

Abstract
The integration of molecular, genomic, and clinical medicine in the post-genome era provides the promise of novel information on genetic variation and pathophysiologic cascades. The current challenge is to translate these discoveries rapidly into viable biomarkers that identify susceptible populations and into the development of precisely targeted therapies. In this article, we describe the application of comparative genomics, microarray platforms, genetic epidemiology, statistical genetics, and bioinformatic approaches within examples of complex pulmonary pathobiology. Our search for candidate genes, which are gene variations that drive susceptibility to and severity of enigmatic acute and chronic lung disorders, provides a logical framework to understand better the evolution of genomic medicine. The dissection of the genetic basis of complex diseases and the development of highly individualized therapies remain lofty but achievable goals.
Increased morbidity and mortality from acute and chronic pulmonary disorders represent sources of enormous health-care expenditures1-6 and mandate better understanding of lung pathobiology. The unraveling of the genetic basis of complex pulmonary diseases such as asthma, emphysema, acute lung inury (ALI), and pulmonary hypertension (PH) provides an opportunity to improve the delivery of targeted therapies that reduce the suffering of patients with these disorders. The challenge, however, historically has been daunting as traditional methods of genetic research, with limited technologies, could only consider the study of individual genes. The complexity of genomic structure and function, disease heterogeneity, the influence of the environment on disease development and progression, and epigenetic mechanisms also contribute to the challenge of mastering the genetic underpinnings of complex lung diseases. Fortunately, in the era after the completion of the Human Genome Project, the availability of multiple high-throughput genomic and genetic technologies now allows elucidation of complex pathophysiology cascades produced by the carefully orchestrated spatial and temporal regulation of tens to hundreds of genes and proteins. In this article, we provide examples of the application of these emerging genomic tools as we integrate genomic knowledge with clinical medicine. We discuss the use of several novel and potentially useful approaches: whole genome linkage analysis scans, conventional candidate gene and single nucleotide polymorphism (SNP)-based approaches, and preclinical animal models of human disease. Ultimately, the promise of the post-genome era is that the increased application of genomic technologies and innovative bioinformatics will translate clinical-based discovery into the personalized medicine experience with the development and identification of novel biomarkers, diagnostic strategies, prognostic indicators, and targets for therapeutic intervention.
PROBING THE GENOME USING LINKAGE ANALYSIS
The genome-wide search for genetic variants that underlie complex diseases consists of narrowing down the genomic regions that are involved in disease susceptibility followed by identification of the relevant gene(s) in this region. Linkage analysis traditionally has been the initial method used to cosegregate common microsatellite markers spanning the genome with a particular phenotypic trait. This method generally has involved the analysis of generations of affected pedigrees or families. If excess sharing of the alleles harboring the microsatellite or a polymorphism are found after genotyping large numbers of affected sibling pairs, linkage is calculated reflecting the probability that the marker or polymorphism in that chromosomal region is sufficiently near the actual gene variant that confers susceptibility to the trait of interest. Once convincing evidence for linkage is observed, the search continues to exactly identify the critical genetic variant within the linked region (often a region of up to millions of base pairs) via finer gene mapping (Fig 1). Linkage analyses detect the presence of a large effect from high penetrance loci and originally were used successfully for mapping of rare disorders of Mendelian origin. For example, one of the first genome-wide linkage studies led to the discovery of the cystic fibrosis (CF) mutation in the CFTR gene (codon deletion at 508).7-10 Genome-wide linkage searches in complex diseases have also led to significant findings in PH with 19 affected individuals from 6 families revealing evidence for linkage with 2 chromosome 2q markers that led to the discovery of mutations in the gene encoding the bone morphogenetic protein receptor type II (BMPR2) (a member of the TGF-beta family) as the cause of familial primary pulmonary hypertension.11,12 Additional linked regions have been localized to chromosome 17p that are associated with lung cysts with spontaneous pneumothorax, which are findings related to families with Birt–Hogg–Dub̀ syndrome, an inherited autosomal genodermatosis also characterized by benign tumors of the hair follicle and renal tumors.13
Fig 1.

Schematic representation of current genomic approaches in the study of complex diseases. Linkage analysis customarily has been used for genome-wide searches for loci responsible for susceptibility to diseases. However, the post-genomic era has generated high-throughput technologies, including RNA-based microarrays and SNP platforms such as Affymetrix and Illumina. These new methods permit the preferential use of genome-wide association studies, which yield candidate genes that can be validated, functionally tested, and ultimately translated to the development of novel diagnostic tests, disease biomarkers, and the selection of patients for innovative therapeutic strategies.
Complex disorders are multifactorial by definition and evolve via the impact of many moderate effect loci. Linkage analysis for complex disorders requires additional analytical power to compensate for single gene-reduced effect size and penetrance. For example, asthma has been screened by over 16 genome-wide scans designed to generate whole genome linkage to map susceptibility genes in large asthma-containing family cohorts. Table I17-39 contains a summary of unique linkage population studies performed to date for multiple phenotypes related to asthma, as well as other pulmonary disorders, including PH and CF. The success of a linkage analysis-based study relies on factors such as the level of complexity of the disease, clarity of phenotype definition, and availability of family members for the study. Each of these factors renders the use of genetic linkage unsuitable for diseases such as ALI (Table I), where the disease is sporadic (lacking an observable pattern of heredity) and an environmental exposure is necessary. The ambiguity of phenotypes also challenges positional cloning analyses and has led to widespread use of intermediary phenotypes (directly measurable or quantitative parameters) that are impaired in a specific disease but that capture the phenotype of interest beyond simple categorical descriptions. These parameters promise greater analytical power in quantitative regression-based linkage models, and they define more precisely genomic regions associated with a complex disease. For example, evidence for linkage in a 20-cM region of chromosome 7p14-p15 was identified for 3 phenotypes: asthma, a high level of IgE (atopy), and the combination of the phenotypes.14 The strongest linkage was observed for high-serum IgE, which is an intermediate quantitative trait related to asthma. However, by mapping genes for only this feature, it is important to recognize that the full extent of the genetic variation that causes asthma is unlikely to be characterized.
Table I.
Genome-wide scans of pulmonary disorders using linkage analysis*
| Disease | Number of studies |
|---|---|
| Asthma14–29 | 16 |
| COPD30 | 1 |
| Lung cancer31, 32 | 1 |
| Tuberculosis33 | 1 |
| Pulmonary hypertension12 | 1 |
| CF10 | 1 |
| Pulmonary fibrosis34, 35 | 2 |
| Spontaneous pneumothorax13 | 1 |
| Sarcoidosis36, 37 | 2 |
| Normal lung function38 | 1 |
| Pulmonary AV malformation39 | 1 |
| Acute lung injury | 0 |
Using the search terms “genome-wide search,” “genome-wide screen,” “genome-wide scan,” with “respiratory,” “lung,” “airway,” “pulmonary,” and the respective diseases listed below, the table displays a list of all genome-wide scans conducted on original linkage populations for the list of pulmonary diseases to date. Over 16 genome-wide scans have been performed to study asthma and asthma-related traits, however, an absence of genome-wide scans exists in the study of acute lung injury (in bold).
Similar phenotype designs have been applied in genetic linkage studies of other pulmonary disorders, such as chronic obstructive pulmonary disease (COPD),30,40-42 where the phenotypes used were categorical (defined as “moderate airflow obstruction” and “chronic bronchitis”)30 and, later, quantitative (spirometric measurements).42 These studies focused on families with severe early-onset disease, which is hypothesized to represent the most severe end of the COPD spectrum. Initial qualitative-based investigations failed to identify linked regions with genome-wide significance, although several marginally linked regions were present.30 Subsequent studies that included additional quantitative phenotypes such as the FEV1/FVC ratio revealed stronger evidence for linkage on chromosome 2q.40,42 A remaining challenge is how best to quantify the severity of lung disease. In CF, for example, although the FEV1% of predicted is commonly accepted as reflecting severity of CF lung disease, a nondiscriminatory comparison requires age-adjusted FEV1%, which can be problematic, as the typical percent predicted correction is based on decline in lung volume with age in “control” subjects without CF.43
GENETIC ASSOCIATION APPROACH
Although genetic linkage is a relatively inexpensive approach to positional cloning, several shortcomings are apparent especially in cases where recruitment of an adequate number of family cohorts is difficult. Linkage studies also typically lack precision and only point to broad genomic regions, typically in the order of many millions of base pairs, where numerous potentially disease-affecting loci reside. The application of linkage studies in oligogenic disorders is problematic in terms of sufficient analytical power because of reduced effect size and penetrance of the susceptibility loci. A conceptual alternative to genetic linkage are genome-wide genetic association studies, which, similar to linkage studies, rely on meiotic recombination to localize susceptibility variants. In contrast to linkage, however, genetic association studies generally rely on the presence of stretches of DNA that recently are coin-herited from the ancestral chromosomes and, therefore, undisturbed by recombination. Because a variable number of polymorphisms are tightly bound together, which are referred to as linkage disequilibrium (LD) or allelic association, analysis of 1 variant provides information about association over several polymorphic variants in LD with each other. Over-representation of alleles or genotypes in specific phenotypic categories is evidence that the marker and the polymorphic variants in LD are highly likely to be associated with the phenotype.
Genetic associations using SNPs in candidate genes have become the standard of association tests and characterize candidate genes or specific genomic regions on a whole genome scale. SNPs have several advantages over microsatellites, which often change gradually over time and are prone to expansion during consecutive meioses. SNPs are more stable on a population scale and are much denser (see dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi). All of these polymorphisms are accessible to the public through several databases, including the dbSNP, the HapMap Project (www.hapmap.org), and the Perlegen (www.perlegen.com). Since typically only 2 alleles are present, both alleles have an easily automatable genotype process and provide more analytical power through a decreased number of degrees of freedom. A variety of study designs can be used by researchers for association studies, including traditional case-control studies, cohort studies, and family studies that consist of affected probands and probands' parents or siblings.
WHOLE GENOME-WIDE ASSOCIATIONS AND HIGH-THROUGHPUT TECHNOLOGIES
A whole genome association analysis is the next step of technological development and was enabled by 2 major developments. The first development was the successful completion of the patterns of human LD in 3 major ethnoracial groups44,45 (HapMap Project), providing novel insights into the scale of human genetic variation and the number of the markers necessary to capture this variation. For the first time, an extensive catalog of more than 3 million SNPs (Fig 2) became available along with information about how these variants cluster together in haplotypes.45
Fig 2.

Explosion in the number of recognized SNPs. As can be observed, the number of SNPs exponentially grew with the mapping of the human genome (gathered from The National Center for Biotechnology Information (NCBI) SNP database).
Key advances in biotechnology also paved the way to whole genome association and linkage studies by developing the simultaneous genotyping of many thousands of SNPs in single assays, lowering resource utilization, labor costs, and processing time. Several platforms such as Affymetrix and Illumina are currently in use, and, as their SNP number increases, are becoming more popular with researchers. These genotyping systems are provided in several versions (10, 100, 500,000, 1 million SNPs) with different template designs. One platform includes a multiple microarray reaction where allele-specific oligonucleotide probes hybridize with their DNA gene target.46 The number of SNPs covered varies with the platform used and are selected arbitrarily on the basis of endonuclease cleavage site availabilities. Other platforms, which are designed on a large number of allele-specific oligonucleotides bound to glass beads, provide different scales of SNP coverage (ranging from custom-made to over 500,000),47,48 where the DNA hybridizes to allele-specific oligonucleotides contained in multiple beads in a hybridization pool. As no prior enzymatic restriction is needed, these platforms incorporate LD patterns in the SNP selection strategy providing more information from less representative SNPs.
Recently, a whole genome SNP linkage scan in 2 large Caucasian families with IPF revealed linkage to chromosome 5p15.34 Within the linked region, sequencing was completed in a candidate gene revealing multiple mutations in telomerase reverse transcriptase, which encodes the catalytic component of telomerase, and 1 heterozygous mutation in TERC, which is the essential RNA component of telomerase. Subjects heterozygous for these mutations demonstrated telomere shortening and an increased susceptibility to developing IPF.34 Association studies at a genomic scale have also been reported recently for several phenotypes. The first lung study that included whole genome SNP arrays identified ORMDL3, which is a gene that strongly predicted early-onset asthma in 3 independent Caucasian cohorts.49 Although this finding will be subjected to the usual scientific scrutiny, ORMLD3 is both important in terms of magnitude of effect in susceptibility and surprising given its obscure functional role and location in a genomic region not previously isolated by several genetic linkage analyses. The infancy of whole genome association studies limits full estimation of the significance of these findings and the potential problems inevitably associated with new methodologies. However, the results published thus far have suggested several totally novel loci and offer the possibility of comprehensively (ie, on a whole genomic scale) probing into the more elusive effects of simultaneous interaction among several genetic loci.
Concomitant testing of a large number of polymorphisms requires large sample sizes to meet stringent criteria for statistical significance. As with other association analyses, results from whole genome associations are expected to be replicated in at least one other independent cohort. The requirement for large cohort sizes, the need for replicates, and the relatively high cost of recruitment with extensive genotyping have convinced many researchers and funding agencies to unite their efforts and join resources in strongly powered study designs. These consortiums have in the past allowed the identification of important loci that underlie common human diseases. For example, the Wellcome Trust Case-Control Consortium recently published findings on 7 diseases,50 (2000 cases for each disease and 3000 healthy British controls), confirmed known disease loci (NOD2 and inflammatory bowel disease) and reported novel genetic loci for susceptibility to type 2 diabetes.51
CANDIDATE GENE APPROACHES
Whole genome association analysis was born out of the need to address the built-in deficiencies observed for the single candidate gene association methodologies. In this latter paradigm, specific genes are selected on the basis of existing knowledge of their function and potential involvement in disease pathogenesis. This feature potentially precludes identification of unsuspected genes or variants that can form the inheritable basis of a disease phenotype, whereas a whole genome association analysis considers a priori probabilities of association to be equal for all genomic regions. Candidate gene association analyses have occasionally produced spurious “positive” association results caused by multiple testing in a large number of small-scale studies with extremely small sample sizes.52,53 Despite these limitations, however, significant evidence supports the utility of a candidate gene approach and its capacity to discover novel genes for association analyses.
Macrophage migration inhibitory factor (MIF), which was originally named for inhibition of random macrophage migration, is an inflammatory cytokine implicated in ALI and acute respiratory distress syndrome (ARDS)54 and represents one powerful example of the potential of a candidate gene association approach. MIF has since been characterized extensively to be involved in a whole host of inflammatory pathways and expressed in a variety of cell types (T cells, epithelium, endothelium)55-58 with increased MIF levels present in the serum and bronchoalveolar lavage fluid of patients with ARDS.54 We examined MIF gene and protein expression in preclinical murine and canine models of ALI (using high tidal volume mechanical ventilation and endotoxin exposure) and in patients with either sepsis or sepsis-induced ALI. MIF gene expression and protein levels were increased significantly in each ALI model, with serum MIF levels significantly higher in patients with either sepsis or ALI compared with healthy control subjects of either African-descent or European-descent Americans.59 To extend the likely role for MIF in ALI and sepsis susceptibility, we next assessed MIF SNPs in both conditions and studied the association of 8 MIF SNPs (all within a 9.7-Kb interval on chromosome 22q11.23) in African-descent and European-descent populations. Genotyping revealed several MIF haplotypes in the 3′ end of the gene that were associated strongly with sepsis and ALI in both populations.59 Thus, our combined functional genomic and genetic approaches suggest that MIF is a relevant molecular target in ALI.
We have also previously studied SNPs in MYLK, the gene encoding myosin light chain kinase (MLCK), a multifunctional protein involved in contraction, inflammation, and apoptosis and whose expression is increased in both asthma and ALI. We sequenced exons, exon–intron boundaries, and 2 Kb of 5′ UTR of MYLK, which revealed 51 SNPs, and evaluated 28 SNPs for potential association with sepsis-associated ALI in a case-control sample of European-descent subjects with sepsis alone, sepsis-associated ALI, or neither (control), and a sample population of African-descent subjects with sepsis and ALI.60 Significant SNP associations were observed with the ALI phenotype in European-descent subjects with unique SNPs also observed in African-descent subjects with ALI. Multiple haplotypic analyses revealed an ALI-specific, risk-conferring haplotype at 5′ of the MYLK gene in both European-descent and African-descent subjects and an additional 3′ region haplotype only in African-descent subjects.60
With the involvement of MYLK in multiple aspects of the inflammatory response, we also considered the possibility that MYLK SNPs are involved in the development of asthma, which is a chronic inflammatory lung disorder. MYLK is located in 3q21.1, which is a region noted by several genome-wide studies to show linkage with asthma and asthma-related phenotypes.14,16,61-65 We investigated 17 MYLK genetic variants in European-descent and African-descent Americans with asthma and identified a single nonsynonymous polymorphism (Pro147Ser) almost entirely restricted to African-descent populations with severe asthma. These results remained highly significant after adjusting for proportions of ancestry, and they may be related causally to the severe asthma phenotype in subjects of African descent,66 results we validated in a Afro-Caribbean population.67 Taken together, these data strongly implicate MYLK genetic variants as risk variants in inflammatory lung disorders, such as ALI and asthma.60 We are currently exploring the potential for MYLK SNPs to contribute to susceptibility to other chronic inflammatory disorders such as inflammatory bowel disease and ischemia reperfusion injuries.
GENE-GENE, GENE-ENVIRONMENT, AND OTHER CONSIDERATIONS
The combination of increased power and the technological advances allowing concomitant multiple genotyping have begun to yield novel information in the challenging area of genetic factor interactions in particular phenotypes. Although gene–gene interactions likely play a major role in vivo, most genetic epidemiologic studies to-date have been limited to the study of the effect of single genes and often of single polymorphisms. Notable success stories of disentanglement of conjoint effects of various genetic loci include identification of interacting genes that affect susceptibility to bronchial asthma and identification of factors that affect disease severity of cystic fibrosis in patients with established high-risk genotypes in the CFTR gene.68
Gene–environment interactions also add to the complexity of and the challenge to our understanding of the genome. Advances in our study designs are conditioned not only by the usual genotyping and analytical constraints but also by the requirement of a solid understanding of the epidemiologic risk factors for the given phenotype. Thus, these interactions should not be considered in isolation as joint effects of different loci can also interact with the surroundings. Despite this constraint, reports of successful identification of such interactions are already being made. McIntire et al69 have described a lower likelihood of the development of atopic sensitization in individuals with a history of Hepatitis A, who are concomitantly predisposed with a specific genetic variant (157insMTTTVP).
Current genetic epidemiologic studies with a SNP-based approach, despite numerous advantages, are powered only to identify common variants present in the general population (as envisaged by the common variant–common disease theory),70 and do not account for multiple variants with rarer individual frequency, which may have considerable phenotypic effects.71,72 Chromosomal structural variation, like sequence variation, plays an important role at the genetic level of both heritable and sporadic human disease. Copy number variation73,74 and variations on the inversion patterns across the general population75 potentially impact biologic processes insufficiently assessed by the currently used methodologies. Furthermore, association studies in a case and control settings are vulnerable to the biasing effects of population heterogeneity (which lead to differences in allele frequencies). Moreover, the common assumption that underlies association studies is that the sample under study is representative of the population at large and the results can thus be extended to a larger number of individuals. Although this assumption can be accurate, it is also possible that hidden biases persist even in the best designed of analyses. A recent study has highlighted the threat of ascertainment bias in genetic association analysis76,77 that can only be made worse by factors that truly affect eligibility of participants in the studies. The advantages of prospective cohort analyses should, therefore, be weighed carefully against other more practical and economical retrospective recruitment of cases and controls. Finally, identification of functional polymorphisms that mechanistically alter disease susceptibility is often difficult in association studies because of the potential presence of extensive LD near the associated markers that span large chromosomal regions and encompass other significantly associated polymorphisms. Given these potential shortcomings of linkage and association studies, the application of functional genomics to link a particular genotype to function and phenotype will require resource-intensive efforts and multidisciplinary expertise in diverse areas such as bioinformatic sequence analysis, in vitro assays, and experimental expertise in animal models of disease.
NOVEL ANIMAL MODELS: CONSOMIC AND CONGENIC RODENTS
The challenges in performing association or linkage studies in humans have resulted in a trend toward using preclinical animal models of disease that minimize confounders and cost. With mapping of both murine and rat genomes, extensive genomic infrastructure already has been established providing excellent models to isolate genetic variants and to control for environmental influences that commonly complicate human studies. Although rodent linkage and association studies have been insightful for a variety of complex diseases,78-80 these approaches are time-consuming and laborious with large populations required to achieve the statistical power necessary to narrow and define quantitative trait loci (stretches of DNA that are linked closely to the genes that underlie the trait in question, QTLs). To address these concerns, the construction of consomic and congenic strains of rats has now been developed and in use.
A consomic rat strain is one in which an entire chromosome is introgressed into the isogenic background of another inbred strain using marker-assisted selection techniques (Fig 3).81,82 Similarly, a congenic strain developed through this method contains only part of the introgressed chromosome, which narrows the region of interest. The foundation for the existing panels of consomic/congenic rats is based on the naturally occurring phenotypic variation between the Brown Norway (BN) rat strain and either the Dahl salt sensitive (SS) or a fawn hooded (FHH) strains. Thus, 2 consomic panels of rats have been constructed in which 21 chromosomes from the BN rat were substituted individually into the genomic background of either a SS or a FHH strain. Using these consomic strains, regions of the genome that harbor the genes responsible for phenotypic differences between rat strains have been mapped and catalogued in a library for 274 physiologic traits that relate to the function of the kidneys, heart, vasculature, and lungs and to levels of circulating substrates and hormones.83 Other advantages of consomics have been reviewed recently81 and detail the platform's ability to develop congenic strains rapidly to fine map the physiologic function and to define the impact of a causal gene region in a permissive genome.
Fig 3.

Schematic representation of the generation of a consomic strain. Parental strains with contrasting phenotypic responses (susceptible vs resistant) are intercrossed to generate a heterozygous F1 population, which is backcrossed with the parental SS strain to produce F2 progeny. F2 rats are then screened for a heterozygous genotype along the target chromosome and, subsequently, backcrossed with the parental strain for 4–8 generations using marker-assisted selection, which results in progeny with an isogenic SS background for all but the target chromosome (in black). These offspring are then mated to produce homozygotes for the chromosome of interest, which are then inbred to produce a stable consomic strain. (modified from Cowley et al82).
We have used consomic rats to study ventilator-associated lung injury (VALI) candidate genes by determining differential rat strain sensitivities to mechanical ventilation.84 For example, introgressions of VALI-sensitive BN chromosomes 13 and 16 into the VALI-resistant SS resulted in significantly increased inflammation (compared with parental SS strain), which suggests that genes residing on BN chromosomes 13 and 16 confer increased sensitivity to high tidal volume ventilation. Interestingly, previous animal studies have identified several VALI/ALI candidate genes (IL-6, PAI1, CCL2, COX2) that map to these chromosomes, which increases their potential role in the VALI phenotype. In addition to our VALI study, other groups have examined respiratory regulation in response to hypoxia and carbon dioxide and variation in the hypercapneic ventilatory response using consomic strains.85 Finally, consomic strains have been employed successfully in the study of pulmonary hypertension86 identifying the PH-resistant nature of the FHH or BN consomic strain (with introgression of BN chromosome 1 into the genomic background of FHH) compared with the PH-sensitive FHH parentals.
EXPRESSION PROFILING AND INNOVATIVE BIOINFORMATICS
The genetic models described above have the merit of studying the presence of DNA factors related to specific phenotypes with the underlying concept that these variants alter proteins that exhibit different adverse quantitative or qualitative properties that cause an impaired phenotype. Studies of transcriptional processes and genome-wide gene expression have the advantages of assessing the final steps of the protein production process in both time and space linked more closely to the phenotype development. Transcriptional changes are not caused uniformly by inheritance with environmental exposure altering gene expression even in the absence of polymorphisms. The mRNA transcription strategies complement other above-described methods and capture a wide range of environmental exposures. Powerful high-throughput gene and protein expression profiling techniques allow the simultaneous and expeditious study of a single cell or a tissue of interest on a genome-wide scale. This technology has the potential to grasp the complexity of human diseases, unencumbered by preexisting gene-based hypotheses, and thus, has expanded the field of genomics. The most common microarray expression profiling experimental design compares 2 conditions (eg, normal vs disease or placebo vs drug). After a series of statistical filtering and normalization for chip design, genes differentially regulated have a greater probability of involvement in the molecular pathway associated with the phenotype or disease, which makes them probable candidate genes and potentially therapeutic targets. Although these differentially regulated genes represent potential mechanisms of disease, cause–effect relationships are not determined easily because they may only participate indirectly in the pathway leading to the disease or may merely reflect the organism's reaction to stress. The utility of microarray technology, since its first application,87 has recently gained widespread popularity in the study of pulmonary diseases as the method becomes more cost effective for the independent investigator.88-95 For example, a transcriptional profile has been characterized for patients with IPF compared against patients with normal lung tissue.96 One of the most distinctive potential candidate genes, matrilysin, was analyzed further with a knockout mouse investigation, which revealed the gene's protective role in bleomycin-induced pulmonary fibrosis.96
We used microarrays in a unilateral canine model of VALI to assess regional cellular and lung responses to local mechanical conditions that potentially contribute to lung injury.97 Highly significant regional differences in gene expression were observed between lung apex/base regions as well as between gravitationally dependent/nondependent regions of the base, with 367 and 1544 genes differentially regulated between these regions, respectively. These comparisons provided a list of potential VALI candidate genes and a powerful hypotheses-building platform for future investigations. This study also demonstrated one of the most common challenges confronting microarray use—the interpretation of large quantities of data generated by deriving a differential set of genes. This problem has fueled the emergence of a new field of bioinformatics that manipulates the large biologic dataset using various statistical, computational, and in silico analyses to produce a smaller, more manageable, set of genes.
In the last 5 years, many novel and insightful techniques have furthered the application of lung genomic strategies. For example, we have combined extensive gene expression profiling studies in both animal (murine, rat, and canine) and human models of ALI with bioinformatic methods linked to a Eukaryote Gene Orthologs database to narrow lists of differentially regulated genes (Fig 4).98 Given that orthologs have evolved from common ancestral genes and are presumed to retain a similar function despite speciation, overlapping responses across species (to a common insult) might serve as a powerful filter to yield information about conserved microarray-derived responses to injury. When orthologous genes that exhibit similar patterns of differential gene expression across all species were selected, the resultant smaller candidate gene pool included genes already under investigation in ALI as well as several novel genes involved in processes and pathways not previously suspected to affect ALI pathogenesis.98
Fig 4.
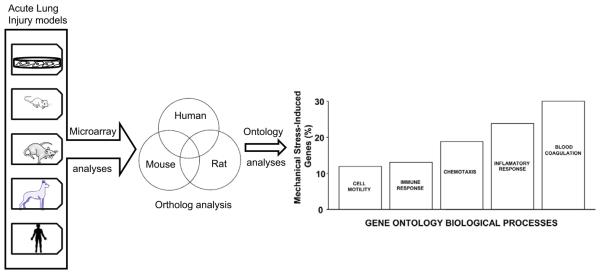
Schematic representation of orthologous approach with microarray analyses to identify ALI-associated candidate genes. Previously reported expression profiling analyses from murine, rat, and canine ALI models as well as from human endothelium exposed to mechanical stretch were analyzed to generate the orthologous gene profiles to a human array. Significantly upregulated or downregulated genes were extracted using an overlap analysis followed by gene ontology classification of the narrowed gene set.77
The combination of experimental results with existing knowledge can also be extremely beneficial as it allows a more informed interpretation of results and provides more focused observations for additional experimental work. Categorizing genes into functional groups ([ie, gene ontologies (GOs)], interaction networks, and biologic pathways can simplify large lists and yield important information about overrepresented pathways involved in the diseased state. We demonstrated recently the bioinformatic power of ontologies in characterizing the genomic response of the multi-kinase inhibitor, sorafenib, in the prevention of 2 rodent PH models (conventional mild hypoxia-based PH and severe PH combining hypoxia with vascular endothelial growth factor receptor blockade, SU5416).99 Extensive expression profiling identified a small set of statistically significant overrepresented GOs and allowed construction of a GO “profile” for each condition. Our novel GO analysis99 implicated 2 overarching GO functional classes including “cell proliferation” and “muscle contraction and development” that represent a unique PH signature profile for sorafenib and provide pathway insights for future PH investigations. This analysis powerfully demonstrates the potential for clinical translation as it relates to personalized patient care.99
In addition to the development of innovative bioinformatic analyses, the integration of high-throughput functional genomic technologies with established candidate gene approaches (positional cloning strategies and animal models of disease) has increased the yield of information from complex disease studies. One example is the identification of a new ALI biomarker, pre-B-cell colony enhancing factor (PBEF). PBEF resides on chromosome 7q22, a genomic site associated with several inflammatory disorders such as inflamma-tory bowel disease, multiple sclerosis, and cystic fibrosis.100-102 A previously obscure gene, we identified marked upregulation of PBEF in microarray analyses of various models of murine and canine ALI and increased gene/protein expression in bronchoalveolar lavage fluid samples from ALI patients.103-105 Given the impressive data that supports the role of PBEF as a novel candidate gene for ALI, we performed a PBEF SNP-based association study104 in ALI subjects of European and African-American descent. Genotyping of 2 PBEF SNPs (positions C-1543T and T-1001G) determined significant associations with sepsis and ALI with the strongest association found with the −1543C/−1001G haplo-type. Although we have found these 2 PBEF SNPs, acceptance of an association or linkage of any specific polymorphism (SNP or insertion/deletion) with a particular pathologic phenotype requires several general criteria, including but not limited to the replication of the genotype-phenotype association/linkage in a fundamentally independent panel with the affected phenotype. As linkage studies are not feasible for ALI, a second report106 recently described a significant association between PBEF SNPs −1543 C/T and −1001T/G and ALI susceptibility in a comparable but separate ALI population. Interestingly, the −1543*−1001 GC haplotype was also associated with increased intensive care unit (ICU) mortality, whereas the TT haplotype at −1543T with fewer ventilator days and decreased ICU mortality.106 Although a causal relationship to a phenotype is not necessarily true for all SNPs, proof of biologic function of a putative polymorphism to a particular phenotype also supports the association. We are unaware of any somatic mutation studies of PBEF as an approach to identify the direct functional consequence, but our prior reports104,105 describe the functional consequences of the 2 PBEF SNPs on promoter activity. Therefore, although the precise pathogenic mechanisms of the gene in ALI remain to be investigated, PBEF represents a powerful example of the utility of integrating global genomic and candidate gene approaches in identifying viable candidate genes and functional polymorphisms (historically considered to be a daunting task with the use of linkage or association analyses alone), and hence, it translates into a novel clinical biomarker.
THE ROAD AHEAD…
The search for genes that influence the susceptibility to complex pulmonary phenotypes remains a challenge. Even in monogenetic lung disorders with Mendelian inheritance patterns (such as CF), many loci likely participate in producing the heterogeneity of clinical outcomes in CF patients with identical F508 mutations.43 In the post-genomic era, however, we are enabled by high-throughput DNA and RNA techniques, which are emerging strengths in the field of bioinformatics, and by increasing availability of genetically manipulated strains of animals for preclinical models of human disease. Although microarray technologies, like other genomic approaches, have inherent weaknesses,107 and steady-state gene expression differences alone cannot determine gene causality for a particular disease, genomic studies have generated tremendous excitement and have moved us closer to dissecting the genetic basis of common yet complex pulmonary disorders.
Translating this information from bench to bedside is a multidisciplinary challenge that requires the consideration of philosophical, economical with cost- benefit analyses, societal, cultural, behavioral, and educational differences between the private and the public sectors, extent of scientific expertise, interdisciplinary communication, and clinical practice. As with most complex diseases, the main effective therapeutic modality at a public health level is probably through preventive medicine, which intuitively mandates the need for more sensitive and specific biomarkers that can identify accurately the at-risk population before the onset of symptoms. However, the positive predictive value of genes with modest effects found from genomic studies may be specific for a phenotype but not sufficiently sensitive for screening the general population. Still, knowledge of the genetic underpinnings of a common disease can provide an array of advantages. Some of these advantages are apparent in the design of clinical trials where predisposed individuals can be tested to assess the role of environmental influences on a phenotype, to test prevention-based therapies, or for prognostication determination, along with other potential avenues. Furthermore, treating a disease with the same drug and dosage across all patient populations is increasingly recognized as deficient in accounting for the genetic architecture of an individual, and the field of pharmacogenetics, which applies pharmacology to polymorphisms, is growing rapidly. Ultimately, these systems-biology types of approaches (genes to communities) will significantly advance efforts to identify common genetic variants that account for complex pulmonary disorders and holds the potential to deliver personalized care to patients with complex lung disorders.
Abbreviations
- ALI
acute lung injury
- ARDS
acute respiratory distress syndrome
- BN
Brown Norway
- CF
cystic fibrosis
- COPD
chronic obstruction pulmonary disease
- FHH
fawn hooded
- GO
gene ontology
- ICU
intensive care unit
- LD
linkage disequilibrium
- MYLK
myosin light chain kinase
- MIF
migration inhibitory factor
- PBEF
pre-B-cell colony enhancing factor
- PH
pulmonary hypertension
- SNP
single nucleotide polymorphism
- SS
salt sensitive
- VALI
ventilator-associated lung injury
REFERENCES
- 1.Braman SS. The global burden of asthma. Chest. 2006;130(Suppl 1):4S–12S. doi: 10.1378/chest.130.1_suppl.4S. [DOI] [PubMed] [Google Scholar]
- 2.Chung F, Barnes N, Allen M, et al. Assessing the burden of respiratory disease in the UK. Respir Med. 2002;96:963–75. doi: 10.1053/rmed.2002.1392. [DOI] [PubMed] [Google Scholar]
- 3.Foster TS, Miller JD, Marton JP, Caloyeras JP, Russell MW, Menzin J. Assessment of the economic burden of COPD in the U.S.: a review and synthesis of the literature. Copd. 2006;3:211–8. doi: 10.1080/15412550601009396. [DOI] [PubMed] [Google Scholar]
- 4.Krauth C, Jalilvand N, Welte T, Busse R. Cystic fibrosis: cost of illness and considerations for the economic evaluation of potential therapies. Pharmacoeconomics. 2003;21:1001–24. doi: 10.2165/00019053-200321140-00002. [DOI] [PubMed] [Google Scholar]
- 5.Molinier L, Combescure C, Chouaid C, et al. Cost of lung cancer: a methodological review. Pharmacoeconomics. 2006;24:651–9. doi: 10.2165/00019053-200624070-00004. [DOI] [PubMed] [Google Scholar]
- 6.Talmor D, Shapiro N, Greenberg D, Stone PW, Neumann PJ. When is critical care medicine cost-effective? A systematic review of the cost-effectiveness literature. Crit Care Med. 2006;34:2738–47. doi: 10.1097/01.CCM.0000241159.18620.AB. [DOI] [PubMed] [Google Scholar]
- 7.Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–65. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 8.Santis G, Osborne L, Knight R, Ramsay M, Williamson R, Hodson M. Cystic fibrosis haplotype association and the delta F508 mutation in adult British CF patients. Hum Genet. 1990;85:424–5. doi: 10.1007/BF02428295. [DOI] [PubMed] [Google Scholar]
- 9.Santis G, Osborne L, Knight RA, Hodson ME. Linked marker haplotypes and the delta F508 mutation in adults with mild pulmonary disease and cystic fibrosis. Lancet. 1990;335:1426–9. doi: 10.1016/0140-6736(90)91448-j. [DOI] [PubMed] [Google Scholar]
- 10.White R, Woodward S, Leppert M, et al. A closely linked genetic marker for cystic fibrosis. Nature. 1985;318:382–4. doi: 10.1038/318382a0. [DOI] [PubMed] [Google Scholar]
- 11.Lane KB, Machado RD, Pauciulo MW, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 12.Nichols WC, Koller DL, Slovis B, et al. Localization of the gene for familial primary pulmonary hypertension to chromosome 2q31-32. Nat Genet. 1997;15:277–80. doi: 10.1038/ng0397-277. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt LS, Warren MB, Nickerson ML, et al. Birt-Hogg-Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001;69:876–82. doi: 10.1086/323744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laitinen T, Daly MJ, Rioux JD, et al. A susceptibility locus for asthma-related traits on chromosome 7 revealed by genome-wide scan in a founder population. Nat Genet. 2001;28:87–91. doi: 10.1038/ng0501-87. [DOI] [PubMed] [Google Scholar]
- 15.Koppelman GH, Stine OC, Xu J, et al. Genome-wide search for atopy susceptibility genes in Dutch families with asthma. J Allergy Clin Immunol. 2002;109:498–506. doi: 10.1067/mai.2002.122235. [DOI] [PubMed] [Google Scholar]
- 16.Pillai SG, Chiano MN, White NJ, et al. A genome-wide search for linkage to asthma phenotypes in the genetics of asthma international network families: evidence for a major susceptibility locus on chromosome 2p. Eur J Hum Genet. 2006;14:307–16. doi: 10.1038/sj.ejhg.5201532. [DOI] [PubMed] [Google Scholar]
- 17.A genome-wide search for asthma susceptibility loci in ethnically diverse populations. The Collaborative Study on the Genetics of Asthma (CSGA) Nat Genet. 1997;15:389–92. doi: 10.1038/ng0497-389. [DOI] [PubMed] [Google Scholar]
- 18.Alcais A, Plancoulaine S, Abel L. An autosome-wide search for loci underlying wheezing age of onset in German asthmatic children identifies a new region of interest on 6q24-q25. Genet Epidemiol. 2001;21(suppl 1):S168–73. doi: 10.1002/gepi.2001.21.s1.s168. [DOI] [PubMed] [Google Scholar]
- 19.Altmuller J, Seidel C, Lee YA, et al. Phenotypic and genetic heterogeneity in a genome-wide linkage study of asthma families. BMC Pulm Med. 2005;5:1. doi: 10.1186/1471-2466-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouzigon E, Dizier MH, Krahenbuhl C, et al. Clustering patterns of LOD scores for asthma-related phenotypes revealed by a genome-wide screen in 295 French EGEA families. Hum Mol Genet. 2004;13:3103–13. doi: 10.1093/hmg/ddh340. [DOI] [PubMed] [Google Scholar]
- 21.Celedon JC, Soto-Quiros ME, Avila L, et al. Significant linkage to airway responsiveness on chromosome 12q24 in families of children with asthma in Costa Rica. Hum Genet. 2007;120:691–9. doi: 10.1007/s00439-006-0255-5. [DOI] [PubMed] [Google Scholar]
- 22.Daniels SE, Bhattacharrya S, James A, et al. A genome-wide search for quantitative trait loci underlying asthma. Nature. 1996;383:247–50. doi: 10.1038/383247a0. [DOI] [PubMed] [Google Scholar]
- 23.Hakonarson H, Bjornsdottir US, Halapi E, et al. A major susceptibility gene for asthma maps to chromosome 14q24. Am J Hum Genet. 2002;71:483–91. doi: 10.1086/342205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malerba G, Trabetti E, Patuzzo C, et al. Candidate genes and a genome-wide search in Italian families with atopic asthmatic children. Clin Exp Allergy. 1999 Dec;29(suppl 4):27–30. [PubMed] [Google Scholar]
- 25.Ober C, Cox NJ, Abney M, et al. Genome-wide search for asthma susceptibility loci in a founder population. The Collaborative Study on the Genetics of Asthma. Hum Mol Genet. 1998;7:1393–8. doi: 10.1093/hmg/7.9.1393. [DOI] [PubMed] [Google Scholar]
- 26.Van Eerdewegh P, Little RD, Dupuis J, et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature. 2002;418:426–30. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- 27.Wjst M, Fischer G, Immervoll T, et al. A genome-wide search for linkage to asthma. German Asthma Genetics Group. Genomics. 1999;58:1–8. doi: 10.1006/geno.1999.5806. [DOI] [PubMed] [Google Scholar]
- 28.Yokouchi Y, Nukaga Y, Shibasaki M, et al. Significant evidence for linkage of mite-sensitive childhood asthma to chromosome 5q31-q33 near the interleukin 12 B locus by a genome-wide search in Japanese families. Genomics. 2000;66:152–60. doi: 10.1006/geno.2000.6201. [DOI] [PubMed] [Google Scholar]
- 29.Xu X, Fang Z, Wang B, et al. A genomewide search for quantitative-trait loci underlying asthma. Am J Hum Genet. 2001;69:1271–7. doi: 10.1086/324650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silverman EK, Mosley JD, Palmer LJ, et al. Genome-wide linkage analysis of severe, early-onset chronic obstructive pulmonary disease: airflow obstruction and chronic bronchitis phenotypes. Hum Mol Genet. 2002;11:623–32. doi: 10.1093/hmg/11.6.623. [DOI] [PubMed] [Google Scholar]
- 31.Bailey-Wilson JE, Amos CI, Pinney SM, et al. A major lung cancer susceptibility locus maps to chromosome 6q23-25. Am J Hum Genet. 2004;75:460–74. doi: 10.1086/423857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Girard L, Zochbauer-Muller S, Virmani AK, Gazdar AF, Minna JD. Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res. 2000;60:4894–906. [PubMed] [Google Scholar]
- 33.Bellamy R, Beyers N, McAdam KP, et al. Genetic susceptibility to tuberculosis in Africans: a genome-wide scan. Proc Natl Acad Sci U S A. 2000;97:8005–9. doi: 10.1073/pnas.140201897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007;104:7552–7. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hodgson U, Pulkkinen V, Dixon M, et al. ELMOD2 is a candidate gene for familial idiopathic pulmonary fibrosis. Am J Hum Genet. 2006;79:149–54. doi: 10.1086/504639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iannuzzi MC, Iyengar SK, Gray-McGuire C, et al. Genome-wide search for sarcoidosis susceptibility genes in African Americans. Genes Immunol. 2005;6:509–18. doi: 10.1038/sj.gene.6364235. [DOI] [PubMed] [Google Scholar]
- 37.Schurmann M, Reichel P, Muller-Myhsok B, Schlaak M, Muller-Quernheim J, Schwinger E. Results from a genome-wide search for predisposing genes in sarcoidosis. Am J Respir Crit Care Med. 2001;164:840–6. doi: 10.1164/ajrccm.164.5.2007056. [DOI] [PubMed] [Google Scholar]
- 38.Joost O, Wilk JB, Cupples LA, et al. Genetic loci influencing lung function: a genome-wide scan in the Framingham Study. Am J Respir Crit Care Med. 2002;165:795–9. doi: 10.1164/ajrccm.165.6.2102057. [DOI] [PubMed] [Google Scholar]
- 39.Cole SG, Begbie ME, Wallace GM, Shovlin CL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42:577–82. doi: 10.1136/jmg.2004.028712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeMeo DL, Celedon JC, Lange C, et al. Genome-wide linkage of forced mid-expiratory flow in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170:1294–301. doi: 10.1164/rccm.200404-524OC. [DOI] [PubMed] [Google Scholar]
- 41.Palmer LJ, Celedon JC, Chapman HA, Speizer FE, Weiss ST, Silverman EK. Genome-wide linkage analysis of bronchodilator responsiveness and post-bronchodilator spirometric phenotypes in chronic obstructive pulmonary disease. Hum Mol Genet. 2003;12:1199–210. doi: 10.1093/hmg/ddg125. [DOI] [PubMed] [Google Scholar]
- 42.Silverman EK, Palmer LJ, Mosley JD, et al. Genomewide linkage analysis of quantitative spirometric phenotypes in severe early-onset chronic obstructive pulmonary disease. Am J Hum Genet. 2002;70:1229–39. doi: 10.1086/340316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyle MP. Strategies for identifying modifier genes in cystic fibrosis. Proc Am Thorac Soc. 2007;4:52–7. doi: 10.1513/pats.200605-129JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 45.International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGall GH, Christians FC. High-density genechip oligonucleotide probe arrays. Adv Biochem Eng Biotechnol. 2002;77:21–42. doi: 10.1007/3-540-45713-5_2. [DOI] [PubMed] [Google Scholar]
- 47.Shen R, Fan JB, Campbell D, et al. High-throughput SNP genotyping on universal bead arrays. Mutat Res. 2005;573:70–82. doi: 10.1016/j.mrfmmm.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 48.Steemers FJ, Gunderson KL. Whole genome genotyping technologies on the BeadArray platform. Biotechnol J. 2007;2:41–9. doi: 10.1002/biot.200600213. [DOI] [PubMed] [Google Scholar]
- 49.Moffatt MF, Kabesch M, Liang L, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–3. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 50.The Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–41. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 53.Ioannidis JP, Trikalinos TA, Ntzani EE, Contopoulos-Ioannidis DG. Genetic associations in large versus small studies: an empirical assessment. Lancet. 2003;361:567–71. doi: 10.1016/S0140-6736(03)12516-0. [DOI] [PubMed] [Google Scholar]
- 54.Donnelly SC, Haslett C, Reid PT, et al. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat Med. 1997;3:320–3. doi: 10.1038/nm0397-320. [DOI] [PubMed] [Google Scholar]
- 55.Baugh JA, Bucala R. Macrophage migration inhibitory factor. Crit Care Med. 2002;30(suppl 1):S27–35. [PubMed] [Google Scholar]
- 56.Calandra T, Bernhagen J, Metz CN, et al. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 57.David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc Natl Acad Sci USA. 1966;56:72–7. doi: 10.1073/pnas.56.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lai KN, Leung JC, Metz CN, Lai FM, Bucala R, Lan HY. Role for macrophage migration inhibitory factor in acute respiratory distress syndrome. J Pathol. 2003;199:496–508. doi: 10.1002/path.1291. [DOI] [PubMed] [Google Scholar]
- 59.Gao L, Flores C, Ma SF, et al. Macrophage migration inhibitory factor in acute lung injury: expression, biomarker, and associations. Transl Res. 2007;150:18–29. doi: 10.1016/j.trsl.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao L, Grant A, Halder I, et al. Novel polymorphisms in the myosin light chain kinase gene confer risk for acute lung injury. Am J Respir Cell Mol Biol. 2006;34:487–95. doi: 10.1165/rcmb.2005-0404OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blumenthal MN, Langefeld CD, Beaty TH, et al. A genome-wide search for allergic response (atopy) genes in three ethnic groups: Collaborative Study on the Genetics of Asthma. Hum Genet. 2004;114:157–64. doi: 10.1007/s00439-003-1030-5. [DOI] [PubMed] [Google Scholar]
- 62.Brasch-Andersen C, Tan Q, Borglum AD, et al. Significant linkage to chromosome 12q24.32-q24.33 and identification of SFRS8 as a possible asthma susceptibility gene. Thorax. 2006;61:874–9. doi: 10.1136/thx.2005.055475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dizier MH, Besse-Schmittler C, Guilloud-Bataille M, et al. Genome screen for asthma and related phenotypes in the French EGEA study. Am J Respir Crit Care Med. 2000;162:1812–8. doi: 10.1164/ajrccm.162.5.2002113. [DOI] [PubMed] [Google Scholar]
- 64.Haagerup A, Bjerke T, Schiotz PO, Binderup HG, Dahl R, Kruse TA. Asthma and atopy—a total genome scan for susceptibility genes. Allergy. 2002;57:680–6. doi: 10.1034/j.1398-9995.2002.23523.x. [DOI] [PubMed] [Google Scholar]
- 65.Kurz T, Altmueller J, Strauch K, et al. A genome-wide screen on the genetics of atopy in a multiethnic European population reveals a major atopy locus on chromosome 3q21.3. Allergy. 2005;60:192–9. doi: 10.1111/j.1398-9995.2005.00646.x. [DOI] [PubMed] [Google Scholar]
- 66.Flores C, Ma SF, Maresso K, Ober C, Garcia JG. A variant of the myosin light chain kinase gene is associated with severe asthma in African Americans. Genet Epidemiol. 2007;31:296–305. doi: 10.1002/gepi.20210. [DOI] [PubMed] [Google Scholar]
- 67.Gao L, Grant AV, Rafaels N, et al. Polymorphisms in the myosin light chain kinase gene that confer risk of severe sepsis are associated with a lower risk of asthma. J Allergy Clin Immunol. 2007;119:1111–8. doi: 10.1016/j.jaci.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 68.Drumm ML, Konstan MW, Schluchter MD, et al. Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443–53. doi: 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- 69.McIntire JJ, Umetsu SE, Macaubas C, et al. Immunology: hepatitis A virus link to atopic disease. Nature. 2003;425:576. doi: 10.1038/425576a. [DOI] [PubMed] [Google Scholar]
- 70.Collins FS, Guyer MS, Charkravarti A. Variations on a theme: cataloging human DNA sequence variation. Science. 1997;278:1580–1. doi: 10.1126/science.278.5343.1580. [DOI] [PubMed] [Google Scholar]
- 71.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69:124–37. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cohen JC, Pertsemlidis A, Fahmi S, et al. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc Natl Acad Sci U S A. 2006;103:1810–5. doi: 10.1073/pnas.0508483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smith AJ, Tsalenko A, Sampas N, et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet. 2007;16:2783–94. doi: 10.1093/hmg/ddm208. [DOI] [PubMed] [Google Scholar]
- 75.Bansal V, Bashir A, Bafna V. Evidence for large inversion polymorphisms in the human genome from HapMap data. Genome Res. 2007;17:219–30. doi: 10.1101/gr.5774507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Manolio TA. Taking our obligations to research participants seriously: disclosing individual results of genetic research. Am J Bioeth. 2006;6:32–4. doi: 10.1080/15265160600935993. author reply W10–2. [DOI] [PubMed] [Google Scholar]
- 77.Manolio TA, Bailey-Wilson JE, Collins FS. Genes, environment and the value of prospective cohort studies. Nat Rev Genet. 2006;7:812–20. doi: 10.1038/nrg1919. [DOI] [PubMed] [Google Scholar]
- 78.Liu P, Wang Y, Vikis H, et al. Candidate lung tumor susceptibility genes identified through whole-genome association analyses in inbred mice. Nat Genet. 2006;38:888–95. doi: 10.1038/ng1849. [DOI] [PubMed] [Google Scholar]
- 79.Ohtsuka Y, Wang XT, Saito J, Ishida T, Munakata M. Genetic linkage analysis of pulmonary fibrotic response to silica in mice. Eur Respir J. 2006;28:1013–9. doi: 10.1183/09031936.06.00132505. [DOI] [PubMed] [Google Scholar]
- 80.Reinhard C, Meyer B, Fuchs H, et al. Genomewide linkage analysis identifies novel genetic Loci for lung function in mice. Am J Respir Crit Care Med. 2005;171:880–8. doi: 10.1164/rccm.200409-1204OC. [DOI] [PubMed] [Google Scholar]
- 81.Cowley AW, Jr, Liang M, Roman RJ, Greene AS, Jacob HJ. Consomic rat model systems for physiological genomics. Acta Physiol Scand. 2004;181:585–92. doi: 10.1111/j.1365-201X.2004.01334.x. [DOI] [PubMed] [Google Scholar]
- 82.Cowley AW, Jr, Roman RJ, Jacob HJ. Application of chromosomal substitution techniques in gene-function discovery. J Physiol. 2004;554:46–55. doi: 10.1113/jphysiol.2003.052613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Malek RL, Wang HY, Kwitek AE, et al. Physiogenomic resources for rat models of heart, lung and blood disorders. Nat Genet. 2006;38:234–9. doi: 10.1038/ng1693. [DOI] [PubMed] [Google Scholar]
- 84.Nonas SA, Vinasco LM, Ma SF, et al. Use of consomic rats for genomic insights into ventilator-associated lung injury. Am J Physiol Lung Cell Mol Physiol. 2007;293:L292–302. doi: 10.1152/ajplung.00481.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dwinell MR, Forster HV, Petersen J, et al. Genetic determinants on rat chromosome 6 modulate variation in the hypercapnic ventilatory response using consomic strains. J Appl Physiol. 2005;98:1630–8. doi: 10.1152/japplphysiol.01148.2004. [DOI] [PubMed] [Google Scholar]
- 86.Bonnet S, Michelakis ED, Porter CJ, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–41. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 87.Wang T, Hopkins D, Schmidt C, et al. Identification of genes differentially over-expressed in lung squamous cell carcinoma using combination of cDNA subtraction and microarray analysis. Oncogene. 2000;19:1519–28. doi: 10.1038/sj.onc.1203457. [DOI] [PubMed] [Google Scholar]
- 88.Tzouvelekis A, Patlakas G, Bouros D. Application of microarray technology in pulmonary diseases. Respir Res. 2004;5:26. doi: 10.1186/1465-9921-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Borczuk AC, Powell CA. Expression profiling and lung cancer development. Proc Am Thorac Soc. 2007;4:127–32. doi: 10.1513/pats.200607-143JG. [DOI] [PubMed] [Google Scholar]
- 90.Bull TM, Coldren CD, Geraci MW, Voelkel NF. Gene expression profiling in pulmonary hypertension. Proc Am Thorac Soc. 2007;4:117–20. doi: 10.1513/pats.200605-128JG. [DOI] [PubMed] [Google Scholar]
- 91.Chen ES, Moller DR. Expression profiling in granulomatous lung disease. Proc Am Thorac Soc. 2007;4:101–7. doi: 10.1513/pats.200607-140JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hansel NN, Diette GB. Gene expression profiling in human asthma. Proc Am Thorac Soc. 2007;4:32–6. doi: 10.1513/pats.200606-132JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lande JD, Patil J, Li N, Berryman TR, King RA, Hertz MI. Novel insights into lung transplant rejection by microarray analysis. Proc Am Thorac Soc. 2007;4:44–51. doi: 10.1513/pats.200605-110JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meyer NJ, Garcia JG. Wading into the genomic pool to unravel acute lung injury genetics. Proc Am Thorac Soc. 2007;4:69–76. doi: 10.1513/pats.200609-157JG. [DOI] [PubMed] [Google Scholar]
- 95.Wurfel MM. Microarray-based analysis of ventilator-induced lung injury. Proc Am Thorac Soc. 2007;4:77–84. doi: 10.1513/pats.200608-149JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zuo F, Kaminski N, Eugui E, et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc Natl Acad Sci U S A. 2002;99:6292–7. doi: 10.1073/pnas.092134099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Simon BA, Easley RB, Grigoryev DN, et al. Microarray analysis of regional cellular responses to local mechanical stress in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L851–61. doi: 10.1152/ajplung.00463.2005. [DOI] [PubMed] [Google Scholar]
- 98.Grigoryev DN, Ma SF, Irizarry RA, Ye SQ, Quackenbush J, Garcia JG. Orthologous gene-expression profiling in multi-species models: search for candidate genes. Genome Biol. 2004;5:R34. doi: 10.1186/gb-2004-5-5-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moreno-Vinasco L, Gomberg-Maitland M, Maitland M, et al. Genomic assessment of a multi- kinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol Genom. 2008 Feb 26; doi: 10.1152/physiolgenomics.00169.2007. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McConkie-Rosell A, Chen YT, Harris D, et al. Mild cystic fibrosis linked to chromosome 7q22 markers with an uncommon haplotype. Ann Intern Med. 1989;111:797–801. doi: 10.7326/0003-4819-111-10-797. [DOI] [PubMed] [Google Scholar]
- 101.Vandenbroeck K, Fiten P, Heggarty S, et al. Chromosome 7q21-22 and multiple sclerosis: evidence for a genetic susceptibility effect in vicinity to the protachykinin-1 gene. J Neuroimmunol. 2002;125:141–8. doi: 10.1016/s0165-5728(02)00023-1. [DOI] [PubMed] [Google Scholar]
- 102.van Heel DA, Fisher SA, Kirby A, Daly MJ, Rioux JD, Lewis CM. Inflammatory bowel disease susceptibility loci defined by genome scan meta-analysis of 1952 affected relative pairs. Hum Mol Genet. 2004;13:763–70. doi: 10.1093/hmg/ddh090. [DOI] [PubMed] [Google Scholar]
- 103.Garcia JG. Searching for candidate genes in acute lung injury: SNPs, Chips and PBEF. Trans Am Clin Climatol Assoc. 2005;116:205–19. [PMC free article] [PubMed] [Google Scholar]
- 104.Ye SQ, Simon BA, Maloney JP, et al. Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med. 2005;171:361–70. doi: 10.1164/rccm.200404-563OC. [DOI] [PubMed] [Google Scholar]
- 105.Ye SQ, Zhang LQ, Adyshev D, et al. Pre-B-cell-colony-enhancing factor is critically involved in thrombin-induced lung endothelial cell barrier dysregulation. Microvasc Res. 2005;70:142–51. doi: 10.1016/j.mvr.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 106.Bajwa EK, Yu CL, Gong MN, Thompson BT, Christiani DC. Pre-B-cell colony-enhancing factor gene polymorphisms and risk of acute respiratory distress syndrome. Crit Care Med. 2007;35:1290–5. doi: 10.1097/01.CCM.0000260243.22758.4F. [DOI] [PubMed] [Google Scholar]
- 107.Chittur SV. DNA microarrays: tools for the 21st Century. Comb Chem High Throughput Screen. 2004;7:531–7. doi: 10.2174/1386207043328454. [DOI] [PubMed] [Google Scholar]