

Abstract
There is mounting evidence to suggest that the epigenetic reprogramming capacity of the oocyte is superior to that of the current factor-based reprogramming approaches and that some factor-reprogrammed induced pluripotent stem cells (iPSCs) retain a degree of epigenetic memory that can influence differentiation capacity and may be linked to the observed expression of immunogenicity genes in iPSC derivatives. One hypothesis for this differential reprogramming capacity is the “chromatin loosening/enhanced reprogramming” concept, as previously described by John Gurdon and Ian Wilmut, as well as others, which postulates that the oocyte possesses factors that loosen the somatic cell chromatin structure, providing the epigenetic and transcriptional regulatory factors more ready access to repressed genes and thereby significantly increasing epigenetic reprogramming. However, to empirically test this hypothesis a list of candidate oocyte reprogramming factors (CORFs) must be ascertained that are significantly expressed in metaphase II oocytes. Previous studies have focused on intraspecies or cross-species transcriptional analysis of up to two different species of oocytes. In this study, we have identified eight CORFs (ARID2, ASF1A, ASF1B, DPPA3, ING3, MSL3, H1FOO, and KDM6B) based on unbiased global transcriptional analysis of oocytes from three different species (human, rhesus monkey, and mouse) that both demonstrate significant (p<0.05, FC>3) expression in oocytes of all three species and have well-established roles in loosening/opening up chromatin structure. We also identified an additional 15 CORFs that fit within our proposed “chromatin opening/fate transformative” (COFT) model. These CORFs may be able to augment Shinya Yamanaka's previously identified reprogramming factors (OCT4, SOX2, KLF4, and cMYC) and potentially facilitate the removal of epigenetic memory in iPSCs and/or reduce the expression of immunogenicity genes in iPSC derivatives, and may have applications in future personalized pluripotent stem cell based therapeutics.
Introduction
Global epigenetic (Bar-Nur et al., 2011; Kim et al., 2010; Kim et al., 2011; Wakayama et al., 2006) analysis provides evidence in support of the hypothesis that the mammalian metaphase II oocyte possesses a superior capacity to epigenetically reprogram somatic cell nuclei towards an embryonic stem cell (ESC)-like state than the current factor-based reprogramming approaches. The significance of this putative incomplete factor-based reprogramming for future patient-specific cellular therapeutics was increased when factor-reprogrammed isogenic cells recently demonstrated a T cell-dependent immune response upon transplantation into a perfectly matched (syngeneic) mouse, a phenomena not seen in syngeneic transplantation of ESCs (Zhao et al., 2011). One hypothesis for this differential reprogramming capacity is that the oocyte possesses specific factors that are lacking in the current factor-based reprogramming approaches and that it may be possible using factors identified from oocytes to recapitulate the oocyte's putative superior epigenetic reprogramming capacity (Fig. 1). John Gurdon, Ian Wilmut, and others have previously suggested that the key reprogramming factors in the oocyte may be involved in loosening somatic chromatin and thereby providing the transcriptional regulatory apparatus access to repressed genes (Gurdon and Wilmut, 2011).
FIG. 1.

CORF-based reprogramming concept. Oocyte-based reprogramming involves placing the somatic cell nucleus into an enucleated metaphase II oocyte. Factors in the ooplasm can induce complete epigenetic reprogramming, and subsequent ESCs derived from oocyte-reprogrammed (somatic cell nuclear transfer) embryos (blastocysts) demonstrate similar epigenetic patterns to ESCs derived from fertilized blastocysts (fESCs). Standard factor-based reprogramming involves exposing the somatic cell nucleus directly to standard iPSC-Fs, such as OCT4, SOX2, KLF4, and cMYC. These factors can induce reprogramming of the somatic cell nucleus into iPSCs, but there is mounting evidence that these cells do not always possess the same epigenetic patterns or differentiation potential as fESCs. The CORF-augmented reprogramming model involves using both iPSC-Fs and CORFs at the same time to investigate if the additional CORF factors will open up the chromatin and significantly increase reprogramming of augmented CORF-iPSCs toward a fESC epigenetic pattern. The CORF-dynamic model involves first using CORFs to reprogram the somatic cell nucleus back into an oocyte-specific (totipotent) epigenetic state, followed by exposure to the iPSC-Fs to differentiate these totipotent cells into CORF-iPSCs to investigate if this approach will significantly increase reprogramming of dynamic CORF-iPSCs toward a fESC epigenetic pattern.
Here, we propose that in addition to oocyte-based factors that open up/loosen chromatin, the key candidate oocyte reprogramming factors (CORFs) may also include factors that promote a transformation in cell fate, which we refer to as the “chromatin opening/fate transformative” (COFT) model. Whether CORFs are selected based exclusively on their known ability to loosen chromatin, using our expanded COFT model, or based on additional considerations, we propose that there are two basic approaches for future usage of CORFs in reprogramming experiments—the CORF-augmented approach and the CORF-dynamic approach. The CORF-augmented reprogramming model involves including CORFs and induced pluripotent stem cell factors (iPSC-Fs), such as OCT4, SOX2, KLF4, and cMYC (Takahashi and Yamanaka, 2006; Takahashi et al., 2007), together with the hypothesis that the CORFs will augment the reprogramming capacity of the iPSC-Fs, either by opening up the chromatin to be more accessible to epigenetic reprogramming and/or through an as of yet unidentified mechanism, and generate an epigenetic and transcriptional landscape that is closer to ESCs. The CORF-dynamic reprogramming model involves using CORFs initially to reprogram the somatic cell back into an oocyte-specific (totipotent) epigenetic state and then transition to iPSC-Fs to differentiate the totipotent cells into pluripotent stem cells that more closely resemble ESCs (Fig. 1). One of the potential benefits of generating CORF-iPSCs that are fully reprogrammed back into an ESC-like epigenetic state is that their derivatives may not express immunogenicity gene expression, as has been observed for iPSC-derivatives (Zhao et al., 2011). However, to test these models empirically, a list of CORFs must be ascertained. The COFT model proposed here suggests that the key oocyte factors will play a role in either remodeling the chromatin architecture to an euchromatic state to be accessed by transcriptional regulators and/or through promotion of a transformation in cellular fate, preferentially toward an oocyte/totipotent or stem cell/pluripotent epigenetic state.
Previous research has focused on identifying CORFs based on intraspecies (Dobson et al., 2004) or interspecies analysis using up to two different species (Kocabas et al., 2006). In this study, global transcriptional meta-analysis was performed on human, rhesus monkey, and mouse metaphase II oocytes in comparison to their respective adult dermal fibroblast transcriptomes, representing the first multispecies global transcriptional analysis of metaphase II oocytes for identification of putative CORFs. We identified a set of 23 CORFs using the COFT model criteria that demonstrated significantly increased expression in oocytes from all three species, and of those 23 CORFs, we propose that eight CORFs (ARID2, ASF1A, ASF1B, DPPA3, ING3, MSL3, H1FOO, and KDM6B) possess a function that most closely correlates with the “chromatin loosening/enhanced reprogramming” concept as previously proposed by Gurdon and Wilmut.
Materials and Methods
Ethics statement
Written approvals for human skin biopsy procedures, human fibroblast derivation, culture, and experimental use was obtained from the Stanford University Institutional Review Board, the Stanford University Stem Cell Research Oversight (SCRO) committee, and written informed consent was obtained from each individual participant. Biopsy material used in this study was obtained and initially analyzed at Stanford University, as previously described (Byrne et al., 2009) and transferred to UCLA through a material transfer agreement. Written approvals for the experiments performed in this study were obtained from the UCLA Institute Biosafety Committee and UCLA SCRO committee.
Cell culture
Human adult dermal fibroblasts were cultured in complete Dulbecco's modified Eagle medium (DMEM)/Mixture F-12, supplemented with 10% fetal bovine serum (FBS), 1× minimal essential medium (MEM) nonessential amino acids, 1× Glutamax, and 100 IU/mL penicillin-streptomycin (all from Invitrogen/Gibco, Grand Island, NY, USA) and maintained at 37°C in a 5% CO2 incubator. Culture medium was changed every 2 days. Cells were allowed to expand to 80–90% confluency before passaging with 0.05% trypsin-EDTA (Invitrogen) and replated at a 1:3 ratio. A large bank of early passage cells was cryopreserved in culture medium supplemented with 10% dimethylsulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA). All research adhered to National Academy of Sciences guidelines. H9 human embryonic stem cells (hESCs) (UCLA Broad Stem Cell Research Center-Stem Cell Core) were cultured in standard ESC conditions, as previously described (Byrne et al., 2009). Briefly, hESC medium consisted of DMEM/F12 supplemented with 20% Knockout Serum Replacement (KSR), 1× Glutamax, 1× nonessential amino acids, 100 IU/mL penicillin-streptomycin (all from Invitrogen), 1× β-mercaptoethanol (Millipore, Billerica, MA, USA), and 10 ng/mL recombinant human basic fibroblast growth factor (bFGF; Globalstem, Rockville, MD, USA).
Global transcriptional meta-analysis
All cultured cells (including adult dermal fibroblasts and human ESCs) were harvested for total mRNA extraction using the High Pure RNA Isolation Kit according to manufacturer's instructions (Roche, Indianapolis, IN, USA). Microarray analysis was carried out as published (Crespo et al., 2012). Briefly, total RNA was used for an Affymetrix Differential Gene Expression Assay Human Genome U133 Plus 2.0 Array (Genoseq UCLA) for global transcriptional analysis using standard Affymetrix protocols (Affymetrix GeneChip Expression Analysis Technical Manual, rev.3. 2001). Uploading and cluster analysis of the CEL files between replicate samples was through GeneSifter (VizX Labs, www.geospiza.com) using the Advanced Upload Method and normalized using the Affymetrix Microarray Analysis Suite (MAS) 5.0 algorithm. All microarray data were deposited in the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/projects/geo/). Affymetrix CEL files for all other human, rhesus monkey, and mouse cells analyzed in this study were obtained from GEO. Each CEL file was generated through analysis of the cells' total RNA hybridized to the species-relevant Affymetrix GeneChip Array, with the U133 Plus 2.0 Arrays used for human cells, the Rhesus Arrays used for rhesus monkey cells, and the 430 2.0 Mouse Arrays used for mouse cells. The following CEL files (with their respective GEO accession numbers) were analyzed and compared in this study: human metaphase II oocytes (GSM304261, GSM304262, GSM136512, GSM136513, GSM136519, GSM136525, GSM288812, GSM288876), human adult dermal fibroblasts (GSM994327, GSM994328, GSM301264, GSM301265, GSM288223, GSM288224, GSM288225, GSM288226), human embryonic stem cells (GSM994321, GSM994322, GSM194307, GSM194308, GSM378813, GSM378818, GSM462819, GSM462820), rhesus monkey metaphase II oocytes (GSM300529, GSM300530), rhesus monkey adult dermal fibroblasts (GSM187389, GSM187390), mouse metaphase II oocytes (GSM132659, GSM132660), and mouse adult dermal fibroblasts (GSM106139, GSM106141).
Data analysis
Each CEL file was uploaded to GeneSifter (VizX Labs, Seattle, WA) using the Advanced Upload Method and normalized using the Affymetrix MAS 5.0 algorithm. Cluster analysis between all human samples was performed through GeneSifter Project Analysis using analysis of variance (ANOVA) statistical analysis (p<0.01, threshold >100, Manhattan distance, ward linkage, and gene row centering). GeneSifter pairwise analysis between oocytes and fibroblasts for each species was performed using all mean normalization and t-test statistical analysis (p<0.01). For each pairwise analysis, between two to eight biological replicates from each cell line or tissue type were used, dependent on availability. Probe sets were considered to be significantly upregulated (compared to the species-specific adult dermal fibroblast baseline) when the p value was <0.01 and fold change was equal or greater than 3. When duplicate probe sets or genes were identified, the duplicates with the lower fold change were removed. Gene ontology analysis for biological processes was performed in GeneSifter on the significantly upregulated probe sets. CORFs were identified on the basis of both demonstrating significantly increased expression in oocytes from all three species and possessing a function that fit within the COFT reprogramming model.
Results
Investigating interexperimental variability
To ensure that interexperimental variability, such as cell line variability and differences manifested in experimental design, would not contribute to false positives in identifying differences in gene expression, analysis of the variability between samples harvested from separate experiments was taken into account. Cluster analysis was performed using eight biological replicates each of human metaphase II oocytes, human adult dermal fibroblasts, and human ESCs. Despite the materials being derived from a number of different experiments, we observed cell-type specific clustering for each of the cell types analyzed across all experiments (Fig. 2A) and clusters of cell-specific gene expression (Fig. 2B), suggesting that interexperimental variability was significantly lower than the intrinsic similarities for cell type–specific transcriptomes. This result was used as the foundation to justify the use of materials obtained from multiple experiments in subsequent pairwise comparison analyses.
FIG. 2.

Global transcriptional analysis of human samples. Global gene expression cluster analysis of human oocyte, ESC, and adult dermal fibroblast samples from different experiments demonstrating cell type–specific clustering and clustering of cell type specific genes.
Identifying putative human CORFs
Before cross-species specific analysis was applied, we established a baseline of upregulated genes that would serve as the foundation for putative CORFs in the human. Pairwise analysis of the eight biological replicates of the human metaphase II oocytes was compared to the eight biological replicates of the human dermal fibroblasts. Gene ontological analysis and filtering was performed on the significantly upregulated genes, and 404 human putative CORFs were identified based on their possession of a function in chromatin remodeling, transcriptional regulation, and/or having previously been associated with a stem cell-like state (see Table S1) (Supplementary Data are available at www.liebertpub.com/cell/).
Cross-species analysis of putative CORFs
In an effort to further narrow down potential CORF candidates, the putative human CORFs identified in the gene ontological analysis were subjected to cross-species analysis to identify overlapping upregulated genes that could be identified as cross-species specific CORFs. Pairwise analysis was repeated for both rhesus monkey and mouse metaphase II oocytes in comparison analysis with their respective adult dermal fibroblasts; and following the same gene ontological filtering, a list of 377 rhesus monkey putative CORFs (Table S2) and 399 mouse CORFs (Table S3) were identified. Cross-species analysis of the various putative CORFs from all species was performed, and 48 species-independent putative CORFs were identified (Fig. 3, Table S4). Background research was performed on these putative CORFs, and a final list of 23 CORFs was identified that met all of the CORF-criteria included in the COFT model (Table 1). Specifically, these 23 factors possessed a function in either remodeling the chromatin architecture to loosen/open it up to be accessed by transcriptional regulators and/or through promotion of a transformation in cellular fate, preferentially toward an oocyte/totipotent or stem cell/pluripotent epigenetic state (Table 1). These 23 factors included factors that remodel and open up chromatin. They include:
FIG. 3.
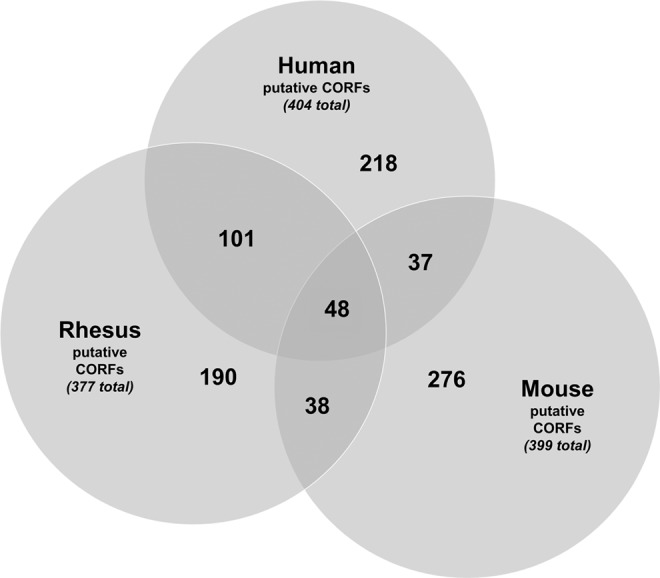
Identification of species-independent putative CORFs. Analysis of overlap between human, rhesus monkey, and mouse putative CORFs.
Table 1.
Candidate Oocyte Reprogramming Factors (CORFs)
| |
Fold change in expressionacompared to fibroblasts in: |
|
|
||
|---|---|---|---|---|---|
| Gene (aka) | Human oocytes | Rhesus oocytes | Mouse oocytes | Function | Reference |
| AFAP1L2 (XB130) | 5.5 | 155 | 130 | Contributes to SRC-regulated transcriptional activation | Xu et al. (2007) |
| ARID2 (BAF200)b | 19 | 8.2 | 13 | Activates gene expression through the PBAF chromatin remodeling complex | Xu et al. (2012) |
| ASF1A (CIA) | 6.6 | 17 | 4.8 | Histone chaperone that cooperates with CAF-1 in pluripotent embryonic cells | Houlard et al. (2006); GeneCards.org (2012) |
| ASF1B (CIA-II) | 4.0 | 12 | 8.8 | Histone chaperone that cooperates with CAF-1 in pluripotent embryonic cells | Houlard et al. (2006); GeneCards.org (2012) |
| BRDT (BRD6) | 7.6 | 120 | 280 | Reorganization of acetylated chromatin in germ cells | Pivot-Pajot et al. (2003) |
| DPPA3 (STELLA) | 370 | 650 | 110 | Critical component involved with altering chromatin structure in the oocyte | Liu et al. (2012) |
| DPPA5 (ESG1) | 457 | 235 | 3.3 | Pluripotency-associated factor | Kim et al. (2005) |
| ERG (p55) | 6.6 | 65 | 120 | Transformation-specific transcription factors | Tsuzuki et al. (2011) |
| FOXK2 (ILF1) | 14 | 6.5 | 4.5 | Promotes AP-1–mediated transcriptional regulation throughout genome | Ji et al. (2012) |
| H1FOO | 320 | 18 | 480 | Replaces linker H1 histones and induces embryonic chromatin structure | Becker et al. (2005) |
| HHEX | 40 | 190 | 7.4 | Transcription factor important for embryonic development | GeneCards.org (2012) |
| ING3 | 11 | 14 | 13 | Transcriptional activation of select genes principally by acetylation of H4 and H2A | GeneCards.org (2012) |
| KDM6B (JMJD3) | 21 | 10 | 17 | Histone demethylase that specifically demethylates 'Lys-27' of histone H3 | Aoto et al. (2008) |
| LEF1 (TCF10) | 61 | 45 | 6.9 | Transcriptionally activates MYC and enhances cell proliferation | GeneCards.org (2012) |
| MLL3 (HALR) | 21 | 31 | 41 | Methylates 'Lys-4' of histone H3 and essential in maintaining stem cell state | Jude et al. (2007) |
| MSL3 | 4.4 | 56 | 7.2 | Acetylation of histone H4 at 'Lys-16', which opens up chromatin | Shogren-Knaak et al. (2006) |
| NCOA3 (SRC-3) | 4.5 | 9.2 | 16 | Remodels chromatin through histone acetyltransferase activity | GeneCards.org (2012) |
| NFATC2 | 10 | 39 | 8.1 | Induces lymphocyte proliferation | Caetano et al. (2002) |
| NR5A2 (FTF) | 3.6 | 94 | 26 | Transcription factor that can replace OCT4 in reprogramming | Heng et al. (2010) |
| POU4F1 (BRN3A) | 13 | 250 | 143 | Transcription factor associated with cancer (acute myeloid leukemia) | Fortier et al. (2010) |
| RPS6KA5 (MSK1) | 9.6 | 89 | 8.4 | Contributes to gene activation by histone phosphorylation | GeneCards.org (2012) |
| TADA2L (ADA2A) | 5.3 | 4.7 | 3.1 | Histone H3 and H4 acetyltransferase activity | (GeneCards.org2012) |
| TAF4B (TAF2C2) | 21 | 9.2 | 4.2 | Gene-selective coactivator in certain cells and activates antiapoptotic genes | GeneCards.org (2012) |
All fold changes represented a significant (p<0.05) three-fold or greater increase in detected gene expression in the relevant species metaphase II oocytes compared to their adult dermal fibroblasts.
The eight genes highlighted in bold represent the CORFs most closely associated with the “Chromatin loosening” concept as proposed by John Gurdon and Ian Wilmut (Gurdon and Wilmut 2011) and others.
ARID2, which plays a key role in activating gene expression through the PBAF chromatin remodeling complex (Xu et al., 2012);
ASF1A and ASF1B, which are histone-remodeling chaperones that cooperate with chromatin assembly factor 1 (CAF-1), which plays a key role in remodeling chromatin in pluripotent embryonic cells (Houlard et al., 2006; GeneCards.org 2012);
BRDT, which plays a role in the reorganization of acetylated chromatin in germ cells (Pivot-Pajot et al., 2003);
DPPA3 and DPPA5, which are pluripotency-associated factors, with DPPA3 in particular playing a known role in altering chromatin structure in oocytes (Kim et al., 2005; Liu et al., 2012);
RPS6KA5, which contributes to gene activation by histone phosphorylation (GeneCards.org 2012);
TADA2L, a component of the ATAC complex, which has histone acetyltransferase (HAT) activity on histones H3 and H4 (GeneCards.org 2012);
ING3, a component of the NuA4 HAT complex that is involved in transcriptional activation of select genes principally by acetylation of nucleosomal histones H4 and H2A (GeneCards.org 2012);
MLL3, which activates transcription through methylation of 'Lys-4' of histone H3 and is essential in maintaining the hematopoietic stem cell state (Jude et al., 2007);
MSL3, a component of the MSL complex that is responsible for the majority of histone H4 acetylation at 'Lys-16', which is implicated in the formation of a more open chromatin structure, specifically by inhibiting the formation of compact 30-nanometer–like fibers and impeding the ability of chromatin to form cross-fiber interactions (Shogren-Knaak et al., 2006);
NCOA3, a nuclear receptor co-activator that displays HAT activity (GeneCards.org 2012);
H1FOO, the oocyte-specific linker histone that has greater mobility than somatic histones and plays a key role in generating the increased instability of the embryonic chromatin structure following fertilization and somatic cell nuclear transfer (Becker et al., 2005); and
KDM6B, a histone demethylase that specifically demethylates 'Lys-27' of histone H3 and thereby prevents the formation of repressive chromatin through polycomb group (PcG) protein complex PRC1 binding (Aoto et al., 2008).
Also included are oocyte-expressed transcription factors that promote global epigenetic transformation and/or reprogramming to a stem cell-like state, such as:
FOXK2, which promotes activation protein 1 (AP-1)-mediated transcriptional regulation throughout the genome (Ji et al., 2012);
NR5A2, a transcription factor that can replace OCT4 in reprogramming somatic cells into iPSCs (Heng et al., 2010);
TAF4B, which functions as a gene-selective co-activator in certain cells and is involved in the activation of antiapoptotic genes (GeneCards.org 2012);
HHEX, a transcription factor important for embryonic development (GeneCards.org 2012);
LEF1, which transcriptionally activates MYC and CCND1 expression and enhances cell proliferation (GeneCards.org 2012);
ERG, a transformation-specific transcription factor that promotes and maintains leukemia (Tsuzuki et al., 2011);
NFATC2, which induces lymphocyte proliferation (Caetano et al., 2002);
POU4F1, a transcription factor associated with cancer (acute myeloid leukemia) (Fortier et al., 2010); and
AFAP1L2, which contributes to SRC-regulated transcriptional activation (Xu et al., 2007).
Discussion
IPSCs have significant promise for cell replacement therapy, but some iPSCs demonstrate epigenetic memory and derivative immunogenicity gene expression that could negatively impact their clinical application. Studies comparing the DNA methylomes of mouse and human iPSCs with their respective species-specific ESCs discovered many iPSC lines retained aberrant iPSC-specific differential methylation patterns, a phenomenon referred to as “epigenetic memory” (Kim et al., 2010; Kim et al., 2011). The epigenetic memory of iPSCs was observed to impair the differentiation capacity of the iPSCs, with iPSCs demonstrating a reduced capacity to differentiate into cells from lineages different to the donor cell type (Kim et al., 2010; Kim et al., 2011). There is significant evidence that most, if not all, iPSC lines can gradually resolve at least some, if not most, of their transcriptional and epigenetic differences with ESCs with increased passaging (Polo et al., 2010). However, it has also been observed that a subset of iPSC lines nevertheless retain epigenetic memory, even following extended passaging (Kim et al., 2011).
How significant a challenge this residual epigenetic memory will pose for future autologous cellular therapeutics is unclear. However, it may be prudent to consider erring on the side of caution and continuing to investigate novel augmented nuclear reprogramming approaches that may be able to both help remove residual epigenetic memory, regardless of passage, as well as potentially augment the nuclear reprogramming process, increasing the overall feasibility of the human iPSC-based approach. There is mounting evidence to suggest that the epigenetic reprogramming capacity of the oocyte is superior to that of the current factor-based reprogramming approaches. One hypothesis for this differential reprogramming capacity postulates that the oocyte possesses factors that loosen the somatic cell chromatin structure, providing the epigenetic and transcriptional regulatory factors more ready access to repressed genes and thereby significantly augmenting epigenetic reprogramming. These CORFs may be able to loosen chromatin during the reprogramming process and thereby result in “CORF-iPSCs” with significantly lower levels of epigenetic memory and derivative immunogenicity gene expression. It is also possible that incorporating CORFs will also speed up the epigenetic reprogramming toward pluripotency and/or enhance the overall percentage of cells that attain pluripotency, although none of these things have been investigated to date. If CORF-iPSCs could be generated with less epigenetic memory and/or lower/no derivative immunogenicity gene expression, this may make them better sources of autologous pluripotent material than the current iPSC-generation approaches and thus represent a potentially transformative impact upon personalized pluripotent stem cell-based regenerative medicine and an important early translational consideration for future iPSC-based therapeutics.
However, the key first step toward the generation of CORF-iPSCs is the identification of putative CORFs. In this study, global transcriptional meta-analysis was performed on human, rhesus monkey, and mouse metaphase II oocytes in comparison to their respective adult dermal fibroblast transcriptomes and a set of 23 CORFs was identified that were shown to have significantly increased expression in oocytes from all three species and to possess a function that fit within the COFT model. Of these 23 CORFs, eight possess a function that most closely correlates with the “chromatin loosening/enhanced reprogramming” hypothesis.
There are several considerations to discuss in regard to this type of multispecies in silico study. First, we used data that were obtained from several different groups, including our own, and a possible concern is that a potentially significant proportion of the transcriptional differences observed will be due to lab-specific variables not shared across multiple research groups. However, this does not appear to have significantly affected the results of this global transcriptional meta-analysis study because we observed cell-type specific clustering for all samples analyzed. The second consideration is that this type of in silico transcriptional analysis approach lacks the primary biological material to perform quantitative RT-PCRs for these CORFs. Thus, these CORFs must be considered as provisional until they are confirmed using RT-PCR. The third consideration is that we have made several underlying assumptions in our CORF-identification approach that may not be correct, including our hypothesis that CORFs will be strongly expressed and will either open chromatin or transform cellular fate.
We acknowledge that it is certainly possible that some very important reprogramming factors may be expressed at relatively low levels undetectable to microarray-based analysis or may not function in a role that opens chromatin or transforms cellular fate. The CORF-identification approach performed in this study may not be optimal, and alternative approaches, such as those that incorporate embryonic stem cells into the meta-analysis (Kocabas et al., 2006) may provide a superior transcriptional approach toward CORF identification. The fourth consideration is that we propose the hypothesis that the underlying reprogramming mechanism will be maintained across oocytes from different mammalian species, specifically human, rhesus monkey, and mouse. However, it is certainly possible that there are species-specific reprogramming molecules that would be eliminated by our multispecies conserved factor-identification approach, and perhaps the previously reported intraspecies analysis (Dobson et al., 2004) would provide a superior CORF-identification approach.
The final consideration of a transcriptional-based approach toward putative CORF identification is that it does not analyze alternatives to messenger RNA (mRNA) transcripts in the oocyte, such as stable long half-life proteins, microRNAs, and intrinsic physical variables of the oocytes, such as a large amount of ooplasm to quickly dilute somatic cell factors posttransplantation (Byrne, 2011). Nevertheless, this study provides an important first step toward a transcriptional-based identification of species-independent CORFs, which represents the first such multispecies transcriptional analysis of potential oocyte-reprogramming factors and an important foundation for testing the CORF-augmented and CORF-dynamic reprogramming models, using either vectors (Takahashi et al., 2007) or synthetic mRNAs (Warren et al., 2010). With regard to the CORF-dynamic reprogramming model, this will be facilitated by the usage of synthetic mRNAs as the mixture that is transfected into the somatic cell can simply be transitioned over time from a CORF mixture toward an iPSC-F mixture.
We propose that an important next step will be the derivation, global epigenetic analysis (Irizarry et al., 2008; Kim et al., 2010; Kim et al., 2011), and derivative immunogenicity gene expression analysis (Zhao et al., 2011) of CORF-iPSCs in comparison with iPSCs generated using standard approaches (Lowry et al., 2008; Park et al., 2008; Takahashi et al., 2007; Yu et al., 2007). If CORF-augmented iPSCs demonstrate significantly lower levels of epigenetic memory and derivative immunogenicity gene expression, we would then propose that CORF-iPSCs may have significant promise for future personalized pluripotent stem cell based therapeutics.
In conclusion, we have identified 23 CORFs that are significantly detected in human, rhesus monkey, and mouse metaphase II oocytes, eight of which are associated with chromatin “loosening.” These CORFs represent a foundation for future research to investigate the CORF-augmented and CORF-dynamic factor-reprogramming approaches and may provide an important step toward generating immune-compatible patient-specific iPSC-based cellular therapeutics.
Supplementary Material
Acknowledgments
This work is supported by funding to J.B. from the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA, The Phelps Family Foundation, and a UCLA Clinical & Translational Science Institute (CTSI) Scholar's Award (NCATS grant no. UL1TR000124).
Author Disclosure Statement
The authors have no conflicts of interest or competing financial interests to report.
References
- Aoto T. Saitoh N., et al. Polycomb group protein-associated chromatin is reproduced in post-mitotic G1 phase and is required for S phase progression. J. Biol. Chem. 2008;283:18905–18915. doi: 10.1074/jbc.M709322200. [DOI] [PubMed] [Google Scholar]
- Bar-Nur O. Russ H.A., et al. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell. 2011;9:17–23. doi: 10.1016/j.stem.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Becker M. Becker A., et al. Differential in vivo binding dynamics of somatic and oocyte-specific linker histones in oocytes and during ES cell nuclear transfer. Mol. Biol. Cell. 2005;16:3887–3895. doi: 10.1091/mbc.E05-04-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne J. Global transcriptional analysis of oocyte-based and factor-based nuclear reprogramming in the nonhuman primate. Cell. Reprogram. 2011;13:473–481. doi: 10.1089/cell.2011.0033. [DOI] [PubMed] [Google Scholar]
- Byrne J.A. Nguyen H.N., et al. Enhanced generation of induced pluripotent stem cells from a subpopulation of human fibroblasts. PLoS One. 2009;4:e7118. doi: 10.1371/journal.pone.0007118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caetano M.S. Vieira-de-Abreu A., et al. NFATC2 transcription factor regulates cell cycle progression during lymphocyte activation: Evidence of its involvement in the control of cyclin gene expression. FASEB J. 2002;16:1940–1942. doi: 10.1096/fj.02-0282fje. [DOI] [PubMed] [Google Scholar]
- Crespo A.V. Awe J.P., et al. Human skin cells that express stage-specific embryonic antigen 3 associate with dermal tissue regeneration. BioResearch Open Access. 2012;1:25–33. doi: 10.1089/biores.2012.0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson A.T. Raja R., et al. The unique transcriptome through day 3 of human preimplantation development. Hum. Mol. Genet. 2004;13:1461–1470. doi: 10.1093/hmg/ddh157. [DOI] [PubMed] [Google Scholar]
- Fortier J.M. Payton J.E., et al. POU4F1 is associated with t(8;21) acute myeloid leukemia and contributes directly to its unique transcriptional signature. Leukemia. 2010;24:950–957. doi: 10.1038/leu.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- “The GeneCards Human Gene Database.”. GeneCards.org. 2012. www.genecards.org/ GeneCards.orgwww.genecards.org/
- Gurdon J.B. Wilmut I. Nuclear transfer to eggs and oocytes. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a002659.a002659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng J.C. Feng B., et al. The nuclear receptor Nr5a2 can replace Oct4 in the reprogramming of murine somatic cells to pluripotent cells. Cell Stem Cell. 2010;6:167–174. doi: 10.1016/j.stem.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Houlard M. Berlivet S., et al. CAF-1 is essential for heterochromatin organization in pluripotent embryonic cells. PLoS Genet. 2006;2:e181. doi: 10.1371/journal.pgen.0020181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R.A. Ladd-Acosta C., et al. Comprehensive high-throughput arrays for relative methylation (CHARM) Genome Res. 2008;18:780–790. doi: 10.1101/gr.7301508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z. Donaldson I.J., et al. The forkhead transcription factor FOXK2 promotes AP-1-mediated transcriptional regulation. Mol. Cell. Biol. 2012;32:385–398. doi: 10.1128/MCB.05504-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jude C.D. Climer L., et al. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1:324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. Doi A., et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. Zhao R., et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011;29:1117–1119. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.K. Suh M.R., et al. Identification of developmental pluripotency associated 5 expression in human pluripotent stem cells. Stem Cells. 2005;23:458–462. doi: 10.1634/stemcells.2004-0245. [DOI] [PubMed] [Google Scholar]
- Kocabas A.M. Crosby J., et al. The transcriptome of human oocytes. Proc. Natl. Acad. Sci. USA. 2006;103:14027–14032. doi: 10.1073/pnas.0603227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.J. Nakamura T., et al. Essential role of DPPA3 for chromatin condensation in mouse oocytogenesis. Biol. Reprod. 2012;86:40. doi: 10.1095/biolreprod.111.095018. [DOI] [PubMed] [Google Scholar]
- Lowry W.E. Richter L., et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc. Natl. Acad. Sci. USA. 2008;105:2883–2888. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.H. Zhao R., et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Pivot-Pajot C. Caron C., et al. Acetylation-dependent chromatin reorganization by BRDT, a testis-specific bromodomain-containing protein. Mol. Cell. Biol. 2003;23:5354–5365. doi: 10.1128/MCB.23.15.5354-5365.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo J.M. Liu S., et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010;28:848–855. doi: 10.1038/nbt.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M. Ishii H., et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311:844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- Takahashi K. Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K. Tanabe K., et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tsuzuki S. Taguchi O., et al. Promotion and maintenance of leukemia by ERG. Blood. 2011;117:3858–3868. doi: 10.1182/blood-2010-11-320515. [DOI] [PubMed] [Google Scholar]
- Wakayama S. Jakt M.L., et al. Equivalency of nuclear transfer-derived embryonic stem cells to those derived from fertilized mouse blastocysts. Stem Cells. 2006;24:2023–2033. doi: 10.1634/stemcells.2005-0537. [DOI] [PubMed] [Google Scholar]
- Warren L. Manos P.D., et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F. Flowers S., et al. Essential role of ARID2 protein-containing SWI/SNF complex in tissue-specific gene expression. J. Biol. Chem. 2012;287:5033–5041. doi: 10.1074/jbc.M111.279968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. Bai X.H., et al. XB130, a novel adaptor protein for signal transduction. J. Biol. Chem. 2007;282:16401–16412. doi: 10.1074/jbc.M701684200. [DOI] [PubMed] [Google Scholar]
- Yu J. Vodyanik M.A., et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zhao T. Zhang Z.-N., et al. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–215. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.