

Abstract
Although replete with cytotoxic machinery, uterine NK (uNK) cells remain tolerant at the maternal-fetal interface. The mechanisms that facilitate the uNK cell tolerance are largely unknown. Here we demonstrate that VEGF C, a pro-angiogenic factor produced by uNK cells, is responsible for their non-cytotoxic activity. VEGF C-producing uNK cells support endovascular processes as demonstrated in a three dimensional co-culture model of capillary tube formation on matrigel. Peripheral blood NK cells fail to produce VEGF C and remain cytotoxic. This response can be reversed by exogenous VEGF C. We show that cytoprotection by VEGF C can be related to induction of the “transporter associated with antigen processing (TAP)-1” expression and MHC class I assembly in target cells. siRNA-mediated silencing of TAP-1 expression abolished the VEGF C-imparted protection. Overall, these results demonstrate that empowerment of uNK cells with angiogenic factors keeps them non-cytotoxic. This phenotype is critical to their pregnancy compatible immuno-vascular role during placentation and fetal development.
Keywords: Reproductive Immunology, Uterine and decidual NK cells, Angiogenesis, Immune Tolerance, VEGF C, Immune-Vascular Interface
Introduction
Maternal immune tolerance of the semi-allograft fetus is a subject of intense debate. Although the decidualized uterus is replete with immune cells, fetal development ensues unhindered and results in normal term delivery. An important aspect of successful pregnancy outcome is that they invoke immune tolerance and de novo angiogenesis, two highly intriguing, indispensable and well choreographed processes (1–3). These processes are central to optimal placentation and involve interactions between immune cells, endothelial cells, and invading trophoblasts. With respect to immune cells, T cell-mediated rejection of the fetus has been shown in mice, but not in humans (4). Moreover, uterine T cells or macrophages may not promote immuno-vascular cross talk with the invading trophoblast lacking expression of HLA (HLA-A, HLA-B, or HLA-D) antigens (1, 5). Thus, it is reasonable to propose that the predominant immune tolerance cross-talk is between uterine NK (uNK) cells and the trophoblast. Interestingly, uNK cells have been uniquely detected in the proximity of endothelial cells and invading trophoblasts associated with spiral arteries undergoing physiological transformation (6, 7).
How uNK cells induce tolerance to the fetal tissue is an important unresolved question, with implications for our understanding of the role of not only uNK cells but peripheral blood NK (pNK) cells in adverse pregnancy outcomes, tumor surveillance, autoimmunity and immunity against infections. In humans, unlike peripheral blood NK cells of cytotoxic CD56dimCD16+ phenotype, the pregnant uterus (decidua) is populated with specialized uNK cells that are of non-cytotoxic CD56brightCD16− phenotype (8–10). These cells are present in the endometrium during the secretory phase of the menstrual cycle and reach peak numbers (60–70%) during first trimester of pregnancy. Thereafter, their numbers decline rapidly (11–13). Although the physiological role of uNK cells remains poorly understood, several groundbreaking observations and views have implicated these cells in decidualization, invasion of the trophoblast, and production of angiogenic factors and chemokines (14–18). Major mechanistic findings on uNK cells have come from animal studies suggesting their role in spiral artery remodeling. NK cell-deficient mice display abnormalities in decidual artery remodeling and trophoblast invasion, possibly due to lack of uNK cell-derived IFN-γ (3, 14). In humans, lack of “physiological transformation” of decidual spiral arteries is thought to be associated with preeclampsia, preterm birth and intrauterine growth restriction (7, 19, 20). In this regard, evidence has been presented for defective interactions between uNK cells and the invading trophoblast in a cohort of preeclampsia patients (7, 21).
Despite their pregnancy compatible role, uNK cells at the maternal-fetal interface harbor a repertoire of natural cytotoxicity receptors (NCRs), cytolytic granules, activating and inhibitory killer-IgG receptors (KIR) (16,22–24). This suggests that these innate cells retain their primordial role as sentinels with an ability to attack target cells in response to “danger signals” in their proximate milieu. Indeed, recent studies from our laboratory and others have shown that uNK cells “switch on” their cytolytic machinery leading to fetal resorption or preterm birth in mice exposed to lipopolysaccharide (LPS) (25, 26) or dsRNA (27). In addition, increased cytolytic CD16+ NK cells have been reported in the endometrium of patients with a history of recurrent spontaneous abortion and implantation failure (28–30). Other studies have shown that uNK cells cultured in vitro can be readily activated with IL-2 to lyse target cells including trophoblasts (31, 32). Thus, the mechanisms that facilitate pregnancy compatible, non-cytotoxic characteristics of uNK cells need to be delineated. We hypothesized that the signature angiogenic machinery of uNK cells was responsible for their non-cytotoxic behavior during normal pregnancy. We show that this is accomplished through uNK cell-produced VEGF C which enhances resistance to lysis of trophoblasts and endothelial cells through induction of the transporter associated with antigen processing (TAP-1) protein.TAP-1 is associated with transport of processed peptides to the endoplasmic reticulum to ascertain functional MHC class I assembly (33).
Materials and Methods
Human subjects
All human studies were done with the approval of the Institutional Review Board of Dartmouth Medical School (Hanover, NH) and Women and Infants Hospital (Providence, RI). Informed consent was obtained from all tissue donors.
Isolation of primary peripheral blood NK (pNK) cells
Mononuclear cells from blood were collected from non-pregnant healthy women 35 ± 5 years of age separated from heparinized blood samples by Ficoll gradient centrifugation as previously described (51). NK cells were further purified using human NK cell isolation kit (Milteyni Biotec, USA) based on depletion of non-NK cells (negative selection) as per the manufacturer's instructions.
Isolation of decidual NK (dNK) cells
First trimester decidua were obtained from pregnant women who underwent elective termination. All subjects were between 7 and 11 weeks gestation. Gestational age was calculated based on last menstrual period or ultrasound at the time of procedure. Decidual tissue was processed for NK cell isolation immediately after extraction as previously described (51). The cells were suspended in RPMI supplemented with 10% fetal calf serum and antibiotics and adjusted to a concentration of 1 × 106 cells/ml. dNK cells were further separated using human NK cell isolation kit (Milteyni Biotec, USA) as described above. Cell viability was confirmed to be >90% by trypan blue staining. The purity of the preparation (>95%) were confirmed by FACS by gating on forward scatter and side scatter dot plots of CD45+ cells to exclude non-cellular debris (51).
Generation of uNK cell clones
CD56brightCD3− uNK cells were isolated from endometrial tissue specimens obtained from women undergoing hysterectomy for various gynecological disorders. Tissue samples used were distal to any pathological changes and were processed to isolate NK cell population as described (38). To generate uNK cell clones, enriched NK cells were cultured in 500 U IL-2/ml for 2–3 days to allow cell expansion in complete media [RPMI 1640 supplemented with 2-mercaptoethanol (50 μM), penicillin (100 U/ml), streptomycin (100 μg/ml), sodium pyruvate (1 mM), nonessential amino acids (0.1 mM), and 5% human serum]. After 2–3 days in culture, uNK cells were cloned using limiting dilution method as described (36). Actively growing cells from wells were analyzed to confirm their NK cell phenotype (CD56+, CD3−), and these uNK clones were expanded and maintained in minimum amounts of IL-2 for the remainder of their culture.
Trophoblast cells and endothelial cells
Immortalized first trimester trophoblast cell line HTR8 with properties of invasive extravillous cytotrophoblasts was established and kindly provided by Dr. Charles Graham (52). HTR8 cells were grown to ~80% confluence in RPMI standard growth medium and used only during eight passages. Human umbilical cord endothelial cells (HUVEC) and human uterine endothelial cells (HUtEC) were obtained from Cambrex (East Rutherford, NJ, USA) and cultured in EBM-2 medium (Cambrex, East Rutherford, NJ, USA). All cells were maintained in standard culture conditions of 5% CO2 at 37°C.
In vitro three-dimensional tube formation assay
We have recently established a three dimensional dual culture system to study endovascular activity involving trophoblasts and endothelial cells (34). This method has been now modified to examine the role of uNK cells in angiogenic processes. Briefly, growth factor-reduced Matrigel (BD Biosciences, San Diego, CA, USA) was thawed overnight at 4°C and mixed to homogeneity. Culture plates (48-well) were coated with 0.1 ml of Matrigel and allowed to gelatinize at 37°C for 30 min. Trophoblasts or endothelial cells (2.5 × 104), labeled with cell tracker green CMFDA or cell tracker red CMTMR (Molecular Probes, Eugene, OR, USA) respectively were co-cultured with pNK or uNK cells (1:1:1) on matrigel coated plates. The spontaneous interaction and endothelial cell-directed tube formation by trophoblasts was monitored and recorded 12–14 hrs after incubation under standard culture conditions using florescence microscopy (Nikon Eclipse TS 100 coupled with CCD camera). The average number of tubes/vacuoles formed was quantified by counting the number of tube like structures formed by connected capillary bridge in four different fields by two independent investigators (34).
Calcein-AM retention assay for cytotoxicity measurement
We determined the cytotoxic ability of uNK cells and pNK cells on target endothelial (HUVEC, HUtEC) or trophoblast (HTR8) cells using calcein-AM retention assay (38). Briefly, adherent confluent target cells (HUVEC, HUtEC or HTR8; 2×103) were plated in a 96 well plate and labeled with 8 mM calcein-AM (Molecular Probes) in serum- and phenol red-free medium (GIBCO) for 40 min at 37°C. Based on pilot experiments, effector cells (pNK or uNK cells) were added at 25:1 ratio to calcein-AM labeled target cells in quadruplicate. Maximal lysis was determined by solubilizing target cells in lysis buffer (0.1% Triton X-100 in PBS, pH 9.0). After the indicated time (2–3 hr) of incubation at 37°C, the assays were terminated by washing the plates twice and the remaining fluorescence was read using a 96 well fluorescence plate reader (SpectraMax Gemini EM, Molecular Devices) with excitation and emission at 485 and 538nm, respectively. Percent specific cytotoxicity was calculated as described (37). To monitor the cytoprotective activity of VEGF C or VEGF A, the target cells were pre-incubated overnight with cytokines (100 ng/ml) and subjected to cytotoxicity assay.
Quantification of uNK cell-secreted factors
We cultured 1×106 uNK cell clones (n=22) individually for 48 hrs in RPMI media containing 5 U/ml recombinant IL-15. Measurement of the concentrations of VEGF A, VEGF C, PlGF, IFN-γ, and TNF-α, in supernatants was completed in triplicate using respective ELISA kits (Quantikine Kits, R&D System, Minneapolis, MN, USA) according to the manufacturers' instructions.
Flow cytometry
We evaluated the surface expression by FACS analysis using flurochrome-conjugated mAb. Cells were harvested and incubated in dark at 4°C for 30 min with antibodies. The antibodies used were anti-class-I HLA (clone W6/32, Abcam), anti-HLA E (MEM-E/08, Abcam), anti-HLA G (MEM-G/9, Abcam), anti-human CD16 (clone 3G8), anti-human CD56, CD45, CD3, NKP46 (BD Biosciences Pharmingen), NKp30, NKp44, NKG2D (BioLegend), VEGF R1 (49560), VEGF R2 (89106), VEGF R3 (54733) (R&D Systems). For HLA-E staining we used the anti-human HLA-E (MEM-E/08) as primary antibody, followed by FITC labeled goat anti-mouse secondary antibody. TAP-1 expression in response to VEGF C (100 ng/ml), IFN-γ (100 ng/ml), VEGF A (100 ng/ml), PlGF (100 ng/ml) or combination of VEGF C and IFN-γ (100 ng/ml each) was carried out by intracellular staining using specific antibody (clone TAP1.28, MBL laboratory, Japan) after fixing and permeablizing the cells as recommended by the manufacturer. Negative controls were performed for each cell type by incubating the cells with isotype-matched antibodies. The stained cells were acquired (10,000 cells) and analyzed by FACS Calibur™ (Becton Dickinson).
siRNA-mediated silencing of TAP-1 in trophoblasts
We transfected HTR8 trophoblast cells in a six well plate with control siRNA (scrambled) or siRNA against TAP-1 (Santa Cruz Biotechnology, Inc.) using Lipofectamine 2000 (Invitrogen) for 12 hours in OptiMEM I reduced serum medium (Invitrogen). After the transfection period, the cells were allowed to recover in complete RPMI media for another 12 hours. Cells were then harvested for FACS analysis, immunobloting or used as targets for cytotoxicity assay.
Western blotting
We determined the protein expression of TAP-1 in HTR8 trophoblast cells treated with recombinant human IFN-γ (100 ng/ml), VEGF C (100 ng/ml R&D System) or combination of both for 24hrs. The cell lysates were prepared in lysis buffer (10 mM Tris HCl, pH 7.6, 50 mM NaCl, 5 mM EDTA, 50 mM NaF, 0.1 mM sodium orthovanadate, 30 mM sodium pyrophosphate, 1% Triton X-100, 10 μg/ml leupeptin, and 10 μg/ml aprotinin) on ice for 30 min. The cell lysates were separated on 12% SDS–polyacrylamide gels and blotted onto PVDF membranes and probed with anti-TAP-1 antibody (clone TAP1.28, MBL laboratory, Japan) (1:200 dilution) in 1% BSA in PBST. Actin was probed using anti-actin antibodies (Chemicon, USA) and used as an internal loading control. The bands were visualized using horseradish peroxidase conjugated secondary antibodies followed by ECL chemiluminescence (Amersham Biosciences, Piscataway, NJ). The protein bands were recorded using Konica SRX 101A developer.
Statistical analysis
P values for a pair of data sets were obtained with the two-tailed Student's t-test.
Results
VEGF C-producing uNK cells facilitate endovascular processes
Recent observations have suggested that human decidual NK cells chemoattract trophoblasts and regulate their invasion and participation in vascular remodeling (16). A central question that still remains to be addressed is what factors regulate the immune and vascular activities of non-cytotoxic uNK cells. To address this, we have taken advantage of primary uNK and pNK cells, and short term cultured clones of these cells to assess their ability to influence vascular remodeling. NK cell clones from peripheral blood or endometrial tissue were established and propagated as described in Methods. We utilized a co-culture model of vascular remodeling that involves trophoblast-endothelial cell-NK cell interactions on matrigel and mimics trophoblast invasion of the spiral arteries during pregnancy (34). Using this approach, we demonstrate that HTR8 cells, representing first trimester extravillous trophoblasts, footprint the endothelial cell-guided capillary like tube structures in response to growth factors from endothelial basal media (EBM2) (Fig.1A). Primary pNK cells disrupt the interaction (tube structure formation) between HTR8 cells and HUVEC endothelial cells (Fig. 1B). On the other hand, decidual NK (dNK) cells facilitate formation of definitive tube structures with intense yellow color, a sign of trophoblast-endothelial cell co-localization (Fig. 1C). NK cells are not seen in these panels as they were not marked with a cell tracker.
Figure 1.

Effect of uNK cell clones and VEGF C on tube formation between endothelial cell and trophoblasts.
Human umbilical vein endothelial cell HUVEC (EC, labeled red) and first trimester extravillous trophoblasts (HTR8, labeled green) were cultured overnight on matrigel in presence or absence of primary pNK, dNK cells or uNK cell clones (unlabeled). The capillary tube formation was recorded as described in Methods. Representative Fig.s of EC-directed tube formation by HTR8 cells (4× magnification) are shown involving: (A) No uNK cells, (B) primary pNK cells (n=4), (C) primary dNK cells (n=3), (D) a disruptive uNK cell clone (n=18), (E) a facilitative uNK cell clone (n=4) are shown. Exogenous addition of VEGF C (100 ng/ml) (F) but not VEGF A (100 ng/ml) (G) rescued the EC-directed tube formation by HTR8 cells in presence of disruptive uNK clones. (H) Average number of tubes/vacuoles formed was quantified in four different fields (4×, magnification). The numbers (bars, ± s.d.) are average of representative experiments in triplicate, assessed by two independent investigators. (I) Average levels of VEGF A, VEGF C and IFN-γ secreted by disruptive and facilitative uNK cell clones. Values are mean ± s.d. of all the samples. ** P<0.05 (Student t test). One representative data set is shown of multiple experiments performed in triplicates.
To further delineate the mechanism(s) for differences between pNK and uNK cells, we employed NK cell clones maintained in the presence of low doses of IL-2. uNK and pNK cell clones were established from 4 different individuals and further characterized as described in Methods. We tested a total of 22 uNK cell clones, 18 of which mimicked primary pNK cells with tube disruption phenotype (data with a representative clone are shown in Fig. 1D), whereas 4 clones facilitated tube formation similar to dNK cells (data with a representative clone are shown in Fig. 1E). A total of 5 pNK cell clones were also tested (data not shown) and the observations were similar to their primary counterparts (Fig. 1B). The average numbers of tubes/vacuoles formed in the presence or absence of “disruptive” or “facilitative” uNK clones were quantified as described in Methods and are presented in Fig. 1H.
Next, we examined production of growth factors (VEGF A, VEGF C, PlGF) and cytokines (IFN-γ, TNF-α) secreted by disruptive and facilitative uNK cell clones. For simplicity, data are shown only for VEGF A, VEGF C and IFN-γ as PlGF and TNF-α were minimally produced as detected by ELISA (Fig. 1I). Although uNK cell clones produced variable amounts of VEGF A and VEGF C, a striking observation was that facilitative uNK cell clones produced 3–4 fold higher levels of VEGF C compared to disruptive clones. Although VEGF C mRNA and protein have previously been shown to be expressed in NK cells from endometrium and decidua (35, 36), the immuno-regulatory role of VEGFs in the uNK cell biology has not been described. On the other hand, no VEGF C mRNA was detected in the human placenta (37), suggesting a unique role of uNK cell-produced VEGF C at the maternal-fetal interface. This prompted us to evaluate exogenous VEGF C and VEGF A for their ability to rescue tube formation in the presence of disruptive uNK cell clones. Interestingly, VEGF C (Fig. 1F), not VEGF A (Fig. 1G), was partially able to rescue tube formation. Quantification of VEGF C-mediated rescue of formation of tube structures is shown in Fig. 1H.
Facilitative and disruptive uNK clones do not differ in their phenotypic features
Cytolytic functions are generally associated with CD56dimCD16+ NK cells (8). To rule out disparate behavior of uNK cell clones due to heterogeneity in phenotypic characteristics, we performed FACS analysis for the surface expression of CD16 and CD56 on all uNK cell clones and compared them with pNK cell clones. CD56 expression on uNK and pNK cell clones was found to be bright and dim, respectively (Fig. 2B), in agreement with published observations (38). All the uNK cell clones (n=22) used in the study were found predominantly to be CD16 null (CD56+CD16−), whereas pNK cell clones (n=5) were mostly CD56+CD16+ (Fig. 2A). In both groups, NK cells were CD45+ and CD3− (data not shown). Since the expression of NCRs is a distinguishing feature of NK cells, we monitored the surface expression of NKP30, NKp44, NKp46 and NKG2D. uNK cell clones not only expressed all NCRs irrespective of their ability to disrupt or support tube formation, but the intensity of their surface expression was significantly higher compared to pNK cell clones (Fig. 2C). In addition, freshly isolated pNK cells lacked expression of NKp44. Thus, it is possible that NKp44 expression is regulated by activation stimuli. Since addition of VEGF C-producing uNK cells or exogenous VEGF C was able to restore capillary tube formation (Fig. 1F), we evaluated the expression of VEGF receptors R1, R2 and R3 on uNK cells. As shown in Fig. 2D, none of the VEGF receptors were expressed on either pNK cells or uNK cell clones, suggesting that angiogenic factors secreted by uNK cells act in a paracrine manner on cells expressing these receptors. For a positive control, HTR8 trophoblast cells were used as these cells are known to express all VEGF receptors (Fig. 2D).
Figure 2.
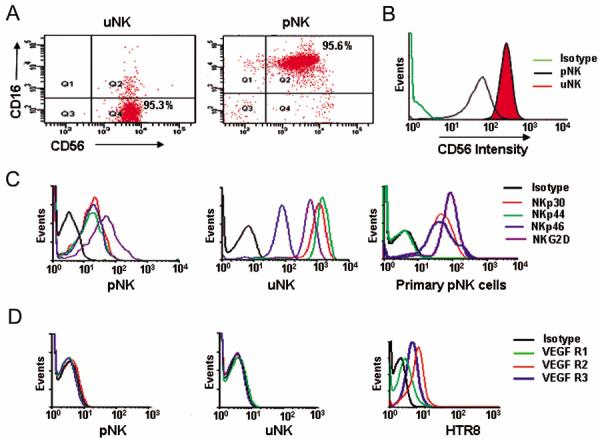
Characterization of uNK and pNK cells.
Representative FACS analysis is shown for the expression of CD56 and CD16 antigens in (A) uNK cell clone and pNK cell clone with CD56 dim and CD56 bright characteristics (B). (C) Presents phenotypic characterization of natural cytotoxicity receptors in pNK cell clones, uNK cell clones and primary pNK cells. (D) A representative phenotypic profile of VEGF receptors in pNK and uNK cell clones as compared to HTR8 trophoblast cells. One representative data set is shown out of four different experiments.
VEGF C rescues target cells from NK cell-mediated cytotoxicity
Disruption of tube formation by uNK cell clones could result from the cytolytic activity of these cells. This prompted us to assess cytolytic activity of uNK cell clones. Using calcien-AM retention assay (39), we evaluated the cytotoxic effects of uNK and pNK cell clones on HUVEC cells, human uterine endothelial cells (HUtEC), and HTR8 cells. Although non-pregnant endometrium does not contain trophoblasts, the use of extravillous HTR8 cells is justified as the same endometrial NK cells are further amplified in the decidua where they encounter invading trophoblasts. Further, since addition of VEGF C was able to restore tube formation (Fig. 1F), we investigated whether VEGF C or VEGF A would be able to rescue target cells from NK cell-mediated killing. To establish a suitable effector:target (E:T) cell ratio, we performed the assay using a wide range of E:T ratios involving a disruptive NK cell clone and target HUVEC or HTR8 cells (Fig. 3A). Based on these data, we used 25:1 E:T ratio for all subsequent experiments involving all disruptive uNK cell clones (n=18) and pNK cell clones (n=5). Fig. 3, B–G shows data for representative uNK or pNK cell clones. Disruptive uNK cell clones demonstrated robust cytolytic activity towards all target cells, HUVEC (Fig. 3B), HUtEC (Fig. 3C) or HTR8 (Fig. 3D), albeit at varying levels. Similarly, pNK cells were able to robustly lyse HUVEC (Fig. 3E) and to lesser extent HTR8 cells (Fig. 3F). Importantly, pre-incubation of target cells overnight with VEGF C (Fig. 3, B–F) but not VEGF A (Fig. 3G) inhibited NK cell-mediated lysis.
Figure 3.
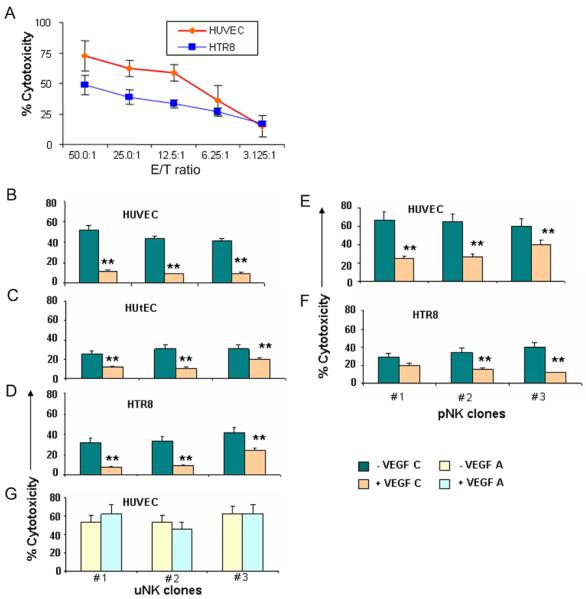
VEGF C rescues target cells from NK cell cytotoxicity.
(A) Percent cytotoxicity of pNK cells on HUVEC, HUtEC and HTR8 cells at different E/T ratios. Values are mean+ s.d. for triplicate reaction. (B–G) indicate cytotoxicity of three representative disruptive uNK cell clones with E/T ratio of 25:1 with HUVEC (B), HUtEC (C), HTR8 (D) as target cells in presence (tan bars) or absence (teal bars) of VEGF C. (E) and (F) represent cytotoxicity of three pNK cell clones against HUVEC and HTR8 cells respectively in presence (tan bars) or absence (teal bars) of VEGF C. (G) shows cytotoxicity of three uNK cell clones on HUVEC in presence (light blue) or absence (light yellow) of VEGF A. ** P<0.05 (Student t test).
Cytoprotection by VEGF C is mediated by up-regulation of TAP-1
Since both cytotoxic and non-cytotoxic uNK cell clones express a similar profile of cytotoxic machinery (Fig. 2C), it is plausible that VEGF C modifies target cells to escape NK cell-mediated killing. In this regard, induction of TAP-1 expression has been shown to be associated with cytoprotection of microvascular endothelial cells from activated NK cells (39, 40). We found that VEGF C up-regulated the expression of TAP-1 in HTR8 cells and HUVEC cells as confirmed by FACS analysis (Fig. 4, A–B) and by Western blotting (Fig. 5B). This induction of TAP-1 was found to be synergistic with IFN-γ on HTR8 cells (Fig. 4A and Fig. 5B). Quantification of percent TAP-1 positive HTR8 or HUVEC cells in response to VEGF C, VEGF A, PlGF and IFN-γ by flow cytometry is presented in Fig. 4B. Both VEGF C and IFN-γ induced TAP-1 expression in HUVEC and HTR8 although IFN-γ was found to be more potent in HUVEC. On the other hand, IFN-γ elicited less activity in trophoblast HTR8 cells (Fig. 4A and Fig. 5B). Importantly, VEGF A and PlGF failed to induce any significant amount of TAP-1 in either cell type (Fig. 4B).
Figure 4.
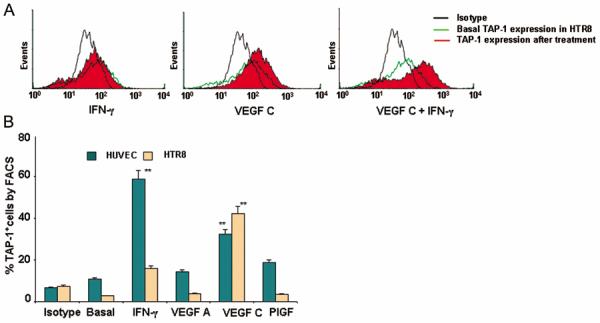
VEGF C induces TAP-1 expression.
(A) Shows intracellular staining and FACS analysis of TAP-1 expression (solid red) in HTR8 cells in response to IFN-γ (100 ng/ml), VEGF C (100 ng/ml) or VEGF C+IFN-γ (100 ng/ml, each) as compared with untreated (green line) and isotype control (black line). One representative experiment is shown out of three performed. (B) Represents the percentage of TAP-1 expressing endothelial (teal bars) and HTR8 cells (tan bars) in response to different treatments. Values are mean ± s.d. of three experiments. Note that VEGF C but not VEGF A or PlGF were able to significantly increase the expression of TAP-1.
Figure 5.

Silencing TAP-1 abrogates VEGF C induced cytoprotection in HTR8 trophoblasts. (A) A representative histogram of TAP-1 knockdown by siRNA as compared with scrambled siRNA, VEGF C treated and untreated HTR8 cells. (B) A representative Western blot analysis indicating TAP-1 induction in response to VEGF C, IFN-γ or their combination. TAP-1 induction is abrogated in TAP-1 siRNA treated HTR8 cells in response to VEGF C, IFN-γ or combination of both. (C) Cytotoxicity of a representative pNK cell clone on TAP-1 siRNA treated HTR8 cells (tan bars) as compared to TAP-1 expressing HTR cells (teal bars) in presence or absence of VEGF C or VEGF C+IFN-γ. Values are mean ± s.d of triplicate reactions. **P<0.05 (Student t test) as compared to respective HTR8 treatment group. #P<0.05 as compared to HTR8 group without VEGF C or IFN-γ treatment. One representative data set is shown out of three experiments performed.
siRNA-mediated knock-down of TAP-1 expression reverses cytotoxicity of uNK cells
To further confirm the role of TAP-1 in the VEGF C-induced non-cytotoxicity profile of uNK cells, we used gene specific siRNA to knock-down TAP-1 expression in HTR8 cells induced by IFN-γ, VEGF C or combination of both. As demonstrated by FACS analysis (Fig. 5A) and Western blotting (Fig. 5B), siRNA treatment of cells significantly blocked induction of TAP-1 by VEGF C, IFN-γ or combination of both. Next, we tested whether siRNA-mediated inhibition of TAP-1 expression would reverse the protection profile of target cells. siRNA treatment abolished the protection imparted by VEGF C or VEGF C+IFN-γ in HTR8 cells against NK cell-mediated lysis as indicated by the calcien-AM cytotoxicity assay (Fig. 5C). This clearly confirms both mechanistic and functional importance of VEGF C in priming targets that otherwise would be susceptible to killing by cytotoxic NK cells.
Induction of TAP-1 by VEGF C leads to increase in MHC class I presentation on target cells
Activation or inhibition of the cytolytic machinery in NK cells is determined by the extent of MHC class I complex on target cells. Because we observed up-regulated TAP-1 expression in HUVEC and HTR8 cells in response to VEGF C, we propose that TAP-1 induction results in greater amounts of MHC class I on treated cells. In this regard, we analyzed HLA-E expression which engages the CD94-NKG2A inhibitory or CD94-NKG2C activating receptors on NK cells (41, 42). Basal expression of HLA-E was low in both HUVEC and HTR8 cells. Interestingly, VEGF C but not VEGF A induced HLA-E expression on HUVECs, and HTR8 trophoblast cells (Fig. 6A) which was consistent with TAP-1 induction (Fig. 4B). Next, we evaluated the surface expression of MHC class I molecules using a pan-class I antibody on HUVEC and HTR8 cells (Fig. 6B). Both cell types exhibited significant MHC class I expression in response to VEGF C but not VEGF A (Fig. 6B). As trophoblasts are not known to express classical MHC class I molecules, the observed expression with pan-antibody possibly reflects binding to induced HLA-C or HLA-G. Thus, VEGF C-induced TAP-1 expression and MHC class I assembly in trophoblasts and endothelial cells could facilitate survival by providing inhibitory signals to uNK cells.
Figure 6.
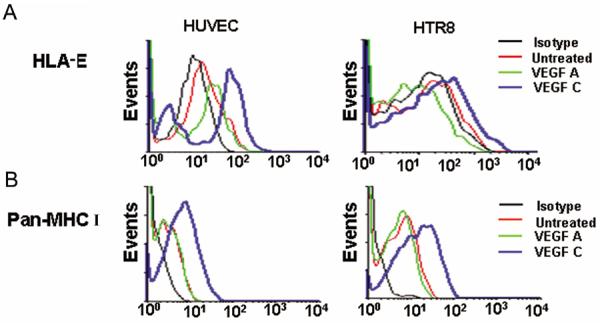
VEGF C induces HLA-E expression.
(A) Show representative histograms for the surface expression of HLA-E in HUVEC and HTR8 in response to VEGF C and VEGF A. (B) Show representative histograms of MHC class I expression in HUVEC and HTR8 cells in response to VEGF C or VEGF A treatments. One representative data is shown out of three experiments performed.
Discussion
Host defense, self-tolerance, and tissue transplantation are major health issues that invoke both innate and adaptive immune responses. However, the immune defense mechanisms of the fetus have challenged the concepts embedded in the traditionally defined pathways of immunity. This is further complicated by “immunologically tolerant” phenotype of the predominant NK cell population in the pregnant uterus and their perceived role in placentation-associated endovascular activity (16, 24). In this study we describe novel mechanisms by which uNK cells maintain non-cytotoxic and pro-angiogenesis status.
Our results provide evidence for a central link between production of VEGF C, a pro-angiogenic factor, by uNK cells and their non-cytotoxic phenotype. Using primary human peripheral blood or endometrial NK cells and panel of clones derived from primary cells, we demonstrate that VEGF C production is a hallmark property of non-cytotoxic uNK cells. We further demonstrate that VEGF C, not VEGF A or PlGF, can protect target endothelial and trophoblast cells from killing by cytotoxic pNK cells. Lack of killing is directly associated with VEGF C-mediated induction of TAP-1 in target cells, a key molecule in the process of MHC class I assembly. This is directly demonstrated by siRNA-mediated knockdown of VEGF C-mediated TAP-1 expression which results in reversal of non-cytotoxic characteristics of uNK cells.
Using a three-dimensional culture system on matrigel to evaluate the endovascular activity of uNK cells, we demonstrate that there are differences in the ability of activated uNK cell clones to facilitate or disrupt the endothelial-trophoblast dual cell interactions mimicking vascular remodeling. While CD56brightCD16− uNK cell clones consistently expressed NCRs, they differed in their cytotoxic profile. This is not surprising because uNK cell clones were propagated in vitro in the presence of activating stimuli. In vivo, most uNK cells are likely to be of non-cytotoxic phenotype. Despite in vitro conditions, several uNK cell clones maintained their non-cytotoxic phenotype. In these clones, the non-cytotoxic phenotype correlated with the production of high amounts of VEGF C. Importantly, exogenous VEGF C could rescue trophoblasts and endothelial cells from traditionally cytotoxic NK cells. Dysregulation in VEGF C production at the maternal-fetal interface could be a signal for poor angiogenesis and pregnancy complications. Reduced expression of VEGF C has been reported in pregnancies experiencing intrauterine growth restriction (IUGR) and preeclampsia (43). Involvement of VEGF C may thus explain non-killer phenotype of uNK cells despite possessing toxic granules and expressing cytotoxicity receptors.
Since our results show that uNK cells do not express receptors for VEGFs, it is tempting to argue that angiogenic factors are produced for a paracrine action on uterine endothelial cells and invading trophoblasts. Our results point to this activity of uNK cell derived VEGF C. VEGF C acts by binding to VEGF R2 and VEGF R3 and triggers intracellular survival signals in tumor cells and promote their invasion (44). In addition to maintaining uNK cell non-cytotoxicity, VEGF C may support survival of endothelial cells (45). Although during pregnancy, intrauterine hormones and factors that upregulate secretions of VEGF C in uNK cells are not known, in vitro studies suggest involvement of cytokines like IL-15 (36).
The non-cytotoxic capacity of NK cells is based on its ability to recognize surface major histocompatibility complex (MHC) class I molecules on target cells that deliver signals to suppress NK cell functions. A lack of engagement of such MHC-specific receptors leads to NK cell-mediated killing (8, 18, 23). Interestingly, Bender and colleagues (39) have shown that endothelial cells exhibited sensitivity to activated peripheral blood NK cells in the absence of expression of TAP-1. IFN-γ was shown to induce TAP-1 in endothelial cells and protect them from peripheral blood NK cell-mediated killing (39). However, our data suggest that VEGF C is robustly produced by non-cytotoxic uNK cells compared to IFN-γ. It is thus proposed that VEGF C is the predominant regulator of TAP-1 expression in the uterus, although it is likely that VEGF C and IFN-γ act in tandem as supported by our observations (Fig. 5B). Interestingly VEGF A or PLGF failed to induce TAP-1 expression (Fig. 4B). TAP-1 is a key factor essential for peptide loading for MHC class I assembly and antigen presentation (33, 46). Silencing TAP-1 expression using siRNA in trophoblasts abolished the cyto-protective activity of VEGF C, confirming the mechanism of action of VEGF C.
Trophoblasts cells do not express classical MHC class I molecules but express HLA-C and unconventional HLA-G and HLA-E molecules (5). By using HLA-E as a molecule of choice, we show that VEGF C enhanced surface expression of HLA-E on first trimester HTR8 trophoblasts. Surface expression of HLA-E normally depends on the recruitment and binding of TAP-dependent classical nonameric peptides with anchoring residues derived from HLA class I signal sequence (47, 48). Interestingly, TAP-1 expression has been shown to be significantly higher in extravillous trophoblasts that are positive for non-polymorphic HLA class I molecules (5, 49, 50). We also detected significant presence of pan-MHC class I on both HTR8 trophoblast cells and HUVECs in response to VEGF C, suggesting that this angiogenic factor is a potent immunoregulator in the uterine microenvironment.
Taken together, these findings support a dual role of VEGF C in immune tolerance and promoting active angiogenesis by uNK cells. Our findings for the first time provide evidence that non-cytotoxicity of uNK cells is directly coupled to their angiogenic translational machinery. uNK cells apparently prepare trophoblasts and endothelial cells to express modest levels of MHC class I molecules to evade killing by a major cytotoxic cell population at the maternal-fetal interface.
Acknowledgments
We thank James Padbury for his thoughtful critique of the manuscript.
Nonstandard abbreviations used
- HUVEC
human umbilical vein endothelial cells
- HUtEC
human uterine endothelial cells
- TAP 1
transporter associated protein-1
- uNK
uterine natural killer cells
- pNK
peripheral blood natural killer cells
- dNK
decidual natural killer cells
- NCR
natural cytotoxicity receptors
- LPS
lipopolysaccharide
- IUGR
intrauterine growth restriction
Footnotes
Disclosures The authors have no financial conflict of interest.
References
- 1.Moffett-King A. Natural killer cells and pregnancy. Nat. Rev. Immunol. 2002;2:656–663. doi: 10.1038/nri886. [DOI] [PubMed] [Google Scholar]
- 2.Norwitz ERD, Schust J, Fisher SJ. Implantation and the survival of early pregnancy. N. Engl. J. Med. 2001;345:1400–1408. doi: 10.1056/NEJMra000763. [DOI] [PubMed] [Google Scholar]
- 3.Croy BA, Esadeg S, Chantakru S, van den Heuvel M, Paffaro VA, He H, Black GP, Ashkar AA, Kiso Y, Zhang J. Update on pathways regulating the activation of uterine natural killer cells, their interactions with decidual spiral arteries and homing of their precursors to the uterus. J Reprod Immunol. 2003;59:175–191. doi: 10.1016/s0165-0378(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 4.Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat. Immunol. 2004;5:266–271. doi: 10.1038/ni1037. [DOI] [PubMed] [Google Scholar]
- 5.Shorter SC, Starkey PM, Ferry BL, Clover LM, Sargent IL, Redman CW. Antigenic heterogeneity of human cytotrophoblast and evidence for the transient expression of MHC class I antigens distinct from HLA-G. Placenta. 1993;14:571–582. doi: 10.1016/s0143-4004(05)80210-3. [DOI] [PubMed] [Google Scholar]
- 6.Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. 2006;27:939–958. doi: 10.1016/j.placenta.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Parham P. NK cells and trophoblasts: partners in pregnancy. J. Exp. Med. 2004;200:951–955. doi: 10.1084/jem.20041783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–640. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 9.Koopman LA, Kopcow HD, Rybalov B, Boyson JE, Orange JS, Schatz F, Masch R, Lockwood CJ, Schachter AD, Park PJ, Strominger JL. Human decidual natural killer cells are a unique NK cell subset with immunomodulatory potential. J Exp Med. 2003;198:1201–1212. doi: 10.1084/jem.20030305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saito S, Nishikawa K, Morii T, Enomoto M, Narita N, Motoyoshi K, Ichijo M. Cytokine production by CD16-CD56bright natural killer cells in the human early pregnancy decidua. Int Immunol. 1993;5:559–563. doi: 10.1093/intimm/5.5.559. [DOI] [PubMed] [Google Scholar]
- 11.Kitaya K, Yamaguchi T, Yasuo T, Okubo T, Honjo H. Post-ovulatory rise of endometrial CD16 (−) natural killer cells: in situ proliferation of residual cells or selective recruitment from circulating peripheral blood? J Reprod Immunol. 2007;76:45–53. doi: 10.1016/j.jri.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Bulmer JN, Lash GE. Human uterine natural killer cells: a reappraisal. Mol. Immunol. 2005;42:511–521. doi: 10.1016/j.molimm.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 13.Kalkunte S, Chichester CO, Gotsch F, Sentman CL, Romero R, Sharma S. Evolution of non-cytotoxic uterine natural killer cells. Am J Reprod Immunol. 2008;59:425–432. doi: 10.1111/j.1600-0897.2008.00595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashkar AA, Di Santo JP, Croy BA. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J Exp Med. 2000;192:259–270. doi: 10.1084/jem.192.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ain R, Canham LN, Soares MJ. Gestation stage-dependent intrauterine trophoblast cell invasion in the rat and mouse: novel endocrine phenotype and regulation. Dev. Biol. 2003;260:176–190. doi: 10.1016/s0012-1606(03)00210-0. [DOI] [PubMed] [Google Scholar]
- 16.Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, Gazit R, Yutkin V, Benharroch D, Porgador A, Keshet E, Yagel S, Mandelboim O. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. 2006;12:1065–1074. doi: 10.1038/nm1452. [DOI] [PubMed] [Google Scholar]
- 17.Le Bouteiller P, Tabiasco J. J. Killers become builders during pregnancy. Nat Med. 2006;12:991–992. doi: 10.1038/nm0906-991. [DOI] [PubMed] [Google Scholar]
- 18.Moffett A, Loke C. Immunology of placentation in eutherian mammals. Nat Rev Immunol. 2006;6:584–594. doi: 10.1038/nri1897. [DOI] [PubMed] [Google Scholar]
- 19.Kim YM, Bujold E, Chaiworapongsa T, Gomez R, Yoon BH, Thaler HT, Rotmensch S, Romero R. Failure of physiologic transformation of the spiral arteries in patients with preterm labor and intact membranes. Am J Obstet Gynecol. 2003;189:1063–1069. doi: 10.1067/s0002-9378(03)00838-x. [DOI] [PubMed] [Google Scholar]
- 20.Sargent IL, Borzychowski AM, Redman CW. NK cells and preeclampsia. J Reprod Immunol. 2007;76:40–44. doi: 10.1016/j.jri.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 21.Hiby SE, Walker JJ, O'shaughnessy KM, Redman CW, Carrington M, Trowsdale J, Moffett A. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J.Exp. Med. 2004;200:957–965. doi: 10.1084/jem.20041214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobs R, Hintzen G, Kemper A, Beul K, Kempf S, Behrens G, Sykora KW, Schmidt RE. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur J Immunol. 2001;31:3121–3127. doi: 10.1002/1521-4141(2001010)31:10<3121::aid-immu3121>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 23.Riley JK, Yokoyama WM. NK cell tolerance and the maternal-fetal interface. Am J Reprod Immunol. 2008;59:371–387. doi: 10.1111/j.1600-0897.2008.00593.x. [DOI] [PubMed] [Google Scholar]
- 24.Kopcow HD, Allan DS, Chen X, Rybalov B, Andzelm MM, Ge B, Strominger JL. Human decidual NK cells form immature activating synapses and are not cytotoxic. Proc Natl Acad Sci U S A. 2005;102:15563–15568. doi: 10.1073/pnas.0507835102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy SP, Fast LD, Hanna NN, Sharma S. Uterine NK cells mediate inflammation-induced fetal demise in IL-10-null mice. J Immunol. 2005;175:4084–4090. doi: 10.4049/jimmunol.175.6.4084. [DOI] [PubMed] [Google Scholar]
- 26.Murphy SP, et al. Evidence for uterine NK cell-dependent etiology of preterm birth. Am J Obstet Gynecol. 2008 in press. [Google Scholar]
- 27.Kinsky R, Delage G, Rosin N, Thang MN, Hoffmann M, Chaouat G. A murine model of NK cell mediated resorption. Am J Reprod Immunol. 1990;23:73–77. doi: 10.1111/j.1600-0897.1990.tb00675.x. [DOI] [PubMed] [Google Scholar]
- 28.Lachapelle MH, Miron P, Hemmings R, Roy DC. Endometrial T, B, and NK cells in patients with recurrent spontaneous abortion. Altered profile and pregnancy outcome. J Immunol. 1996;156:4027–4034. [PubMed] [Google Scholar]
- 29.Quenby S, Bates M, Doig T, Brewster J, Lewis-Jones DI, Johnson PM, Vince G. Pre-implantation endometrial leukocytes in women with recurrent miscarriage. Hum Reprod. 1999;14:2386–2391. doi: 10.1093/humrep/14.9.2386. [DOI] [PubMed] [Google Scholar]
- 30.Kwak JY, Beaman KD, Gilman-Sachs A, Ruiz JE, Schewitz D, Beer AE. Up-regulated expression of CD56+, CD56+/CD16+, and CD19+ cells in peripheral blood lymphocytes in pregnant women with recurrent pregnancy losses. Am J Reprod Immunol. 1995;34:93–99. doi: 10.1111/j.1600-0897.1995.tb00924.x. [DOI] [PubMed] [Google Scholar]
- 31.King A, Loke YW. Human trophoblast and JEG choriocarcinoma cells are sensitive to lysis by IL-2-stimulated decidual NK cells. Cell Immunol. 1990;129:435–448. doi: 10.1016/0008-8749(90)90219-h. [DOI] [PubMed] [Google Scholar]
- 32.Abadía-Molina AC, Ruiz C, Montes MJ, King A, Loke YW, Olivares EG. Immune phenotype and cytotoxic activity of lymphocytes from human term decidua against trophoblast. J Reprod Immunol. 1996;31:109–123. doi: 10.1016/0165-0378(96)00965-5. [DOI] [PubMed] [Google Scholar]
- 33.Cox JH, Yewdell JW, Eisenlohr LC, Johnson PR, Bennink JR. Antigen presentation requires transport of MHC class I molecules from the endoplasmic reticulum. Science. 1990;247:715–718. doi: 10.1126/science.2137259. [DOI] [PubMed] [Google Scholar]
- 34.Kalkunte S, Zhongbin L, Tewari N, Chichester C, Romero R, Padbury J, Sharma S. In vitro and in vivo evidence for lack of endovascular remodeling by third trimester trophoblasts. Placenta. 2008;29:871–878. doi: 10.1016/j.placenta.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lash GE, Schiessl B, Kirkley M, Innes BA, Cooper A, Searle RF, Robson SC, Bulmer JN. Expression of angiogenic growth factors by uterine natural killer cells during early pregnancy. J Leukoc Biol. 2006;80:572–580. doi: 10.1189/jlb.0406250. [DOI] [PubMed] [Google Scholar]
- 36.Li XF. Angiogenic growth factor messenger ribonucleic acids in uterine natural killer cells. J Clin Endocrinol.Metab. 2001;86:1823–1834. doi: 10.1210/jcem.86.4.7418. [DOI] [PubMed] [Google Scholar]
- 37.Clark DE, Smith SK, Licence D, Evans AL, Charnock-Jones DS. Comparison of expression patterns for placenta growth factor, vascular endothelial growth factor (VEGF), VEGF-B and VEGF-C in the human placenta throughout gestation. J Endocrinol. 1998;159:459–467. doi: 10.1677/joe.0.1590459. [DOI] [PubMed] [Google Scholar]
- 38.Eriksson M, Meadows SK, Wira CR, Sentman CL. Unique phenotype of human uterine NK cells and their regulation by endogenous TGF-beta. J Leukoc Biol. 2004;76:667–675. doi: 10.1189/jlb.0204090. [DOI] [PubMed] [Google Scholar]
- 39.Ayalon O, Hughes EA, Cresswell P, Lee J, O'Donnell L, Pardi R, Bender JR. Induction of transporter associated with antigen processing by interferon gamma confers endothelial cell cytoprotection against natural killer-mediated lysis. Proc Natl Acad Sci U S A. 1998;95:2435–2440. doi: 10.1073/pnas.95.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma W, Lehner PJ, Cresswell P, Pober JS, Johnson DR. Interferon-gamma rapidly increases peptide transporter (TAP) subunit expression and peptide transport capacity in endothelial cells. J Biol Chem. 1997;272:16585–16590. doi: 10.1074/jbc.272.26.16585. [DOI] [PubMed] [Google Scholar]
- 41.King A, Allan DS, Bowen M, Powis SJ, Joseph S, Verma S, Hiby SE, McMichael AJ, Loke YW, Braud VM. HLA-E is expressed on trophoblast and interacts with CD94/NKG2 receptors on decidual NK cells. Eur J Immunol. 2000;30:1623–1631. doi: 10.1002/1521-4141(200006)30:6<1623::AID-IMMU1623>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 42.Braud VM, Allan DS, O'Callaghan CA, Soderstrom K, D'Andrea A, Ogg GS, Lazetic S, Young NT, Bell JI, Phillips JH, Lanier LL, McMichael AJ. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795–799. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- 43.Dunk C, Ahmed A. Expression of VEGF-C and activation of its receptors VEGFR-2 and VEGFR-3 in trophoblast. Histol Histopathol. 2001;16:359–375. doi: 10.14670/HH-16.359. [DOI] [PubMed] [Google Scholar]
- 44.Ueda M, Hung YC, Terai Y, Kanda K, Kanemura M, Futakuchi H, Yamaguchi H, Akise D, Yasuda M, Ueki M. Vascular endothelial growth factor-C expression and invasive phenotype in ovarian carcinomas. Clin Cancer Res. 2005;11:3225–3232. doi: 10.1158/1078-0432.CCR-04-1148. [DOI] [PubMed] [Google Scholar]
- 45.Zhou Y, Bellingard V, Feng KT, McMaster M, Fisher SJ. Human cytotrophoblasts promote endothelial survival and vascular remodeling through secretion of Ang2, PlGF, and VEGF-C. Dev Biol. 2003;263:114–125. doi: 10.1016/s0012-1606(03)00449-4. [DOI] [PubMed] [Google Scholar]
- 46.Goldszmid RS, Bafica A, Jankovic D, Feng CG, Caspar P, Winkler-Pickett R, Trinchieri G, Sher A. TAP-1 indirectly regulates CD4+ T cell priming in Toxoplasma gondii infection by controlling NK cell IFN-gamma production. J Exp Med. 2007;204:2591–2602. doi: 10.1084/jem.20070634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee N, Goodlett DR, Ishitani A, Marquardt H, Geraghty DE. HLA-E surface expression depends on binding of TAP-dependent peptides derived from certain HLA class I signal sequences. J Immunol. 1998;160:4951–4960. [PubMed] [Google Scholar]
- 48.Coupel S, Moreau A, Hamidou M, Horejsi V, Soulillou JP, Charreau B. Expression and release of soluble HLA-E is an immuno-regulatory feature of endothelial cell activation. Blood. 2007;109:2806–2814. doi: 10.1182/blood-2006-06-030213. [DOI] [PubMed] [Google Scholar]
- 49.Roby KF, Gershon D, Hunt JS. Expression of the transporter for antigen processing-1 (TAP-1) gene in subpopulations of human trophoblast cells. Placenta. 1996;17:27–32. doi: 10.1016/s0143-4004(05)80640-x. [DOI] [PubMed] [Google Scholar]
- 50.Clover LM, Sargent IL, Townsend A, Tampé R, Redman CW. Expression of TAP1 by human trophoblast. Eur J Immunol. 1995;25:543–553. doi: 10.1002/eji.1830250236. [DOI] [PubMed] [Google Scholar]
- 51.Plevyak M, Hanna N, Mayer S, Murphy S, Pinar H, Fast L, Ekerfelt C, Ernerudh J, Berg G, Matthiesen L, Sharma S. Deficiency of decidual IL-10 in first trimester missed abortion: a lack of correlation with the decidual immune cell profile. Am J Reprod Immunol. 2002;47:242–250. doi: 10.1034/j.1600-0897.2002.01060.x. [DOI] [PubMed] [Google Scholar]
- 52.Graham CH, Hawley TS, Hawley RG, MacDougall JR, Kerbel RS, Khoo N, Lala PK. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp. Cell. Res. 1993;206:204–211. doi: 10.1006/excr.1993.1139. [DOI] [PubMed] [Google Scholar]