kämper N, Day PM, Nowak T, Selinka H-C, Florin L, Bolscher J, Hilbig L, Schiller JT, Sapp M. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J. Virol. 2006;80:759–768. doi: 10.1128/JVI.80.2.759-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchodkar SB, Levine B. Viruses and autophagy. Rev. Med. Virol. 2009;19:359–378. doi: 10.1002/rmv.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laniosz V, Dabydeen SA, Havens MA, Meneses PI. Human papillomavirus type 16 infection of human keratinocytes requires clathrin and caveolin-1 and is Brefeldin A sensitive. J. Virol. 2009;83:8221. doi: 10.1128/JVI.00576-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack HID, Münger K. Modulation of autophagy-like processes by tumor viruses. Cells. 2012;1:204–247. doi: 10.3390/cells1030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers C, Mayer TJ, Ozbun MA. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA. J. Virol. 1997;71:7381–7386. doi: 10.1128/jvi.71.10.7381-7386.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004;36:2491–2502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Modis Y, Trus BL, Harrison SC. Atomic model of the papillomavirus capsid. EMBO J. 2002;21:4754–4762. doi: 10.1093/emboj/cdf494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orvedahl A, MacPherson S, Sumpter R, Tallóczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbun MA. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 2002;76:11291–11300. doi: 10.1128/JVI.76.22.11291-11300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- Pyeon D, Lambert PF, Ahlquist P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc. Natl. Acad. Sci. USA. 2005;102:9311–9316. doi: 10.1073/pnas.0504020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009;5:e1000318. doi: 10.1371/journal.ppat.1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Imarisio S, Sarkar S, O’Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J. Cell Sci. 2008;121:1649–1660. doi: 10.1242/jcs.025726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012;8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selinka H-C, Giroglou T, Nowak T, Christensen ND, Sapp M. Further evidence that papillomavirus capsids exist in two distinct conformations. J. Virol. 2003;77:12961–12967. doi: 10.1128/JVI.77.24.12961-12967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selinka H-C, Giroglou T, Sapp M. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology. 2002;299:279–287. doi: 10.1006/viro.2001.1493. [DOI] [PubMed] [Google Scholar]
- Smith JL, Campos SK, Wandinger-Ness A, Ozbun MA. Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J. Virol. 2008;82:9505–9512. doi: 10.1128/JVI.01014-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. Clathrin- and caveolin-independent entry of human papillomavirus type 16—involvement of tetraspanin-enriched microdomains (TEMs) PLoS ONE. 2008;3:e3313. doi: 10.1371/journal.pone.0003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W-C, Chao T-C, Huang Y-L, Weng S-C, Jeng K-S, Lai MMC. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011;85:10561–10571. doi: 10.1128/JVI.00173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- Yang R, Day PM, Yutzy WH, Lin K-Y, Hung C-F, Roden RBS. Cell surface-binding motifs of L2 that facilitate papillomavirus infection. J. Virol. 2003;77:3531–3541. doi: 10.1128/JVI.77.6.3531-3541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M, Christoforidis S, McBride HM, Burgoyne RD. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 1999a;397:621–625. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- Zerial M, Nielsen E, Severin F, Backer JM, Hyman AA. Rab5 regulates motility of early endosomes on microtubules. Nat. Cell Biol. 1999b;1:376–382. doi: 10.1038/14075. [DOI] [PubMed] [Google Scholar]
- Zhou X, Wang L, Hasegawa H, Amin P, Han B-X, Kaneko S, He Y, Wang F. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc. Natl. Acad. Sci. USA. 2010;107:9424–9429. doi: 10.1073/pnas.0914725107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.