

Abstract
The link between Akt activation and gluconeogenic repression remains unclear despite many years of investigation and remarkable progress. Rodgers and colleagues now introduce us to the Clk2 kinase, an Akt substrate that can directly phosphorylate and inhibit PGC-1α, blunting hepatic glucose production.
A decade after its cloning, PGC-1α’s role as a master regulator of mitochondrial biogenesis and gluconeogenesis is undisputed (Rodgers et al., 2008). Initially identified as a cofactor for the nuclear receptor PPARγ (Puigserver et al., 1998), PGC-1α is the founding member of a small family of coactivators, also formed by PGC-1β and PRC, involved in the regulation of metabolism in general, and mitochondrial function in particular (Rodgers et al., 2008). Recent work has shown that PGC-1α activity is controlled by an ever-increasing number of post-translational modifications (PTMs). These PTMs are induced by multiple hormonal, metabolic and stress signals, and can be considered integrative nodes that fine-tune the activity of PGC-1α (see Figure). In this regard, PGC-1α can be phosphorylated on different residues by several kinases, including p38MAPK, AMPK and Akt (Rodgers et al., 2008). Similar PTMs, however, do not necessarily have similar effects on PGC-1α. For example, phosphoyrlation of PGC-1α on Thr177 and Ser538 by AMPK activates PGC-1α, while phosphorylation by Akt on Ser570 is inhibitory (see Figure). PGC-1α activity is also regulated by acetylation, of up to 13 lysine residues (Rodgers et al., 2005). When acetylated by GCN5 (Lerin et al., 2006), PGC-1α activity is low, while deacetylation by SIRT1 robustly increases its activity (Rodgers et al., 2008). In this issue of Cell Metabolism, Rodgers et al. deepen our knowledge of PGC1α regulation by finding a new modification that critically controls PGC-1α’s ability to regulate the gluconeogenic gene program
Figure 1. Post-translational modifications (PTMs) of PGC-1α.
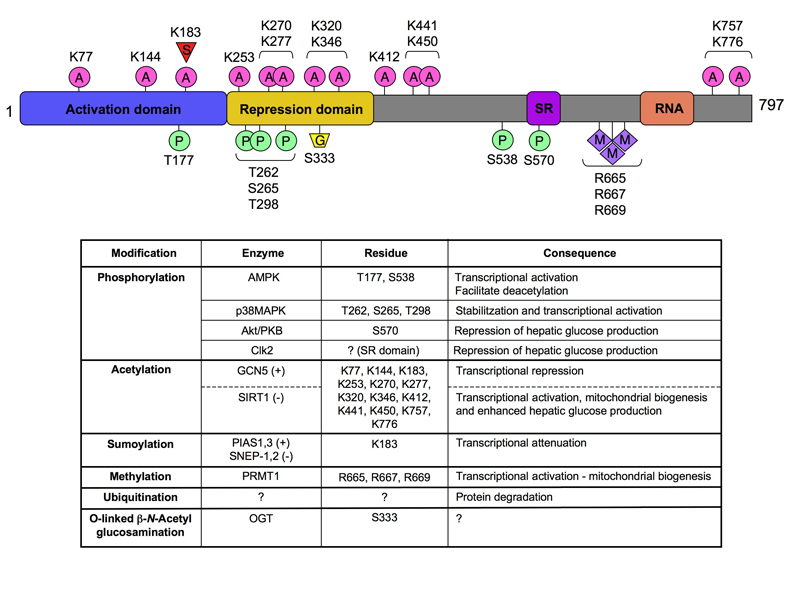
The architecture of PGC-1α shows how PTMs occur across the span of the different protein domains. The table below summarizes the relationship between the various modifications and their biological output. A: acetylation; G: O-linked β-N-acetylglucosamine; M: methylation; P: phosphorylation; S: sumoylation; SR: serine-arginine rich domain; RNA: RNA binding domain.
During fasting, PGC-1α promotes the expression of gluconeogenic genes by coactivating a number of transcription factors, including the FOXOs, HNF4α or CREB. This implies that PGC-1α activity has to be quickly turned off upon food intake, an effect that could be achieved if PGC-1α was responsive to insulin. Now, work by Rodgers et al. confirms that this is in fact the case (Rodgers et al., 2009). First, the authors identify Clk2 (Cdc2-like kinase) as a Akt target. Clk2 belongs to an evolutionary conserved family of protein kinases that phosphorylate SR domains of splicing factors (Colwill et al., 1996). The function and regulation of the Clks, however, are still largely a mystery. The Puigserver lab now demonstrates that Clk2 protein levels are exquisitely regulated during fasting/refeeding cycles through changes in Clk2 protein stability. Insulin action increases Clk2 protein stability, and therefore activity, through phosphorylation of Clk2 on Thr343 by Akt. Furthermore, Clk2 can also autophosphorylate, amplifying and perpetuating Clk2 activity once Akt activity is shut down. Looking downstream, Rodgers et al. show that Clk2 phosphorylates the SR domain of PGC-1α. This domain is pivotal to PGC-1α function, as it mediates insulin responsiveness, contains an Akt phosphorylation site (Li et al., 2007) and constitutes the region of interaction with FOXO transcription factors (Puigserver et al., 2003). Phosphorylation of the SR domain by Clk2 renders PGC-1α unable to specifically coactivate FOXO and other nuclear receptors controlling the gluconeogenic program. Interestingly, SR phosphorylation by Clk2 does not affect PGC-1α coactivation of transcription factors involved in mitochondrial biogenesis, such as NRF-1. The suppressive role of Clk2 on hepatic glucose output is elegantly supported by data showing that overexpression of Clk2 induces fasting hypoglycemia and ameliorates hyperglycaemia in db/db mice. Based on this observation, the authors postulate that Clk2 acts as a “long-acting” insulin-induced gluconeogenic repressor that sustains the inhibition of glucose production during refeeding once Akt is downregulated.
It is interesting to note that Akt can also inhibit PGC-1α through direct phosphorylation on Ser570 (Li et al., 2007), a residue located in the SR domain, the region which is also phosphorylated by Clk2. The fact that two effector kinases promote similar actions, raises the question of whether the observed effects are simply due to regulation of Akt by Clk2. It was therefore reassuring to see that Clk2 does not phosphorylate Ser570, the residue targeted by Akt (Li et al., 2007). In fact, Clk2 can repress the activity of the PGC-1α S570A mutant, which is not responsive to Akt, indicating that Akt and Clk2 may use different mechanisms to inhibit PGC-1α’s gluconeogenic activity. Akt and Clk2 could therefore inhibit PGC-1α in a complementary fashion in different physiological settings and phases, a point that warrants future investigation. Furthermore, to ensure robust repression of gluconeogenesis, Akt activation simultaneously impacts on diverse gluconeogenic regulators other than PGC-1α, such as FOXO1 and CRTC2. Hence, it would not be surprising if the actions of Clk2 expand beyond PGC-1α.
While this work constitutes a major leap in our understanding of the regulation of glucose production, it also raises new questions. For example, cross-regulation between Clk2 and Akt might indeed exist, as the authors show that artificial modulation of Clk2 levels leads to an inverse effect on Akt activity. For example, knock-down of Clk2 increases Akt Ser473 and PGC-1α Ser570 phosphorylation, yet results in an apparently contradictory increase in PGC-1α activity. This feedback loop between Clk2 and Akt makes the physiological implications of manipulating these pathways difficult to predict. Another interesting point will be to understand how Clk2 is shut down once Akt triggers Clk2 auto-regulation. It will also be challenging to elucidate the implications of the insulin/Akt/Clk2 axis in non-gluconeogenic tissues. Answering these questions might help to further validate Clk2 as a pharmacological target, as the results by Rodgers et al. suggest that artificial activation of liver Clk2 could be used as a glucose lowering strategy.
Altogether, the phosphorylation of PGC-1α by Clk2 adds yet another layer of regulation to the biology of PGC-1α. It will be interesting to understand the cross-talk between the wide spectrum of PGC-1α PTMs. The fact that the phosphorylation of PGC-1α by AMPK regulates PGC-1α deacetylation (Canto et al., 2009), indicates that such interactions might not happen independently but rather act in coordination. Also, the sheer variety of PTMs calls into question their role as global functional switches; rather, they may constitute a fine-tuning mechanism that drives PGC-1α activity towards specific gene sets by, for example, allowing or obstructing interaction with specific transcription factors. The fact that Clk2 phosphorylation only attenuates the action of PGC-1α on gluconeogenesis, leaving its effect on mitochondrial genes intact, supports this line of thinking. Consequently, the alteration of specific PTMs in PGC-1α might be an attractive strategy in the management of disease, as it would allow targeting of selective gene sets and functions, refining current concepts aimed at inducing PGC-1α expression.
REFERENCES
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. Embo J. 1996;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1alpha. Cell Metab. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]