

Summary
Background:
Heparin induced thrombocytopenia (HIT) is a serious complication associated with heparin use. HIT usually develops between 5–14 days after starting heparin. Delayed-onset HIT can still occur 9–45 days after heparin had been discontinued. In patients with delayed HIT, the patient might be admitted to the hospital for new thrombosis and reexposure to heparin further worsens the patient’s condition.
Case Report:
Our patient is a 71-year old female readmitted for worsening dyspnea 2 weeks after she was discharged from the hospital. On her previous hospitalization, she was diagnosed with bronchiolitis obliterans organizing pneumonia (BOOP). She had received prophylactic doses of LMWH. Dyspnea was initially thought to be secondary to CHF exacerbation secondary to atrial fibrillation with rapid ventricular response. She was started on a heparin. However, the patient’s clinical condition deteriorated and she needed to be intubated. Her platelet counts also decreased rapidly. After CT angiography of the chest showed pulmonary embolism, HIT was strongly considered. All forms of heparin were discontinued and argatroban was started. However, the patient did not improve and she subsequently expired on the 7th hospital day. Heparin-induced antibodies came back positive that same day.
Conclusions:
HIT is an immune-mediated disorder characterized by formation of antibodies against heparin-platelet factor 4 complex. The major clinical presentation of HIT is arterial and venous thrombosis. Once HIT is suspected, immediate cessation of any form of heparin is needed. Alternative anticoagulation must be started. Early treatment decreases the incidence of new thrombosis and stroke, and improves survival and cost savings.
Keywords: heparin induced thrombocytopenia, thrombosis, argatroban, leupridin
Background
Heparin is a one of the most commonly used parenteral therapeutic agents. Heparin induced thrombocytopenia (HIT) is a serious complication associated with heparin use. Heparin is used for anticoagulation and it normally does not affect the platelets. When patients develop HIT, which is the effect of the formation of antibodies to the heparin and platelet factor 4 complex, acute venous or arterial thrombosis can occur as well as thrombocytopenia. The classical signs and symptoms of HIT develop between 5–14 days after starting heparin. However, delayed-onset HIT can still occur 9–45 days after heparin had been discontinued. In patients with delayed HIT, the patient might be admitted to the hospital for new thrombosis, which would normally be treated with heparin. However. reexposure to heparin further worsens the patient’s condition and this prompts the clinician to suspect HIT. Laboratory tests used for confirmation of suspected HIT includes measurement of HIT antibodies and serotonin release assay.
Case Report
Our patient is a 71 year-old Caucasian female with past medical history of hypertension, interstitial lung disease, rheumatoid arthritis, congestive heart failure, recently diagnosed with bronchiolitis obliterans organizing pneumonia (BOOP) and Pseudomonas pneumonia treated with IV antibiotics on previous hospital admission two weeks prior to this current emergency department visit. She was brought in from the subacute rehabilitation facility because of a one week history of progressive shortness of breath. The dyspnea was initially felt on exertion, and gradually progressed to occur even at rest. There is accompanying nonproductive cough, palpitations and orthopnea. The patient denied any paroxysmal nocturnal dyspnea, fever, chills, hemoptysis, chest pain, syncope or leg swelling. She was receiving prednisone 60 mg PO daily and cefepime 2 g IV daily for pseudomonas pneumonia at the subacute facility. Initial vital signs are as follows: blood pressure was 100/60, respiratory rate was 30, heart rate 160, temp 98.7°F. Oxygen saturation was 95% on 50% FiO2 by venturi mask. The patient was in mild distress but she was awake, alert, and oriented to time, place, and person. The patient had jugular venous distention, irregularly irregular heart rate, holosystolic murmur heard best at the apex, and had bibasilar crackles on lung examination. The patient’s abdominal, extremity and neurologic exams are unremarkable. ECG done showed atrial fibrillation at a rate of 186 per minute. Chest x-ray showed bilateral interstitial markings, but did not show any infiltrate, consolidation or effusion. CBC showed leukocytosis with WBC count of 23,000 with 8% bands. Platelets were 140,000. The patient’s brain natriuretic peptide level was 1628. Cardiac enzymes and electrolytes were within normal limits. The patient has slightly elevated creatinine at 1.28, with BUN of 61. Initial assessment was acute exacerbation of CHF secondary to atrial fibrillation. The patient was admitted to the ICU and was started on heparin drip for anticoagulation and metoprolol for rate control of her atrial fibrillation. Prednisone was continued for BOOP. ABG the next day showed respiratory alkalosis with an A-a gradient of 70. The patient’s platelets dropped to 90,000. The hemoglobin also dropped from 13.1 to 10.5. CT of the chest was done without contrast because the patient was considered high-risk for contrast-induced nephropathy. It showed moderate fibrotic changes throughout the lung fields with preferential involvement of lower lobes with interval decrease in the previously noted diffuse areas of ground glass densities. 2D-echocardiogram showed left ventricular ejection fraction of 60–65% with moderately-elevated pulmonary systolic pressure (50–55 mmHg). The patient went into respiratory distress and was intubated on the 2nd day of admission. Her platelets further decreased to 78,000. Heparin drip is still being given as a form of anticoagulation for the patient’s atrial fibrillation. At this point, it was decided to perform a CT angiography of the chest which showed elongated filling defect in the distal left pulmonary artery and its branches to the left upper lobes (Figure 1). Since the patient is already on the heparin drip, no management changes were done. On the 3rd hospital day the patient’s platelets still continued to drop to 58,000. On the 6th hospital day, heparin was changed to argatroban because heparin-induced thrombocytopenia was finally suspected. However, the patient continued to deteriorate and she expired on the 7th hospital day. That day, heparin-induced antibodies became positive with an optical density of 2.40.
Figure 1.
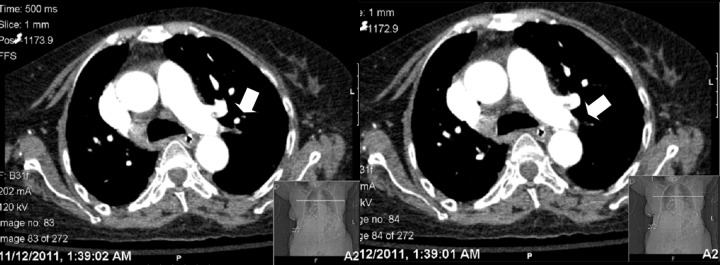
CT Angiogram of the chest demonstrating an embolus on distal left pulmonary artery and its branches to the left upper lobes (depicted by arrows).
Discussion
HIT is an immune-mediated disorder characterized by formation of antibodies against heparin-platelet factor 4 (PF4) complex. Heparin binds with PF4 and forms a highly immunologic complex on the surface of platelets. IgG antibodies form against these complexes in susceptible patients. It takes five days for these antibodies to form and react unless the patient has recent exposure to heparin a few months prior. Binding of antibodies to heparin-PF4 on the surface of platelets leads to activation of platelets and destruction by the reticuloendothelial system leading to development of thrombocytopenia. Activation of platelets also causes generation of procoagulant microparticles which is responsible for the formation of platelet-rich arterial as well as venous blood clots.
HIT presents with fall in platelets counts, classically between 5–14 days of heparin exposure. The platelet count is usually greater than 20,000 and spontaneous bleeding is very rare. However, rapid-onset HIT presents much earlier, even after only a day of heparin exposure. It occurs in patients who had recent exposure to heparin in the past 3 months. They have residual heparin-PF4 antibodies in the circulation. Another form of HIT, the delayed-onset type, leads to thrombocytopenia and thrombosis few days or weeks after heparin had been stopped. Usually, patients with delayed HIT had recently received heparin and had been discharged from the hospital after a benign course during which HIT might have been unrecognized. These patients later on present with objectively proven arterial or venous thrombosis and develop thrombocytopenia after reexposure to heparin.
The major clinical presentation of HIT is arterial as well as venous thrombosis. However, venous thrombosis is more common than arterial thrombosis. Venous thrombosis can occur as DVT, pulmonary embolism, and rarely cerebral venous thrombosis. On the other hand, arterial thrombosis presents with stroke, limb ischemia, and myocardial infarction. Heparin induced thrombocytopenia can lead to extension of already diagnosed blood clots. HIT can also present as skin necrosis at the site of heparin injection and rarely, systemic anaphylactoid reactions.
HIT develops in 1–5% of patients exposed to heparin. Its development depends on the type of heparin used, duration of heparin exposure, source of heparin, route of heparin used, patient population (patients undergoing orthopedic and cardiovascular procedures are among the highest risk), prior heparin exposure and gender. Patients given unfractionated heparin are at more risk of developing HIT than those who are given LMWH [1,2]. HIT also develops with higher frequency in female patients than in males, and in surgical patients as compared to medical patients [3–5]. Switching from unfractionated heparin to LMWH for thromboprophylaxis decreased the incidence of HIT in surgical patients but the incidence remained same in medical patients [1]. One study compared the development of HIT antibodies and HIT in patients undergoing orthopedic and cardiovascular surgery. It showed that IgG antibodies associated with HIT were more likely to form in patients undergoing cardiac surgery than in orthopedic patients. However, among the patients in whom the antibodies did form, HIT was more likely to develop in the orthopedic patients than in the cardiac surgery patients [6]. One prospective cohort study showed that the incidence of HIT is significantly higher in patients with prior exposure of UFH or LMWH [7].
Patients with HIT usually experience one of the following five sequlae: wellness, new thrombosis, amputation, stroke or death.
Earlier recognition of HIT is important for proper management of this life threatening condition and prevention of thrombosis, limb amputation and death. Diagnosis of HIT can be challenging and requires a high index of suspicion to ensure prompt recognition. The initial diagnosis of HIT is based on clinical presentation, heparin exposure, development of thrombocytopenia with or without thrombosis. In real clinical practice immediate action is not taken on first suspicion of possible HIT.
Pretest scoring on the probability of HIT is available. It is based on the timing and severity of thrombocytopenia, presence of new thrombosis and other explanation for thrombocytopenia. Furthermore, a number of laboratory tests are available and are used to confirm the diagnosis of HIT. Solid phase ELISA immunoassay is the screening test used for suspected HIT. This test measures antibodies against Heparin-PF4 complex. This is a highly sensitive test, and a negative test almost rules out HIT. However, it less specific and has a moderate positive predictive value. Functional assays have higher specificity. The 14C-serotonin release assay is the gold standard for the diagnosis of HIT. This is very expensive, technically demanding, and is not widely available. Another available assay is the heparin-induced platelet aggregation assay.
Treatment with alternative anticoagulants should be started once the diagnosis of HIT is suspected on clinical grounds and it should not be delayed until specific confirmatory laboratory results are available [5]. Discontinuation of heparin alone is not sufficient because HIT is a hypercoaguable condition and the patient needs to be started on appropriate alternative non-heparinoid anticoagulants. Immediate withholding of heparin alone in patients with HIT is associated with thrombosis rates of 10–15% by 2 days, 40% by 1 week and 52% by 1 month [3]. Not starting alternative anticoagulant treatment after stopping heparin while waiting for confirmatory test results for HIT is associated with 6.1% rate per day of thromboembolic complication [5]. The mortality rate for HIT with thrombosis is found to be 20–30% [8,9]. Furthermore, platelet transfusions are relatively contraindicated in HIT because it increases the risk of bleeding and new thrombotic events. Transfusing platelets in HIT patients is deemed as “adding fuel to the fire”.
Immediate cessation of all kinds of heparin including heparin flushes and heparin-bonded catheters is the first intervention in suspected or proven HIT because continuation of such agents worsens the patients’ clinical condition. A number of alternative anticoagulants are recommended and are FDA approved for the treatment of HIT. Lepirudin and argatroban are FDA approved non-heparin direct thrombin inhibitors used for the prophylaxis and treatment of HIT.
Clinical trials have shown that the rate of new thrombosis, stroke and death is lesser for patients with HIT with or without thrombosis who have received early argatroban treatment compared to heparin cessation alone [10]. One retrospective analysis has compared the cost-effectiveness of early versus late argatroban treatment in HIT patients with or without thrombosis. Cost data were estimated for hospital days, diagnostic tests, heparin treatment, argatroban treatment, major hemorrhagic events, amputations, new thrombosis, stroke or death. It showed that patients who received early argatroban along with heparin cessation within 48 hours of onset of thrombocytopenia have significantly improved outcomes and lower costs compared to the patients who received argatroban along with heparin cessation after 48 hours of heparin therapy. Early treatment of HIT with argatroban is beneficial clinically as well as in cost savings compared to delayed treatment with argatroban and heparin cessation therapy alone [11].
Argatroban for the treatment of HIT has been studied in two trials. These trials showed that new thrombosis was significantly reduced in patients with isolated HIT when argatroban is used. The standard starting dose is 2 micrograms/kg per minute and the dose is adjusted to maintain APTT between 1.5 to 3.0 times the baseline. Dosage adjustment is required for hepatic dysfunction but not for renal impairment. Argatroban clinical trials showed that delayed treatment of HIT with argatroban had outcomes no better than without argatroban treatment.
Lepirudin is also another nonheparin hirudin analogue. The HIT-1 trial has shown that lepirudin significantly reduced thromboembolic complications and limb amputations. Dosage adjustment is required in renal impairment.
Bivalirudin is also a hirudin analog and a direct thrombin inhibitor. It is FDA-approved only for patients with HIT or at risk of HIT who under goes primary coronary intervention.
Patients with HIT require long term anticoagulation with low dose warfarin (<6mg) for 3–6 months. However, warfarin should not be started unless the patient has been adequately anticoagulated with direct thrombin inhibitors and the INR has been >2 for at least 2 days and the platelet count has gone up to 100,000–150,000. Vitamin K should be given to patients who have already received warfarin without overlapping with direct thrombin inhibitors. HIT may cause depletion of protein C and early initiation of warfarin exacerbates the protein C deficiency. This leads to increased risk of thrombosis and warfarin-induced limb gangrene. High dose warfarin treatment should also be avoided.
Conclusions
HIT is a prothrombotic adverse reaction associated with heparin exposure caused by formation of antibodies against heparin platelet factor-4 complex, which causes destruction and activation of platelets and the release of procoagulant particles. Arterial and venous thrombosis are the main manifestations of HIT. High index of clinical suspicion is required for prompt recognition of this syndrome. Immediate cessation of any form of heparin is needed and the alternative anticoagulation must be started. Early treatment decreases the incidence of new thrombosis and stroke, and improves survival and cost savings.
References:
- 1.Gruel Y, Pouplard C, Nguyen P, et al. Biological and clinical features of low-molecular-weight heparin-induced thrombocytopenia. Br J Haematol. 2003;121:786–92. doi: 10.1046/j.1365-2141.2003.04363.x. [DOI] [PubMed] [Google Scholar]
- 2.Warkentin TE, Levine MN, Hirsh J, et al. Heparin-induced thrombocytopenia in patients treated with low-molecular-weight heparin or unfractionated heparin. N Engl J Med. 1995;332:1330–35. doi: 10.1056/NEJM199505183322003. [DOI] [PubMed] [Google Scholar]
- 3.Warkentin TE, Kelton JG. A 14-year study of heparin induced thrombocytopenia. Am J Med. 1996;101:502. doi: 10.1016/s0002-9343(96)00258-6. [DOI] [PubMed] [Google Scholar]
- 4.Warkentin TE. Heparin induced thrombocytopenia: pathogenesis and management. Br J Haematol. 2003;121:535–55. doi: 10.1046/j.1365-2141.2003.04334.x. [DOI] [PubMed] [Google Scholar]
- 2.Warkentin T, Greinacher A. Heparin-Induced thrombocytopenia: recognition, treatment and prevention: the Seventh ACCP Conference on Antithrombotic and Thrombolytic therapy. Chest. 2004;126(Suppl.3):311S–37S. doi: 10.1378/chest.126.3_suppl.311S. [DOI] [PubMed] [Google Scholar]
- 5.Shuster TA, Silliman R, Coats RD, et al. Heparin induced thrombocytopenia: twenty-nine years later. J Vasc Surg. 2003;38:1316–22. doi: 10.1016/s0741-5214(03)00769-9. [DOI] [PubMed] [Google Scholar]
- 6.Warkentin TE, Sheppard JA, Horsewood P, et al. Impact of the patient population on the risk for heparin-induced thrombocytopenia. Blood. 2000;96:1703–8. [PubMed] [Google Scholar]
- 7.Prandoni P, Siragusa S, Girolami B, Fabris F. The incidence of heparin-induced thrombocytopenia in medical patients treated with low molecular weight heparin: a prospective cohort study. Blood. 2005;106:3049–54. doi: 10.1182/blood-2005-03-0912. [DOI] [PubMed] [Google Scholar]
- 8.Hirsh J, Heddle N, Kelton J. Treatment of heparin-induced thrombocytopenia: a critical review. Arch Intern Med. 2004;164:361–69. doi: 10.1001/archinte.164.4.361. [DOI] [PubMed] [Google Scholar]
- 9.Lewis B, Wallis D, Berkowitz S, et al. Argatroban anticoagulant therapy in patients with heparin-induced thrombocytopenia. Circulation. 2001;103:1838–43. doi: 10.1161/01.cir.103.14.1838. [DOI] [PubMed] [Google Scholar]
- 10.LaMonte MP, Brown MP, Hursting MJ. Stroke in patient with heparin-induced thrombocytopenia and the effect of argatroban therapy. Crit Care Med. 2004;32:976–80. doi: 10.1097/01.ccm.0000119426.34340.e2. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg ARJ, Kim R, Tang B. The Cost-Effectiveness of Argatroban Treatment in Heparin-Induced Thrombocytopenia: The Effect of Early Versus Delayed Treatment. Cardiol Rev. 2006;14(1):7–13. doi: 10.1097/01.crd.0000164011.17381.6f. [DOI] [PubMed] [Google Scholar]