

Summary
Background:
Simultaneous or consequent development of multiple solid tumors might be faced in some patients, especially the young. These tumors might be related to certain hereditary cancer syndromes or certain genetic predispositions.
Case Report:
We present the case of a 19-year-old woman with metastatic breast cancer to the contralateral axillary lymph node, associated with simultaneous osteosarcoma of the left lower femur. As she did not fit into any of the familial cancer syndromes, genetic predisposition was suspected. We detected MLH1 and MSH2 promotor methylation (PM), microsatellite instability (MSI), and different mutational events in both tumors. BRCA1 gene mutations were detected in the breast tumor, with reduced mRNA expression of BRCA1&2. ERCC1, MLH1 and MSH2, especially in OS, and RRM1 was overexpressed in both tumors.
Conclusions:
Aberrations in MMR genes could explain simultaneous or consequent development of multiple solid tumors, especially in a young patient. We recommend detecting these defects, close follow-up for those patients, and genetic counseling for their family members. Further studies in a larger population are essential to support our results.
Keywords: osteosarcoma, breast cancer, double primary, mismatch repair genes
Background
Breast cancer has great genetic heterogeneity, most probably influenced by the contribution of combined variations in steroid hormones, metabolism, cell growth/apoptosis, and DNA repair genes. Genetic variations in DNA repair may impact repair functions, DNA damage, and breast cancer risk [1]. Available evidence indicates that the majority of cancers show instability in specific sequence motifs of dinucleotide repeats. This phenotype of microsatellite instability (MSI) is commonly observed in DNA mismatch repair (MMR) pathway defects [2]. A decreased activity of MMR confers a mutator phenotype, by which the rate of spontaneous mutation is greatly elevated [3]. In addition, DNA repair mechanisms greatly affect the response to cytotoxic treatments, including radiation and chemotherapy, that target cellular DNA [4].
Case Report
A 19-year-old woman presented to the outpatient clinic of the National Cancer Institute (NCI), Cairo University, with a painless right breast mass of 5 months duration, which had progressively increased in size. Three months following appearance of the breast mass, the patient complained of pain related to the left knee, then appearance of a mass in the left knee. The patient denied any personal or family history of malignancy. Upon examination, there was a huge mass (10×12 cm), involving almost the whole of the right breast and invading the overlying skin, causing nipple ulceration. The mass was associated with bilateral axillary lymph nodes enlargement, hard and amalgamated on the ipsilateral side, while mobile and firm on the contralateral one (Figure 1). The clinical and radiological characteristics of the breast mass were highly suspicious of malignancy. Biopsy from the mass revealed invasive duct carcinoma grade II with scattered lympho-vascular permeation (Figure 2A,B). Immunostaining for estrogen and progesterone receptors were positive (ER+, PR +), and Her-2/neu was overexpressed (score 3). Fine-needle aspiration cytology (FNAC) revealed metastatic deposits in the ipsilateral and the contralateral axillary nodes.
Figure 1.
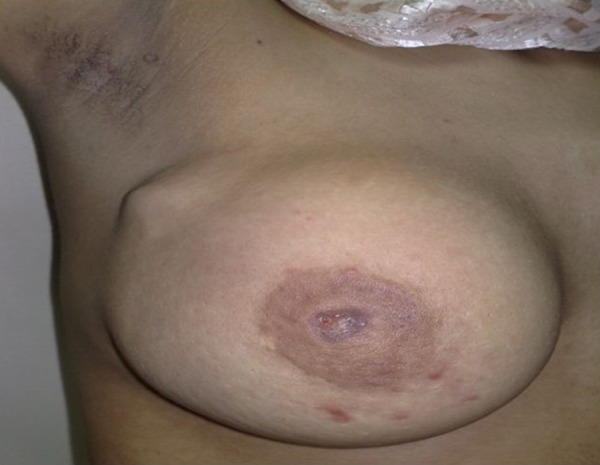
The breast mass involving almost the whole breast with ulcerated nipple.
Figure 2.

(A) The tumor area from the breast mass showing invasive groups of malignant moderately differentiated ductal cells (x), (B) the same tumor area showing positive immunostaining of the malignant ductal cells for Her 2 neu (+3) (×200), (C) a microscopic picture of the bony lesion showing malignant spindle cells associated with deposition of osteoid, which supports the diagnosis of osteosarcoma, and (D) negative immunostaining for cytokeratin CK in the malignant osteoblasts.
Plain X-ray of the chest and liver ultrasonography were free of metastatic deposits, while bone scan showed a single area of increased uptake in the supracondylar region of the left femur, but the rest of the skeleton was free. Local X-ray of the left femur showed an irregular, dense, ill-defined, osteosclerotic lesion, with possible soft tissue extension (Figure 3), which was confirmed by MRI. Radiological features of the lesion raised the suspicion of second primary malignant lesion rather than breast cancer, so biopsy from the bone lesion was taken. Pathological examination showed a malignant neoplasm formed of proliferating pleomorphic large cells having scanty cytoplasm, and hyperchromatic nuclei with related deposition of thin lace-like branching osteoid matrix and negative immunostaining for CK (Figure 2C,D), excluding breast origin and confirming the diagnosis of conventional osteosarcoma GII, osteoblastic type. Hence, the final diagnosis was a case with double primary tumors: a) invasive duct carcinoma of the right breast, metastatic to contra-lateral axillary LNs (T4b N2a M1), and b) osteosarcoma of the LT femur (Enneking stage IIB). Chemotherapy treatment was started with 1 cycle of FEC 100 regimen (cyclophosphamide, epirubicin & 5-fluorouracil), which was associated with regression of the breast mass, but progression of the osteosarcoma. The patient was then shifted to another regimen, which has activity in both types of tumors (docetaxel 70 mg/m2 D1, Cisplatin 70 mg/m2 D1 & Epirubicin 80 mg/m2 D1), and she showed a good partial clinical response in both lesions.
Figure 3.
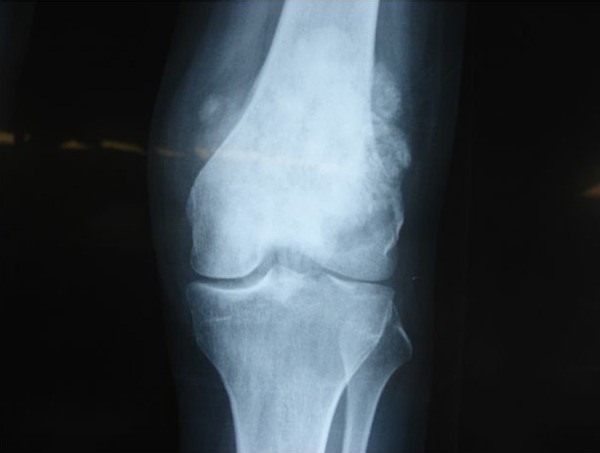
X-ray of the left femur showing an ill defined irregular osteoscelorotic lesion in the supracondylar region with soft tissue extension.
Ten micron thick sections were obtained from paraffin blocks of both biopsies (breast invasive duct carcinoma & Lt femur osteosarcoma) and were subjected to genetic testing to search for mismatch repair (MMR) genes aberrations as a highly possible explanation for the multiple, early-onset neoplasms in the patient. DNA and RNA extraction was done using commercially available DNA& RNA extraction kits (Qiagen) according to manufacturer’s protocols. Extracted DNA was used to detect: a) promoter hypermethylation (PM) in BRCA1, BRCA2, MLH1, MSH2 and GPB6 [5], b) mutations and microsatellite instability (MSI) of MLH1 and MSH2 [6], c) mutations in exons 5–9 of the p53 and exons 6, 7, 11 of BRCA-1 genes by polymerase chain reaction (PCR)/sequencing [7,8], d) loss of heterozygosity (LOH) in exons 6, 7,11 of BRCA-1 [8], and e) mutations of K12/13-ras by PCR/RFLP [9]. The RNA was used to detect BRCA1&2, ERCC1, RRM1, MSH2, MLH1 and MSH6 expression by quantitative real time PCR [5,10,11]. Her2/neu gene amplification in breast and osteosarcoma was determined by chromogen in situ hybridization (CISH) with Invitrogen, Spot Light HER2 detection kit according to manufacturer’s instructions.
Her2/neu gene amplification was detected in the breast tumor (BT) but not in the osteosarcoma (OS). P53 mutations were detected in BT and OS, though at different codons (Figure 4), while K13-ras mutations was detected in OS only (Figure 5). BRCA1 gene mutations were concentrated at exons 11 and 1, whereas LOH concentrated at exons 11 followed by 6 (Tables 1, 2). No mutations were detected in BRCA2 gene, though reduced mRNA expression of BRCA1&2 (Figure 6) was evident in both tumor samples. Similarly, ERCC1, MLH1, MSH2 mRNA expression was reduced in both tumors, especially in the OS, whereas MSH6 was normally expressed and RRM1 was overexpressed in both tumors. MLH1 and MSH2 showed PM in both tumors (Figure 7), MSI in BT, and different mutational events in both tumors (Tables 1, 2 & Figure 8).
Figure 4.

Sequence analysis of the p53 gene for the two lesions showing mutation in the p53 gene (GCC to ACC) in the osteosarcoma (OS) but not in breast invasive duct carcinoma (IDC).
Figure 5.

An ethedium bromide-stained gel showing the wild type of codon 12-ras (125bp band) both in OS and IDC. Mutation in codon 13-ras was detected in OS (157bp band) only whereas the IDC showed no mutations.
Table 1.
Summary of mutational analysis of the studied genes in breast tumor.
| Gene | Exon | Codon | Nucleotide change | Mutation status |
|---|---|---|---|---|
| hMSH2 | 8 | 453 | A1358-------T | Splice site------30bp deletion |
| 12 | SD of exon12 | Del of 11bp at 2005 | Splice defect | |
| 15 | 878 | 2633 del (AG) | Frameshift | |
|
| ||||
| MLH1 | 12 | 397 | 1190 del (T) | Frameshift |
|
| ||||
| BRCA1 | 11 | 64 | TGT-GGT | Missense |
| 392 | 1294–1333 del | Frameshift | ||
| 1252 | 3875–3878 del | Frameshift | ||
| 1656 | 5085–5103 del | Frameshift | ||
| 1773 | ACCC-ACCCC (ins) | Frameshift | ||
|
| ||||
| P53 | 4 | 72 | GGC-CCC | Missense |
| 5 | 138 | GCC-ACC | Missense | |
| 5 | 163 | TAC-AAC | Missense | |
| 5 | 161 | 1 base pair insertion | Frameshift | |
| 6 | 209 | AGA-ACA | Frameshift | |
| 7 | 245 | GGC-AGC | Missense | |
| 8 | 280 | AGA-ACA | Frameshift | |
Table 2.
Summary of mutational analysis of the studied genes in osteosarcoma.
| Gene | Exon | Codon | Nucleotide change | Mutation status |
|---|---|---|---|---|
| hMSH2 | 1 | 45 | 134 del of 29bp | frameshift |
| 8 | 453 | A1358-------T | Splice site------30bp deletion | |
| 15 | 878 | 2633 del (AG) | Frameshift | |
| 7 | 389 | C1165-----T | Arg------Stop codon | |
|
| ||||
| MLH1 | 12 | 397 | 1190 del (T) | Frameshift |
|
| ||||
| BRCA1 | 11 | 64 | TGT-GGT | Missense |
| 392 | 1294–1333 del | Frameshift | ||
| 1443 | CGA-GGA | Missense | ||
| 1773 | ACCC-ACCCC (ins) | Frameshift | ||
|
| ||||
| P53 | 4 | 72 | GGC-CCC | Missense |
| 5 | 126 | TAC-TAG | STOP CODON | |
| 5 | 175 | CGC-CAC | Missense | |
| 5 | 163 | TAC-AAC | Missense | |
| 5 | 161 | 1bp insertion | Frameshift | |
| 6 | 205 | TAT-CAT | Missense | |
| 7 | 241 | TCC-TTC | Missense | |
| 7 | 245 | GGC-AGC | Missense | |
| 8 | 3′+GT-GG | Frameshift | ||
Figure 6.

An ethedium bromide-stained gel for the RT-PCR showing reduction/loss of BRCA1, BRCA2, MLH1, MSH2 mRNA in the IDC as well as in the OS samples. BRCA2 showed reduced expression in OS only while MSH6 was normally expressed.
Figure 7.

Methylation specific PCR showing promoter hypermethylation (PM) of MLH1 and MSH2 genes both in the IDC and OS samples.
Figure 8.
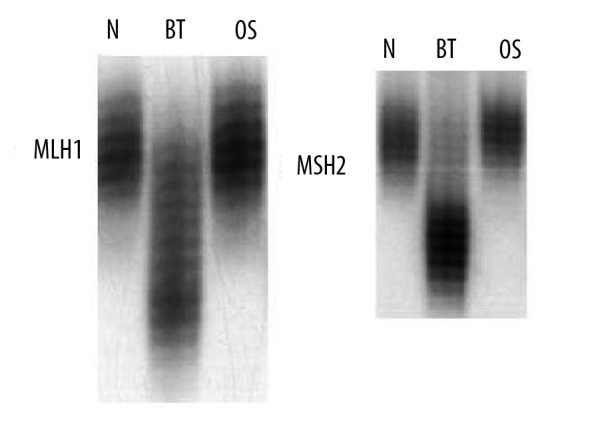
Microsatellite instability (MSI) from the IDC and the OS showing MSI in the MLH1 and MSH2 gene loci in the IDC but not in OS.
Discussion
In the present case, a 19-year-old woman attended the NCI clinics, with breast and femoral bone lesions that proved to be invasive duct carcinoma (IDC) and osteosarcoma (OS); respectively. A genetic predisposition was highly suspected, though the patient did not fit into any of the well-known genetic syndromes such as Li-Fraumeni (LF), ataxia telangiectasia (AT) or Lynch syndrome (LS). Although in LS various organs and organ systems may be affected by malignancies in addition to colorectal and endometrial cancer [12,13], it usually affects individuals in the 4th or 5th decade; but occasionally patients may be younger (in the 3rd decade) or older (in the 6th or 7th decade) [14]. However, LS is characterized by a germline heterozygous defect or mutations in one of the mismatch repair (MMR) genes (MLH1, MSH2, MSH6) transmitted in an autosomal-dominant manner. In our case, the young age at presentation and the negative family history led us to exclude LS, but the early onset and the multiplicity of the lesions raised a high possibility of MMR gene defects. Therefore, we sought to assess for MMR genes aberrations as a possible cause for this case with multiple, early onset malignancies.
MMR gene defects have been previously reported in solid tumors, including sporadic and familial breast cancer and OS [15,16]. Our patient showed reduced mRNA of the assessed MMR genes in both tumor samples; mainly MLH1, MSH2 and MSH6. However, there was a difference between both lesions, since OS showed marked reduction of MLH1, MSH2 compared to BT, whereas MSH6 was much reduced in the BT sample. This reduction in the expression of MLH1, MSH2 and MSH6 could be induced by aberrant PM, MSI, mutations or more than one defect.
Our mutational analysis showed a difference in the mutation pattern in both lesions, but several mutational events were detected in both lesions. In general, the OS revealed a slightly higher mutation frequency (MF) than the BT, especially for MSH2 and p53 genes, suggesting that the OS might have developed later than the BT as a consequence of accumulating genetic damage due to ineffective MMR, although the patient presented with both lesions. On the other hand, BRCA1 showed more mutations in the BT. The mutations reported here have been previously reported in other solid and hematological tumors [17,18].
Recently, other DNA repair genes have been identified that can contribute to tumor development and progression. Among these are the ERCC1 (excision repair cross-complementing 1) and BRCA1, which are key genes in NER, double-strand break repair, and mismatch repair, as well as the ribonucleotide reductase M1 (RRM1), which is involved in DNA synthesis, catalyzing the biosynthesis of DNA from the corresponding ribonucleotides, and involved in gemcitabine resistance [10]. Using real-time quantitative PCR, we found that BRCA1&2, and ERCC1 were markedly reduced in BT and OS, but again to a higher extent in OS, whereas RRM1 was overexpressed in both tumors. BRCA1 gene aberrations could be attributed to mutations, microsatellite instability, and/or loss of heterozygosity (LOH), which is in accordance with previous reports in the literature, whereas reduced BRCA2 and increased RRM1 expression are probably induced by post-translation modification [6,10,18,19].
As a consequence of the multiple defects in the key MMR genes reported in our case, other genetic errors were expected. Therefore, we assessed both tumors (BT&OS) for mutations in some other genes that are frequently mutated in solid tumors and we were able to detect several mutational events in K12/13-ras and p53 genes in BT and OS, although at different sites (Tables 1, 2). This confirmed our assumption that other genetic errors could be present as a consequence of MMR genes defects, although not detected here, and this could have led to the development of other malignancies if the patients survived longer.
Conclusions
We conclude that MMR gene defects through mutations, PM, MSI or other mechanisms could explain simultaneous or consequent development of multiple solid tumors, especially in a young patient. These tumors might not reveal (exploit) the full picture of certain hereditary cancer syndromes, but it might confer genetic predisposition. With time, there is increased probability to acquire more genetic damage, and consequently, there is a high possibility of developing more tumors and/or increasing aggression and resistance of the existing tumors to the current treatment modalities. Therefore, it is important for clinicians and geneticists to suspect MMR gene defects and to recommend techniques for detection of these defects, especially when dealing with multiple tumors in a young patient, and particularly in patients who have small families or hypomorphic mutations (mutations leading to an abnormal protein, even with residual function). It is also important to recommend close follow-up for those patients and genetic counseling for their family members to detect genetic abnormalities in MMR genes and other genes, which are linked to the familial cancer syndromes, as well as for early detection of any malignancy that might develop.
References:
- 1.Smith TR, Levine EA, Freimanis RI, et al. Polygenic model of DNA repair genetic polymorphisms in human breast cancer risk. Carcinogenesis. 2008;29:2132–38. doi: 10.1093/carcin/bgn193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 3.Conde J, Silva SN, Azevedo AP, et al. Association of common variants in mismatch repair genes and breast cancer susceptibility: a multigene study. BMC Cancer. 2009;9:344. doi: 10.1186/1471-2407-9-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowe BP, Glazer PM. Emergence of rationally designed therapeutic strategies for breast cancer targeting DNA repair mechanisms Breast Cancer Research. 2010;12:203. doi: 10.1186/bcr2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.An J, Wei Q, Liu Z, Lu, et al. Messenger RNA expression and methylation of candidate tumor-suppressor genes and risk of ovarian cancer – a case-control analysis. Int J Mol Epidemiol Genet. 2010;1:1–10. [PMC free article] [PubMed] [Google Scholar]
- 6.Poley JW, Wagner A, Hoogmans MM, et al. Biallelic germline mutations of mismatch-repair genes: A Possible Cause for Multiple Pediatric Malignancies. Cancer. 2007;109:2349–56. doi: 10.1002/cncr.22697. [DOI] [PubMed] [Google Scholar]
- 7.Fujita M, Inoue M, Tanizawa O, et al. Alteration of the p53 gene in human primary cervical carcinoma with and without papilloma virus infection. Cancer Res. 1992;52:5323–28. [PubMed] [Google Scholar]
- 8.Hondow HL, Fox SB, Mitchell G, et al. A high throughput protocol for mutation scanning of the BRCA1 and BRCA2 genes. BMC Cancer. 2011;11:265. doi: 10.1186/1471-2407-11-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popović Hadzija M, Korolija M, Jakić Razumović J, et al. K-Ras and Dpc4 Mutations in Chronic Pancreatitis: Case Series. Croat Med J. 2007;48:218–24. [PMC free article] [PubMed] [Google Scholar]
- 10.Bartolucci R, Wei J, Sanchez JJ, et al. XPG mRNA Expression Levels Modulate Prognosis in Resected Non–Small-Cell Lung Cancer in Conjunction with BRCA1 and ERCC1 Expression. Clin Lung Cancer. 2009;10:47–52. doi: 10.3816/CLC.2009.n.007. [DOI] [PubMed] [Google Scholar]
- 11.Zekri AR, Bahnassy AA, Abdel-Wahab SA, et al. Expression of pro- and anti-inflammatory cytokines in relation to apoptotic genes in Egyptian liver disease patients associated with HCV-genotype-4. J Gastroenterol Hepatol. 2009;24:416–28. doi: 10.1111/j.1440-1746.2008.05699.x. [DOI] [PubMed] [Google Scholar]
- 12.Lynch HT, Smyrk T. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review. Cancer. 1996;78:1149–67. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1149::AID-CNCR1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 13.Aarnio M, Mecklin JP, Aaltonen LA, et al. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer. 1995;64:430–33. doi: 10.1002/ijc.2910640613. [DOI] [PubMed] [Google Scholar]
- 14.Vasen HF, Wijnen JT, Menko FH, et al. Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. Gastroenterology. 1996;110:1020–27. doi: 10.1053/gast.1996.v110.pm8612988. [DOI] [PubMed] [Google Scholar]
- 15.Futreal PA, Liu Q, Shattuck-Eidens D, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994;266:120–22. doi: 10.1126/science.7939630. [DOI] [PubMed] [Google Scholar]
- 16.Siclari VA, Qin L. Targeting the osteosarcoma cancer stem cell. J Orthop Surg Res. 2010;5:78. doi: 10.1186/1749-799X-5-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Jong AE, Van Puijenbroek M, Hendriks Y, et al. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10:972–80. doi: 10.1158/1078-0432.ccr-0956-3. [DOI] [PubMed] [Google Scholar]
- 18.De Jong AE, Morreau H, Van Puijenbroek M, et al. The role of mismatch repair gene defects in the development of adenomas in patients with HNPCC. Gastroenterology. 2004;126:42–48. doi: 10.1053/j.gastro.2003.10.043. [DOI] [PubMed] [Google Scholar]
- 19.Ewald IP, Ribeiro PL, Palmero EI, et al. Genomic rearrangements in BRCA1 and BRCA2: A literature review. Genet Mol Biol. 2009;32:437–46. doi: 10.1590/S1415-47572009005000049. [DOI] [PMC free article] [PubMed] [Google Scholar]