

Abstract
5-Fluorouracil (5-FU) is a widely utilized cancer chemotherapeutic that causes DNA damage via two mechanisms. Its active metabolite inhibits thymidylate synthase, which deprives cells of TTP and causes the introduction of uracil in DNA. Also, 5-FU is directly incorporated into DNA. Both uracil and 5-FU in DNA are recognized by uracil-DNA glycosylases (UDGs), which initiate base excision repair. UNG and SMUG1 are the two human UDGs most likely to combat the genomic incorporation of uracil and 5-FU during replication. In this study, we examined the roles of UNG and SMUG1 in the initial cellular response to 5-FU and compared continuous exposure to a 24 hour exposure followed by incubation in drug-free media, which mimics what occurs clinically. Loss of UNG did not alter cellular sensitivity to 5-FU in two human cell lines, despite its predominant biochemical activity for uracil and 5-FU in DNA. Loss of SMUG1 corresponded with >2-fold increase in sensitivity to 5-FU, but only with a 24 h treatment followed by recovery. There was no difference between SMUG1 proficient and depleted cells following continuous exposure. We observed that 5-FU treatment induced an enhanced S-phase arrest and CHK1 activation plus an increase in the formation of strand breaks and alkali-labile sites in all sublines. However, SMUG1-depleted cells showed a prolonged S-phase arrest, a transient increase in DNA double-strand breaks following 5-FU treatment and an altered phosphorylation of CHK1 following removal of drug. Collectively, the results suggest that SMUG1 has a role in the resumption of replication following 5-FU treatment.
1. INTRODUCTION
Inhibition of thymidylate synthase (TS) continues to be a mainstay chemotherapeutic strategy to treat several types of solid tumors. 5-fluorouracil (5-FU) and its prodrug capecitabine are metabolized to FdUMP (5′-fluorodeoxyuridylate), which functions as a suicide inhibitor of TS. Several antifolates including raltitrexed (RTX) and pemetrexed inhibit thymidylate synthesis by competing with the methyl donor, N5,N10-methylene tetrahydrofolate, in the reaction catalyzed by TS. Depletion of thymidylate pools by TS inhibitors results in an increase in genomic uracil [1]. Uracil or 5-FU in DNA is recognized by uracil DNA glycosylase (UDG)-initiated base excision repair (BER) [2]. However, investigations of the precise contributions of individual BER components have not drawn simple conclusions regarding the contribution of BER to the cellular sensitivity to TS inhibitors. Among the four known mammalian UDGs capable of removing uracil and 5-FU from DNA, nuclear UNG has received the most attention because it is the dominant UDG activity in cells, is up-regulated in S-phase, and co-localizes with replication foci [3]. Yet, most studies that have directly tested the role of UNG have concluded that it does not contribute to differences in cellular sensitivity to any TS inhibitor [4–9]. Examination of the other three mammalian UDGs reveals a complex picture. One report of a SMUG1 knockdown in mouse embryonic fibroblasts (MEFs) noted an accumulation of genomic 5-FU and heightened cellular sensitivity to 5-FU [4], while recent reports showed no SMUG1-dependent differences in human cells [8] or more recently derived MEFs [10]. Knockouts/knockdowns of the TDG or MBD4 glycosylases revealed a resistance to 5-FU [11,12].
We previously showed that TS inhibition in UNG-inhibited cells results in significant elevation of genomic uracil, yet did not alter cytotoxicity associated with TS inhibition [7]. These earlier studies were focused on genomic uracil, not 5-FU, and thus predominantly utilized the antifolate RTX. Also note that nearly all previous studies have employed a continuous exposure to 5-FU or other TS inhibitors, and it is known that prolonged exposure to TS inhibitors can induce cell death via multiple means [13]. In the current study, the mechanism of action of 5-FU was investigated with specific attention focused on the role of UNG and SMUG1 in CHK1 activation, strand break formation and resolution.
2. MATERIAL & METHODS
2.1 Drugs and Cell Culture
Raltitrexed (RTX) was generously supplied by AstraZeneca, U.K. 5-Fluorouracil was purchased from Sigma (St. Louis, MO). Comet Assay Slides (4250-050) were purchased from Trevigen (Gaithersburg, MD). Anti-β-actin was from Abcam (Cambridge, MA). Anti-Chk1, anti-p-Chk1 (Ser345), anti-Caspase-3, and anti-cleaved-Caspase-3 were purchased from Cell Signaling Technology (Boston, MA). Horseradish peroxidase-conjugated secondary antibodies were from GE-Amersham. LN428 sublines were cultured in α-Eagle’s MEM without ribonucleosides (ATCC, Manassas, VA) and supplemented with 10% heat-inactivated fetal bovine serum, glutamine (Sigma, St. Louis MO), 1% penicillin/streptomycin (Sigma), and gentamycin (Sigma). The LN428 glioblastoma cells (wild-type, WT) have been described by us previously [14,15]. The LN428/UNG-KD and LN428/SMUG1-KD cells, were generated by lentiviral-mediated expression of shRNA, as described [14,15]. Lentiviruses were prepared in collaboration with the UPCI Lentiviral facility. Lentiviral particles were generated by co-transfection of 4 plasmids [the shuttle vector plus three packaging plasmids: pMD2.g(VSVG), pVSV-REV and PMDLg/pRRE] into 293-FT cells using FuGene 6 Transfection Reagent. Lentiviral transduction was performed by seeding 6.0 × 104 cells into a 60 mm dish 24 h before transduction. Cells were transduced for 18 h at 32°C and then cultured for 72 h at 37°C. Cells were then selected by culturing in growth media with 1.0 μg/mL puromycin [14,15] to yield the pooled populations that were evaluated herein. Single-cell clones were then derived by limiting-dilution cloning in 96-well plates [14,15]. The stable single-cell derived LN428 glycosylase-KD cell lines described here are available from Trevigen, Inc (Gaithersburg, MD). Level of KD was determined by qRT-PCR [14,15].
2.2 Generation of SMUG1 knockdown in 293 cells
The pLKO.1-Puro lentivirus plasmid vector containing MISSION™ shRNA against SMUG1 (NM_014311) and a negative control vector containing no shRNA insert (SHC002) was purchased from Sigma. The plasmids were transfected into HEK293 cells using a GenePulser Xcell electroporator according to the manufacturer’s protocol for HEK293 (Bio-Rad, Hercules, CA). At 24 h post-transfection the media was replaced with DMEM containing 1 μg/ml puromycin. The cells were expanded for 14 days with a change of media every 2 days. The clones were analyzed for SMUG1 expression by western blotting.
2.3 Cell proliferation
Cell survival was measured using the MTS assay (Promega, Wisconsin, USA) following either 24 h drug treatment plus 3 days incubation in drug-free media or continuous exposure for 4 days. Cells in exponential growth were plated at a concentration of 1 × 104 per well 24 h prior to drug treatments. Drugs were added in fresh medium, 6 wells per sample. Following treatments, 20 μL of MTS was added, and the plate was incubated for an additional 3 h at 37 °C. The absorbance values at 490 nm were collected and cell viability was calculated as a percentage compared to untreated control cells.
2.4 Western blot analysis
Cells were treated with drugs for indicated times. Whole cell protein extract was prepared from each cell line using lysis buffer containing Complete™ mini protease inhibitor cocktail (Roche Life Sciences, NJ, USA) and the phosphatase inhibitors sodium orthovanadate and sodium fluoride (Sigma). Electrophoresis and blotting was performed as previously described [7,16–18].
2.5 Cell-cycle analysis by flow cytometry
Cells (5 × 104) were seeded onto 100 mm dishes. After 48 h, medium containing the appropriate dose of 5-FU was added to the treatment plates, while medium without drug was added to control plates. After drug treatment and incubation in drug free media, adherent and floating cells were collected, fixed by chilled 100% ethanol, and stored at 4 °C until processed for analysis. Fixed cells were stained with PI/RNase A solution containing propidium iodide (50 μg/mL), RNase A (0.1 mg/mL) and 1% BSA in PBS. Cell-cycle analyses were performed using a Beckman Coulter FC 500 cytometer (Beckman Coulter, CA, USA) and data quantified using ModFit LT software version 3.1 (Verity Software House, ME, USA).
2.6 Uracil DNA glycosylase (UDG) activity
UDG activity for the cell extracts of LN428 sublines was measured using an oligonucleotide-based assay [7]. The oligodeoxynucleotide contained a single 5-FU substrate (5′-HEX)-[GACTACTACATG(FdU)TTGCCGACCATT-3′] (Midland Certified Reagents, Midlands, TX). The excision assay was carried out by incubation of the duplex oligo with 2 μg of cell extracts in buffer (20 mM Tris,100 mM KCl, 5 mM EDTA, 1 mM EGTA, 5 mM 2-mercaptoethanol, pH 7.2) at 37 °C for the times listed. Purified Ugi was obtained from New England Biolabs. The reaction products were separated on a denaturing 20% polyacrylamide gel. The gels were visualized and quantified with a Bio-Rad Molecular Imager® FX and Quantity One® software.
2.7 Comet Assay
DNA fragmentation was detected using the comet assay as described [17]. Briefly, following treatment and harvesting, cells were re-suspended in PBS, and mixed with 1% low gelling type VII agarose at 40 °C, and pipetted onto pre-coated slides (Trevigen, Gaithersberg, MD). Once the agarose solidified, slides were submerged in ice-cold lysis buffer (1% Triton X-100, 100 mM Na2EDTA, 2.5 M NaCl, 10 mM Tris, pH 10.5). After lysis, slides were washed and submerged in alkali buffer (50 mM NaOH, 1 mM Na2EDTA, pH 13) for 45 min. Electrophoresis was for 25 min at 0.6 V/cm. Slides were washed with neutralization buffer (0.5 M Tris–HCl, pH 7.5), followed by PBS, then dried overnight. The slides were rehydrated with distilled water and stained with Sybr® Green (Invitrogen). Comets were visualized with a Nikon E600 microscope. The endpoint measured was the DNA moment using the LAI automated comet analysis software from Loats Associates (Westminster, MD, USA). The neutral comet assay was performed as above except lysis was carried out overnight at 4 °C. Following lysis, the cells were washed with neutral electrophoresis buffer (150 mM NaCl, 10 mM EDTA, 10 mM Tris) and electrophoresed for 45 min at 1 V/cm. The slides were then processed as described above.
2.8 Statistical Analysis
Means, standard error and standard deviation were calculated using Microsoft Excel or GraphPad Prism software. Significance of data was analyzed using the paired t-test or one-way ANOVA analysis. P-value < 0.05 was considered as statistically significant.
3. RESULTS
3.1 Knockdown of SMUG1 results in a higher sensitivity to 5-FU when treatment is followed by incubation in drug free media
Knockdowns of UNG and SMUG1 were generated in LN428 glioblastoma cells. Measurement of mRNA from the pooled populations revealed that the respective messages were reduced by 75% in the LN428/SMUG1-KD cells and 55% in the LN428/UNG-KD cells (Fig. 1A). Single cell cloning of the UNG-KD cells yielded significantly greater UNG depletion (91% knockdown) but to avoid clonal effects, this cell clone was not used herein. The excision of 5-FU from an oligodeoxynucleotide was measured in cell free extracts from parental (WT), UNG knockdown (LN428/UNG-KD) and SMUG1 knockdown (LN428/SMUG1-KD) cells. The results show that excision of 5-FU is decreased in UNG-KD and SMUG1-KD cells by 30% and 40%, respectively (Fig. 1B), which corresponded with the reduction in mRNA. Inhibition of UNG activity in the cell extracts by co-incubation with Ugi further reduced the excision of 5-FU, suggesting that UNG was responsible for residual 5-FU excision activity seen in extracts from each of the sublines.
Figure 1.
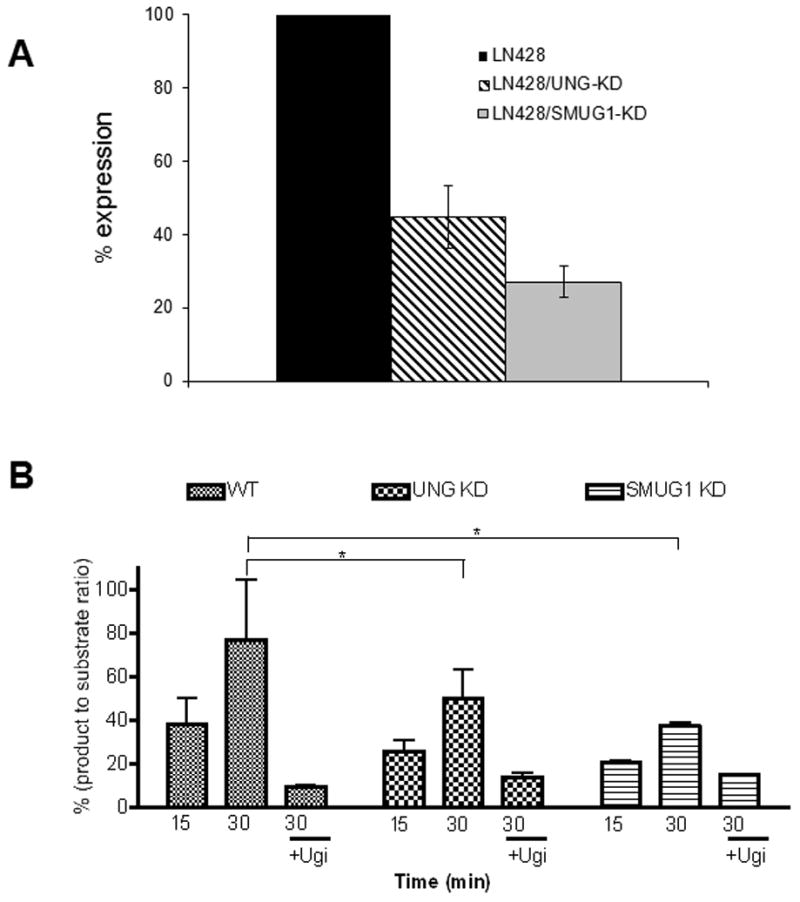
A) Relative expression of SMUG1 mRNA in SMUG1-KD cells and UNG mRNA in UNG-KD cells compared to parental (WT) LN428 cells as measured by RT-PCR. Error bars represent standard error of the mean. B) Excision of 5-FU from a synthetic oligodeoxynucleotide. Whole cell protein extracts (5 μg) from parental (WT), UNG-KD and SMUG1-KD cells were incubated with the oligo for 15 or 30 min alone or in the presence of the Ugi peptide inhibitor of UNG (+ Ugi). Error bars represent standard error of the mean. Statistical significance was measured by ANOVA analysis followed by Bonferroni’s multiple comparison test (* p<0.05).
The 5-FU treatment regimen of 24 h exposure followed by incubation in drug-free media sensitized the LN428/SMUG1-KD cells compared to UNG KD and wild-type cells (Fig. 2A). Specifically, the IC50 of 5-FU in the SMUG1 knockdown cells (20 μM) is 2.5 fold lower than wild-type cells (50 μM), while sensitivity of the UNG knockdowns was intermediate (35 μM). However, there was no difference in sensitization between the cell lines with a continuous exposure to 5-FU (Supplementary Fig. 1). The cytotoxicity of the antifolate RTX was also measured in the LN428 cells to determine the influence of TS inhibition without 5-FU incorporation. There was no difference in sensitivity to RTX among the SMUG1 or UNG knockdown cells compared to wild-type (data not shown). These results suggest that the increased sensitivity seen in SMUG1-deficient cells is specific to 5-FU-mediated lesions in DNA and to a response mediated during an attempted restart of replication (see the next sections). A SMUG1 knockdown was also examined in HEK293 cells to determine the sensitization of a SMUG1 knockdown in an unrelated cell line and to compare with our previous studies in UNG-inhibited cells, in which we saw no effect of UNG [7]. The efficiency of SMUG1 knockdown was 75% as measured by Western blotting (Supplementary Fig. 2A). SMUG1 knockdown resulted in a greater sensitivity to 5-FU with a treatment regimen of 24 h exposure followed by incubation in drug free medium (Supplementary Fig. 2B). Specifically, the IC50 of 5-FU in SMUG1-KD cells (8 μM) was >2-fold lower than in wild-type 293 cells (20 μM). No further studies were conducted in the 293 cells because the activities of UNG proteins in the two cell lines were inhibited via two different methods (Ugi-peptide mediated inhibition vs siRNA KD). Note at the higher concentrations of 5-FU, there was no further killing, which is commonly observed in response to TS inhibitors [4,7,17,19], and is caused by salvage of thymidine [20,21].
Figure 2.
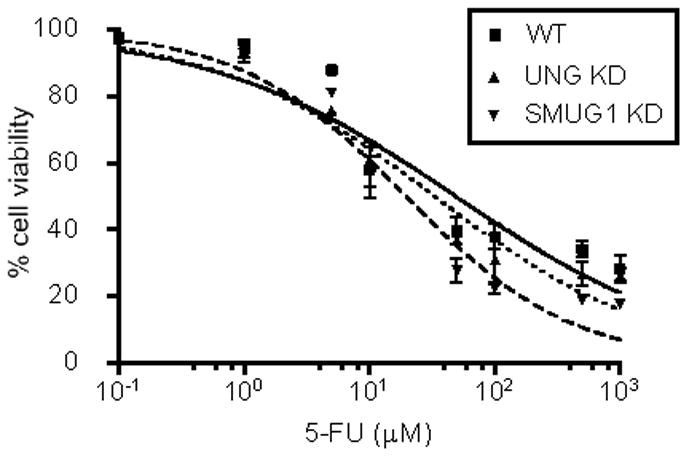
Cytotoxicity of 5-FU in LN428 sublines. Parental LN428 (WT) cells are represented by a filled square and solid line, UNG knockdowns are represented by an open triangle and short dashed line, and SMUG1 knockdowns are represented by an inverted triangle and long dashed line. Cells were treated with increasing doses of 5-FU for 24 h followed by 3 days recovery in drug-free medium. Following recovery, viable cells were measured using the MTS assay (Materials and Methods). Each plot represents % cell viability relative to the untreated control for each cell line. Each data point represents an average of three independent experiments with means calculated from triplicate values in each experiment. The error bars represent standard error of the mean. Statistical significance was determined using a paired t-test (P<0.05).
3.2 Prolonged S-phase cell cycle arrest with 5-FU in SMUG1-KD cells
TS inhibitors induce an early S-phase arrest [7,16,17], but the source of the arrest, nucleotide depletion or DNA damage, is unresolved. The cell cycle distribution of the LN428 sublines after 5-FU treatment was assessed by flow cytometry to examine the response to treatment followed by incubation in drug-free media. Untreated cells showed similar cell cycle profiles for the parental (WT), the UNG-depleted, and SMUG1-depleted cells (Fig. 3A). Treatment with 5-FU for 24 h resulted in a sharp S-phase arrest. LN428/SMUG1-KD cells in particular did not show a measurable G1 peak whereas a G1 population was still evident in parental (26 %) and UNG knockdowns (10 %) (Fig. 3B). Differences in cell cycle distribution between the three sublines were more pronounced during the subsequent incubation in the absence of drug. Incubation in drug-free media for 24 h resulted in the resumption of cell cycle progression in parental cells, as gauged by the re-appearance of G1 and G2 populations comprising >50% of the total population (Fig. 3C). In contrast, >80% of the UNG and LN428/SMUG1-KD cells remained in S phase (Fig. 3C). By 48 h post-drug exposure and incubation in drug-free media, the G1 population in parental and LN428/UNG-KD cells was 40% and 20%, respectively, while there was no measureable G1 peak in the LN428/SMUG1-KD cells, and 80% of LN428/SMUG1-KD remained in S phase (Fig. 3D).
Figure 3.
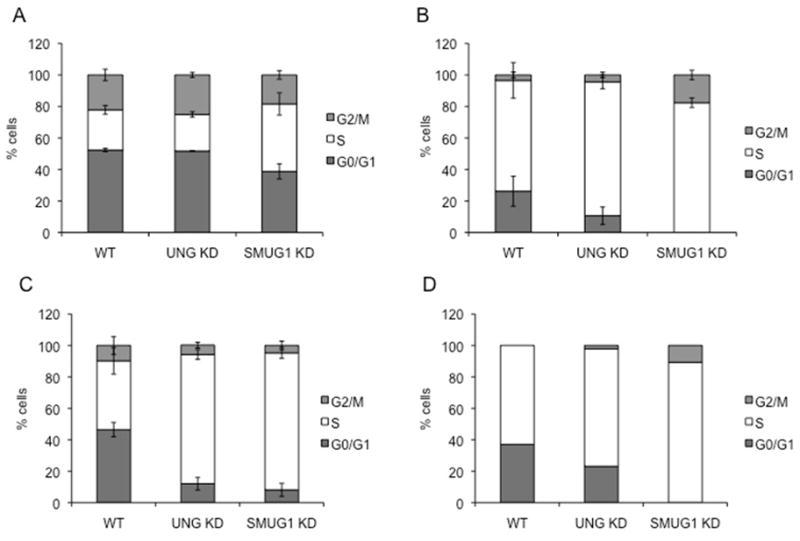
Cell cycle distribution of parental LN428 (WT, left), LN428/UNG-KD (central) and LN428/SMUG1-KD (right) cells. The percentage of cells in G0/G1, S, and G2/M phases of the cell cycle was calculated using Modfit software (Materials and Methods). A) Untreated cells. B) Cells treated with 50 μM 5-FU for 24 h. C) Cells treated with 5-FU for 24 h followed by 24 h incubation in drug-free medium. D) Cells treated with 5-FU for 24 h followed by 48 h incubation in drug-free medium. The error bars represent standard error of the mean.
3.3 CHK1 phosphorylation and dephosphorylation following recovery from 5-FU treatment
Because the data in Fig. 3 suggested that there were differences in resumption of S-phase during recovery from 5-FU treatment in the wild-type cells, the activation of a crucial checkpoint kinase, CHK1, was measured. 5-FU induced S-phase arrest has been shown to be dependent upon CHK1 activation [22]. The dynamics of CHK1 activation via phosphorylation of Ser345 was measured at time points during the 24 h treatment of 5-FU and during recovery in drug-free media. As shown in Fig. 4, CHK1 phosphorylation was not measurably induced by 5-FU at time points between 1 and 12 h. However, p-CHK1 was induced by 5-FU treatment by 24 h in all three sublines, and p-CHK1 was induced to a greater extent in the LN428/UNG-KD and LN428/SMUG1-KD cells (11 and 16-fold respectively, normalized to total CHK1). The results suggest that 5-FU treatment induces a checkpoint damage response and the absence of UNG or SMUG1 glycosylases heightens the activation. Following 24 h incubation in drug-free media, differences in CHK1 phosphorylation in the SMUG1 cells became apparent (Fig. 5). Whereas p-CHK1 remained elevated in the wild-type and LN428/UNG-KD cells at 24 h recovery (3.8 and 4.6-fold increase respectively, normalized to total CHK1), p-CHK1 levels in LN428/SMUG1-KD cells returned to near basal levels (1.2-fold higher compared to untreated cells) (Fig. 5). Loss of p-CHK1 was also seen in WT and LN428/UNG-KD cells, although it was delayed (36 h) and less pronounced (2.9 and 3.7-fold higher, respectively, compared to untreated cells). Interestingly, recovery time points of 36 h (LN428/SMUG1-KD) and 48 h (WT and LN428/UNG-KD) displayed a re-induction in p-CHK1 (Fig. 5). This re-induction of p-CHK1, which was recently observed by others following treatment with TS inhibitors [12], suggests that a time-dependent dephosphorylation and rephosphorylation of CHK1 was occurring during an attempted recovery from 5-FU treatment. CHK2 activation in response to 5-FU treatments was also examined. Although a moderate increase in p-CHK2 levels was observed following TS inhibition, there was no difference in response among the WT, UNG and SMUG1 depleted cells (data not shown).
Figure 4.
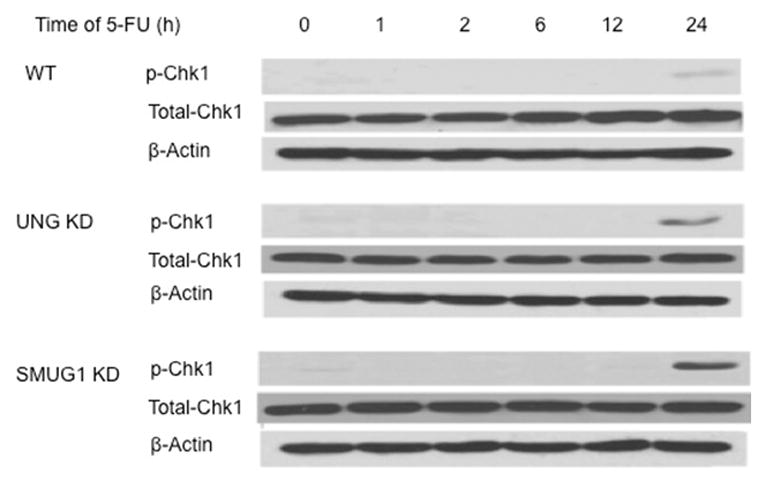
Western blot analysis of p-Chk1 (Ser345) and total Chk1 protein in whole cell lysates of WT (top panels), LN428/UNG-KD (middle panels), and LN428/SMUG1-KD (bottom panels) cells following treatment with 50 μM 5-FU for the indicated times. β-Actin was used for the loading control. The experiment shown is representative of 3 independent experiments.
Figure 5.
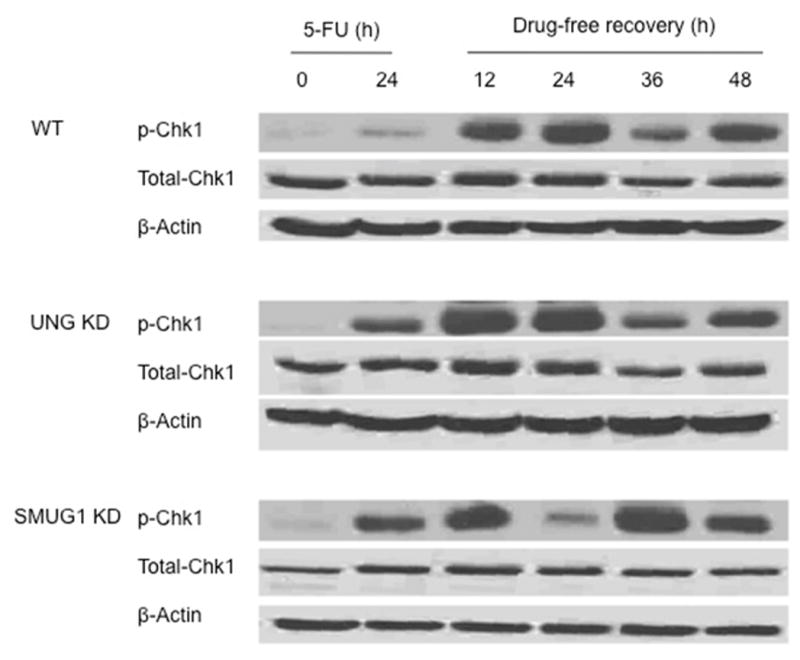
Western blot analysis of p-Chk1(Ser345) and total Chk1 protein in whole cell lysates of WT (top panels), LN428/UNG-KD (middle panels), and LN428/SMUG1-KD (bottom panels) cells following treatment with 50 μM 5-FU for 24 h and recovery in drug free medium for the times indicated. β-Actin was used for the loading control. The experiment shown is representative of 3 independent experiments.
3.4 Generation of strand breaks following 5-FU treatment
Cytotoxicity from TS inhibitors has been shown to be associated with accumulation of DNA strand breaks [17,23]. However, the contribution of specific DNA glycosylases to SSBs or DSBs has not been directly examined with regards to genomic 5-FU. We utilized alkaline single-cell gel electrophoresis (comet assay) to measure the formation and resolution of strand breaks and alkali-labile sites. As shown in Fig. 6a, there was an approximately 2-fold increase in DNA moment of WT cells and 1.5-fold increase in DNA moment of LN428/UNG-KD and LN428/SMUG1-KD cells after 12 h of 5-FU treatment. The initial increase in DNA moment at 12 h suggests the presence of BER intermediates. Following 24 h treatment with 5-FU there was a decline in the DNA moment of the WT cells, but an increase in LN428/UNG-KD and LN428/SMUG1-KD cells. The decline in DNA moment at the 24 h point in WT cells likely represents a resolution of BER intermediates, whereas the elevation of the DNA moment in LN428/UNG-KD and LN428/SMUG1-KD cells likely represents a persistence of intermediates and/or damage, corresponding to the increased p-CHK1 seen at this same time point in these cells.
Figure 6.
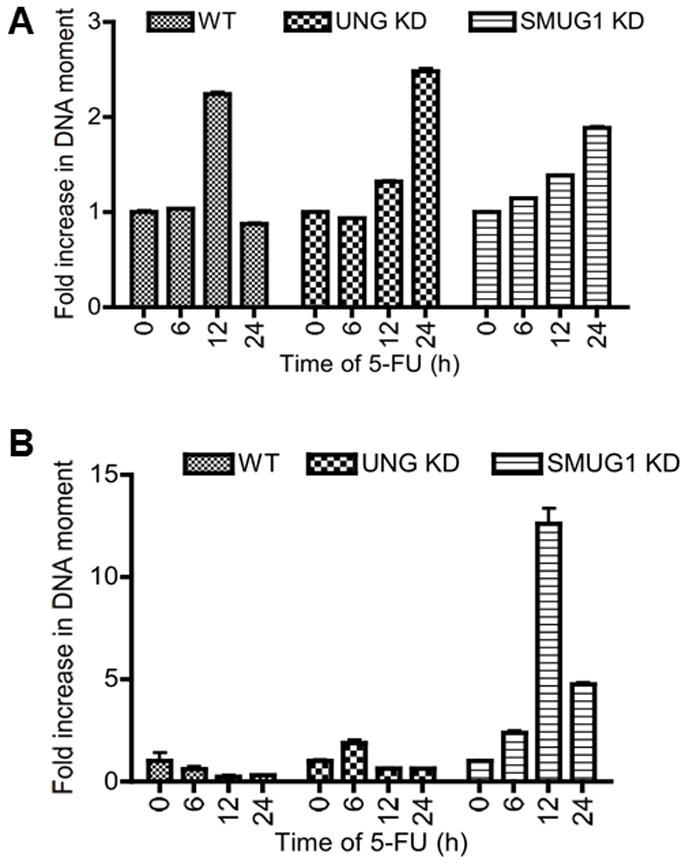
DNA damage after 5-FU treatment as measured by the comet assay in the parental (WT), LN428/UNG-KD and LN428/SMUG1-KD lines. A) Alkaline electrophoresis. B) Neutral electrophoresis. Cells were treated with 50 μM 5-FU for the times indicated and processed as described in Materials and Methods. The y-axis represents fold increase in DNA moment compared to untreated cells (0 h time point). The data represents an average of three independent experiments and 100 cells measured per sample for each experiment. The error bars represent standard error of the mean.
The neutral comet assay was utilized to examine the influence of UNG and SMUG1 activity on the formation of DSBs. Strikingly, a significant elevation of DNA moment was observed at only 12 h of 5-FU treatment in LN428/SMUG1-KD cells (Fig. 6b). There were no corresponding increases in DSBs in the WT and LN428/UNG-KD cells. Interestingly, following 24 h exposure to 5-FU, there was a decrease in the DNA moment in LN428/SMUG1-KD cells relative to 12 h that indicates resolution of the DSBs. However, the extent of DNA DSB formation at 24 h is still 4-fold higher than the untreated controls (Fig. 6b). The results suggest that 5-FU incorporation in DNA can cause DSBs. To confirm that the DSBs observed were not generated due to the execution of apoptosis, we monitored caspase-3 activation following 5-FU treatments and recovery. We failed to observe caspase-3 cleavage in LN428/SMUG1-KD cells when monitored up to 48 h incubation in drug-free media (data not shown). Thus, DSBs generated from 5-FU treatments in LN428/SMUG1-KD cells were not a consequence of apoptotic DNA fragmentation.
4. DISCUSSION
Intra-cellularly, 5-FU is readily converted into FUTP and FdUTP, which can result in RNA and DNA incorporation, respectively. The relative contributions of RNA/DNA incorporation and inhibition of TS to the cytotoxic mechanism of action for fluoropyrimidines has been debated for decades. Because TS inhibition results in an increase in the intracellular dUTP/TTP ratio [24], treatment with 5-FU results in the potential generation of two DNA base lesions, U and 5-FU. However, there appears to be wide variation among cell lines regarding the metabolism of these related yet distinct lesions. Biochemical evidence demonstrates that 5-FU in DNA can be recognized by four different UDGs as well as by mismatch repair proteins [2]. Because TS inhibitors induce an S-phase arrest, it was reasonable to presume that UNG and SMUG1 are initial responders because of their expression during S-phase. Yet, a number of studies in different mammalian cell lines have shown that UNG plays essentially no role in toxicity caused by TS inhibitors [4–9]. One consideration that has received little attention thus far is the possibility that a threshold amount of uracil incorporated into DNA during replication may be just as toxic to mammalian cells as BER intermediates generated by UDG activity. Indeed, this has been shown to be the case in E. coli and S. cerevisiae, in which conditional mutants that lack dUTPase are inviable even in the absence of their respective UNG activities [25,26]. These reports certainly suggest that widespread incorporation of genomic uracil is problematic irrespective of UDG-initiated BER. Evidence from studies in which dUTPase activity was altered also suggest a threshold dependent effect of genomic uracil [27,28]. A threshold effect also offers a plausible explanation for the lack of influence of UNG activity on cellular responses to TS inhibitors in mammalian cells, particularly in studies employing continuous exposures.
Our results with continuous treatments agree with recently published reports [8,10], namely that UNG and SMUG1 status do not influence cytotoxicity from prolonged 5-FU exposure. Other studies have shown that abrogation of CHK1 increases apoptotic cell death from 5-FU, and CHK1-dependent S-phase slowing can protect against cell death from 5-FU [22,29]. As expected, 5-FU induced a sharp S-phase arrest and p-CHK1 activation with no apparent differences between the three sublines. This data suggests that the initial response to replication stress is the same in each subline and therefore is not dependent on SMUG1 or UNG. We speculate that the initial damage signal induced by TS inhibitors is replication stress independent of BER. However, SMUG1 knockdown cells were sensitized to 5-FU when treatment is followed by incubation in the absence of drug, which mimics what occurs clinically. Interestingly, CHK1 is dephosphorylated and then rephosphorylated at later time points in the absence of drug (Fig. 5), even though the majority of cells remain in S-phase (Fig. 3d). Experiments in synchronized cells would provide more definitive proof, but note that the two most common means of synchronizing cells, by serum deprivation or thymidine block, alter the availability of salvageable thymidine in dramatically opposing directions and hence would affect the response to TS inhibitors. Note the turnover of p-Chk1 is within 12 h (Fig. 5), which strongly argues that the changes are not occurring as a result of cells exiting S-phase and completing a cell cycle. Moreover, the altered kinetics of CHK1 phosphorylation is most pronounced in the SMUG1 knockdown cells. Kunz et al. observed a round of CHK1 phosphorylation, dephospyhorylation and re-phosphorylation in wild-type murine embryonic fibroblasts, which they suggested was dependent on TDG [12]. The authors noted that more cells had accumulated in S-phase at 24 h recovery following 5-FU treatment despite a decrease in overall Chk1 phosphorylation at the same time point. In this context, the de-phosphorylation of CHK1 does not represent a bona fide “recovery”, but likely an attempt of the cells to restart replication. We speculate that the differences relate to downstream consequences of repair attempts following the initial arrest in the absence of SMUG1 as it relates to strand breaks.
FdUrd and TS-directed antifolates clearly are capable of inducing DSBs, yet the precise contributions of collapsed replication forks and/or repair intermediates remains unknown [16,18,23,30]. Note that the strand breaks measured here and in other studies, in addition to damage, represent DNA repair intermediates, which themselves are single strand breaks or AP sites that are converted to strand breaks in the presence of alkali. Double strand breaks induced by TS inhibitors and as detected by the comet assay might occur by one of two means: stalled replication forks that eventually collapse, or by a replication fork encountering a BER single strand break intermediate, which then would be repaired by HR [18,30,31]. Increased strand breaks and cell death from 5-FU treatments in SMUG1-inhibited cells during recovery could be a manifestation of differences in AP site metabolism. Following substrate recognition and turnover, SMUG1 binds to the AP-site and inhibits its cleavage by APE-1 [32]. In contrast to SMUG1, UNG was found to stimulate APE-1 cleavage activity [32]. It is tempting to speculate that in the absence of SMUG1, UNG activity specifically during replication restart results in unprotected AP sites. Note that a mutant of APE that binds but does not cleave AP sites acts in a dominant negative fashion in sensitizing cells to 5-FU and FdUrd [33]. In other words, futile cycling during recovery from drug treatment might be heavily influenced by the efficiency with which the lesions are processed by BER, which itself is dependent on which glycosylase initiates the repair. Future studies require a close look at the metabolism and repair of U and 5-FU in clinically relevant dosing schedules that delve into the mechanism of replication restart following thymidylate deprivation.
Supplementary Material
Acknowledgments
This research was supported in part by a grant from the NIH/NCRR to the Center for Colon Cancer Research (P20 RR17698) and from the NIH/NCI to MDW (R01 CA100450). Dr. Sondra Berger is acknowledged for helpful discussions. This work was also supported by grants from the National Institutes of Health (NIH) [GM087798; CA148629; ES019498] to RWS. Support was also provided by the University of Pittsburgh Department of Pharmacology and Chemical Biology with a Molecular Pharmacology Fellowship to DS. Support for the UPCI Lentiviral Facility was provided by the Cancer Center Support Grant from the National Institutes of Health [CA047904].
Abbreviations
- 5-FU
5-fluorouracil
- FdUMP
5-fluoro-2′-deoxyuridylate
- FdUrd
5-fluoro-2′-deoxyuridine
- RTX
Raltitrexed
- TS
thymidylate synthase
- TMP
thymidylate
- dUMP
deoxyuridylate
- SSB
single strand break
- DSB
double strand break
- UDG
uracil DNA glycosylase
- UNG
nuclear UDG encoded by the UNG locus
- SMUG1
single-strand-selective monofunctional DNA glycosylase
- BER
base excision repair
Footnotes
Disclosure of Potential Conflicts of interest
RWS is a scientific consultant for Trevigen, Inc. The remaining authors state that there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berger SH, Pittman DL, Wyatt MD. Uracil in DNA: consequences for carcinogenesis and chemotherapy. Biochem Pharmacol. 2008;67:697–706. doi: 10.1016/j.bcp.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wyatt MD, Wilson DM., 3rd Participation of DNA repair in the response to 5-fluorouracil. Cell Mol Life Sci. 2009;66:788–799. doi: 10.1007/s00018-008-8557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krokan HE, Drablos F, Slupphaug G. Uracil in DNA--occurrence, consequences and repair. Oncogene. 2002;21:8935–8948. doi: 10.1038/sj.onc.1205996. [DOI] [PubMed] [Google Scholar]
- 4.An Q, Robins P, Lindahl T, Barnes DE. 5-Fluorouracil incorporated into DNA is excised by the smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007;67:940–945. doi: 10.1158/0008-5472.CAN-06-2960. [DOI] [PubMed] [Google Scholar]
- 5.Andersen S, Heine T, Sneve R, König I, Krokan HE, Epe B, Nilsen H. Incorporation of dUMP into DNA is a major source of spontaneous DNA damage, while excision of uracil is not required for cytotoxicity of fluoropyrimidines in mouse embryonic fibroblasts. Carcinogenesis. 2005;26:547–555. doi: 10.1093/carcin/bgh347. [DOI] [PubMed] [Google Scholar]
- 6.Grogan BC, Parker JB, Guminski AF, Stivers JT. Effect of the Thymidylate Synthase Inhibitors on dUTP and TTP Pool Levels and the Activities of DNA Repair Glycosylases on Uracil and 5-Fluorouracil in DNA. Biochemistry. 2011;50:618–627. doi: 10.1021/bi102046h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo Y, Walla M, Wyatt MD. Uracil incorporation into genomic DNA does not predict toxicity caused by chemotherapeutic inhibition of thymidylate synthase. DNA Repair (Amst) 2008;7:162–169. doi: 10.1016/j.dnarep.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pettersen HS, Visnes T, Vagbo CB, Svaasand EK, Doseth B, Slupphaug G, Kavli B, Krokan HE. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011;39:8430–8444. doi: 10.1093/nar/gkr563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welsh SJ, Hobbs S, Aherne GW. Expression of uracil DNA glycosylase (UDG) does not affect cellular sensitivity to thymidylate synthase (TS) inhibition. Eur J Cancer. 2003;39:378–387. doi: 10.1016/s0959-8049(02)00610-x. [DOI] [PubMed] [Google Scholar]
- 10.Kemmerich K, Dingler FA, Rada C, Neuberger MS. Germline ablation of SMUG1 DNA glycosylase causes loss of 5-hydroxymethyluracil- and UNG-backup uracil-excision activities and increases cancer predisposition of Ung−/−Msh2−/− mice. Nucleic Acids Res. 2012;40:6016–6025. doi: 10.1093/nar/gks259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, Skalka AM, Meropol NJ, Alberti C, Larue L, Bellacosa A. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci U S A. 2003;100:15071–15076. doi: 10.1073/pnas.2334585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunz C, Focke F, Saito Y, Schuermann D, Lettieri T, Selfridge J, Schar P. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol. 2009;7:e91. doi: 10.1371/journal.pbio.1000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbour KW, Berger FG. Cell death in response to antimetabolites directed at thymidylate synthase. Cancer Chemother Pharmacol. 2008;61:189–201. doi: 10.1007/s00280-007-0461-4. [DOI] [PubMed] [Google Scholar]
- 14.Goellner EM, Grimme B, Brown AR, Lin YC, Wang XH, Sugrue KF, Mitchell L, Trivedi RN, Tang JB, Sobol RW. Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair. Cancer Res. 2011;71:2308–2317. doi: 10.1158/0008-5472.CAN-10-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang JB, Svilar D, Trivedi RN, Wang XH, Goellner EM, Moore B, Hamilton RL, Banze LA, Brown AR, Sobol RW. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro Oncol. 2011;13:471–486. doi: 10.1093/neuonc/nor011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li L, Berger SH, Wyatt MD. Involvement of base excision repair in response to therapy targeted at thymidylate synthase. Mol Cancer Ther. 2004;3:747–753. [PubMed] [Google Scholar]
- 17.Li L, Connor EE, Berger SH, Wyatt MD. Determination of apoptosis, uracil incorporation, DNA strand breaks, and sister chromatid exchanges under conditions of thymidylate deprivation in a model of BER deficiency. Biochem Pharmacol. 2005;70:1458–1468. doi: 10.1016/j.bcp.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Waldman AS, Wyatt MD. DNA damage and homologous recombination signaling induced by thymidylate deprivation. Biochem Pharmacol. 2008;76:987–996. doi: 10.1016/j.bcp.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyers M, Wagner MW, Mazurek A, Schmutte C, Fishel R, Boothman DA. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J Biol Chem. 2005;280:5516–5526. doi: 10.1074/jbc.M412105200. [DOI] [PubMed] [Google Scholar]
- 20.Curtin NJ, Harris AL, Aherne GW. Mechanism of cell death following thymidylate synthase inhibition: 2′-deoxyuridine-5′-triphosphate accumulation, DNA damage, and growth inhibition following exposure to CB3717 and dipyridamole. Cancer Res. 1991;51:2346–2352. [PubMed] [Google Scholar]
- 21.Lehman NL, Danenberg PV. Modulation of RTX cytotoxicity by thymidine and dipyridamole in vitro: implications for chemotherapy. Cancer Chemother Pharmacol. 2000;45:142–148. doi: 10.1007/s002800050022. [DOI] [PubMed] [Google Scholar]
- 22.Robinson HM, Jones R, Walker M, Zachos G, Brown R, Cassidy J, Gillespie DA. Chk1-dependent slowing of S-phase progression protects DT40 B-lymphoma cells against killing by the nucleoside analogue 5-fluorouracil. Oncogene. 2006;25:5359–5369. doi: 10.1038/sj.onc.1209532. [DOI] [PubMed] [Google Scholar]
- 23.Canman CE, Tang HY, Normolle DP, Lawrence TS, Maybaum J. Variations in patterns of DNA damage induced in human colorectal tumor cells by 5-fluorodeoxyuridine: implications for mechanisms of resistance and cytotoxicity. Proc Natl Acad Sci USA. 1992;89:10474–10478. doi: 10.1073/pnas.89.21.10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aherne GW, Brown S. The role of uracil misincroporation in thymineless death. In: Jackman AL, editor. Anticancer Drug Development Guide: Antifolate Drugs in Cancer Therapy. Humana Press Inc; Totowa, NJ: 1999. pp. 409–421. [Google Scholar]
- 25.el-Hajj HH, Wang L, Weiss B. Multiple mutant of Escherichia coli synthesizing virtually thymineless DNA during limited growth. J Bacteriol. 1992;174:4450–4456. doi: 10.1128/jb.174.13.4450-4456.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gadsden MH, McIntosh EM, Game JC, Wilson PJ, Haynes RH. dUTP pyrophosphatase is an essential enzyme in Saccharomyces cerevisiae. Embo J. 1993;12:4425–4431. doi: 10.1002/j.1460-2075.1993.tb06127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koehler SE, Ladner RD. Small interfering RNA-mediated suppression of dUTPase sensitizes cancer cell lines to thymidylate synthase inhibition. Mol Pharmacol. 2004;66:620–626. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 28.Webley SD, Hardcastle A, Ladner RD, Jackman AL, Aherne GW. Deoxyuridine triphosphatase (dUTPase) expression and sensitivity to the thymidylate synthase (TS) inhibitor ZD9331. Br J Cancer. 2000;83:792–799. doi: 10.1054/bjoc.2000.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao Z, Xue J, Sowin TJ, Rosenberg SH, Zhang H. A novel mechanism of checkpoint abrogation conferred by Chk1 downregulation. Oncogene. 2005;24:1403–1411. doi: 10.1038/sj.onc.1208309. [DOI] [PubMed] [Google Scholar]
- 30.Waldman BC, Wang Y, Kilaru K, Yang Z, Bhasin A, Wyatt MD, Waldman AS. Induction of Intrachromosomal Homologous Recombination in Human Cells by Raltitrexed, an Inhibitor of Thymidylate Synthase. DNA Repair. 2008;7:1624–1635. doi: 10.1016/j.dnarep.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Z, Waldman AS, Wyatt MD. Expression and regulation of RAD51 mediate cellular responses to chemotherapeutics. Biochem Pharmacol. 2012;83:741–746. doi: 10.1016/j.bcp.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettersen HS, Sundheim O, Gilljam KM, Slupphaug G, Krokan HE, Kavli B. Uracil-DNA glycosylases SMUG1 and UNG2 coordinate the initial steps of base excision repair by distinct mechanisms. Nucleic Acids Res. 2007;35:3879–3892. doi: 10.1093/nar/gkm372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNeill DR, Lam W, DeWeese TL, Cheng YC, Wilson DM., 3rd Impairment of APE1 function enhances cellular sensitivity to clinically relevant alkylators and antimetabolites. Mol Cancer Res. 2009;7:897–906. doi: 10.1158/1541-7786.MCR-08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.