

Abstract
Forced exogenous gene expression has been well characterized as an effective method for directing both cellular differentiation and dedifferentiation. However, transgene expression is not amenable for therapeutic application due to potential insertional mutagenesis. Protein-based techniques provide a safe alternative, but current protein delivery methods are quite limited by labor-intensive purification processes, low protein yield, and inefficient intracellular targeting. Such limitations may be overcome by using a naturally occurring bacterial protein injection system, called the type III secretion system (T3SS), which injects bacterial proteins directly into the eukaryotic cell cytoplasm. Using a genetically attenuated strain of Pseudomonas aeruginosa, we have previously described the ability of this system to easily deliver a high quantity of protein to both differentiated and pluripotent cells. MyoD is a key muscle regulatory factor, the overexpression of which is able to induce transdifferentiation of numerous cell types into functional myocytes. Here we demonstrate transient injection of MyoD protein by P. aeruginosa to be sufficient to induce myogenic conversion of mouse embryonic fibroblasts. In addition to clear morphological changes, muscle-specific gene expression has been observed both at mRNA and protein levels. These studies serve as a foundation for the bacterial delivery of transcription factors to efficiently modulate concentration-dependent and temporal activation of gene expression that directs cell fate without jeopardizing genomic integrity.
Introduction
Forced exogenous gene expression has been well characterized as an effective method for directing both cellular differentiation and dedifferentiation (Chambers and Studer, 2011; Cherry and Daley, 2012; Yamanaka and Blau, 2010). However, this approach has relied heavily on the use of transgene expression to alter endogenous lineage-specific gene expression patterns. Given the potential for insertional- and recombination-mediated mutagenesis associated with such DNA-based techniques, the cells derived by these methods have limited clinical applicability. Transient protein delivery offers a safe and effective alternative, without the concern for long-term genomic stability. Although numerous protein delivery procedures exist, each is limited by labor-intensive methods of protein purification, low protein yield, or inefficient intracellular targeting.
In a recent study, we described the ability of the Pseudomonas aeruginosa type III secretion system (T3SS) to overcome these limitations and not only produce, but also abundantly inject, nuclear targeting proteins directly into the cytoplasm of both pluripotent and differentiated cells (Bichsel et al., 2011). The T3SS is a transmembrane complex that has a needle-like projection from the Gram-negative bacterial cell membrane and serves to translocate bacterial toxins directly into the eukaryotic host cells (Coburn et al., 2007; Cornelis, 2006; Hayes et al., 2010). Each type III secreted protein is directed for injection by an amino-terminal secretion signal sequence. In characterizing this system for delivery of exogenous proteins, we found that translational fusion of the first 54 amino acids of the P. aeruginosa exotoxin ExoS with Cre recombinase was optimal for directing this protein for type III–mediated injection (Bichsel et al., 2011). Using a genetically attenuated strain of P. aeruginosa expressing this ExoS54–Cre fusion protein, we were able to incite efficient Cre-dependent recombination in reporter cells, indicating that the addition of the signal sequence did not affect the nuclear localization or the biological function of this enzyme and that the injected host cells did not suffer significant cytotoxicity (Bichsel et al., 2011). Additionally, the bacterial infection and T3SS-mediated protein injection did not affect the basic cell biology or pluripotency of the recipient cells (Bichsel et al., 2011; Yu et al., 2009), a finding that is critical for the application of this system for directed stem cell differentiation. These results demonstrate the ease with which such a bacterial system can be used to deliver eukaryotic transcription factors to alter endogenous gene expression.
MyoD is a basic helix–loop–helix DNA-binding protein, which activates transcription at many muscle-specific promoters and has often been referred to as the master regulator of myogenesis for its ability to transdifferentiate a variety of cell types, including fibroblasts, to a myogenic lineage (Tapscott et al., 1988; Weintraub et al., 1989). Traditionally, myogenic conversion has been achieved by constitutive viral transgene expression, which is effective but leaves a genomic footprint. Here we report transient T3SS-mediated injection of human MyoD protein by P. aeruginosa to be sufficient to incite muscle-specific gene expression in mouse embryonic fibroblasts and induce terminal myogenic conversion.
These studies serve as a foundation for the bacterial delivery of transcription factors to efficiently modulate concentration-dependent and temporal activation of gene expression to direct cell fate without jeopardizing genomic integrity, fulfilling the basic safety requirement for medical application of cell replacement therapies.
Materials and Methods
Bacterial strains and plasmids
Strains and plasmid constructs used in this study are listed in Table 1. P. aeruginosa PAK-J with deletions in the type III secreted exotoxins (PAK-JΔexoSexoTexoY; ΔSTY) or deletion of an essential pore-forming protein required for type III-mediated protein injection (ΔpopD) have previously been described (Bichsel et al., 2011). Each strain was electroporated with pExoS54-Flag-MyoD or pExoS54. Human myoD constructs were generated using primers listed in Table 2. Antibiotics were used at a final concentration of 150 μg carbenicillin/mL for plasmid section.
Table 1.
Strains and Plasmids Used
| Strains | Description | Ref. |
|---|---|---|
| PAK-J | PAK derivative with elevated T3SS activity | (Bichsel et al., 2011) |
| PAK-JΔSTY | PAK-J deleted of exoS, exoT, and exoY | (Bichsel et al., 2011) |
| PAK- JΔpopD | PAK-J deleted of popD | (Bichsel et al., 2011) |
| PAK- JΔexsA | PAK-J delete of exsA | This study |
| Plasmids | ||
| pUCP20 | Pseudomonas–E.coli shuttle vector | (West et al., 1994) |
| pExoS54 | pUCP20 with exoS54 | (Bichsel et al., 2011) |
| pExoS54-Flag-MyoD | pExoS54 fused with FLAG tagged myoD | This study |
| pTYF-EF1α | Lenviral expression vector | (Zaiss et al., 2002) |
| pTYF-EF1α-MyoD | pTYF-EF1α containing myoD gene | This study |
| pTYF-EF1α-ExoS54-MyoD | pTYF-EF1α containing exoS54–myoD fusion gene | This study |
Table 2.
Reverse Transcriptase PCR Primer Sets
| Gene | Forward | Reverse |
|---|---|---|
| myoD | 5′-TGGGTTCCCTGTTCTGTGTCGC-3′ | 5′-AGACCACCAACGCTGATCGC-3′ |
| myoG | 5′-CTGGGGACCCCTGAGCA-3′ | 5′-ATCGCGCTCCTCCTGGT-3′ |
| desmin | 5′-TCTCCCGTGTTCCCT-3′ | 5′-ATACGAGCTAGAGTGGCA-3′ |
| mhc | 5′-AGAGCTGACGTGCCTCAATG-3′ | 5′-ATGCCTCTTCTTGCCCTTGT-3′ |
| gapdh | 5′-CCATCACCATCTTCCAGGAG-3′ | 5′-GCATGGACTGTG GTCATGAG-3′ |
Cell culture
C3HT10½ mouse embryonic fibroblasts were grown in Dulbecco's Modified Eagle Medium (DMEM; Gibco) supplemented with 15% fetal bovine serum (FBS) for growth and maintenance. Cells were infected in DMEM containing 5% heat-inactivated FBS, and subsequently changed to DMEM containing 2% horse serum (HS) for differentiation. All cells were cultured at 37°C with 5% CO2 and supplemented with penicillin, streptomycin, and amphotericin (Cellgro). Ciprofloxacin was added to a final concentration of 20 μg/mL for bacterial clearance.
Lentiviral expression of MyoD and ExoS54–MyoD
The human myoD and exoS54-flag-myoD gene sequences were cloned into the lentiviral vector pTYF-EF1α (Zaiss et al., 2002) and viral particles were produced as previously described. Equivalent viral supernatants [2×107 plaque-forming units (pfu)] were incubated with C3HT10½ fibroblasts for 24 h. The virus was then removed and replaced with DMEM containing 2% HS. Cells were monitored over time for myogenesis, fixed, and immunostained for desmin expression at 6 days postinfection (dpi).
Protein injection assay
Mouse embryonic fibroblasts (MEF) were plated at 70% confluence in medium without antibiotics. P. aeruginosa strains were grown at 37°C in Luria broth containing carbenicillin until reaching an optical density (OD600) of 0.8. Cells were infected with bacteria at a multiplicity of infection (MOI) of 100 for 3 h. Infections were cleared by removing the bacteria washing cells three times in phosphate-buffered saline (PBS)] and replacing the medium with DMEM containing 2% HS and ciprofloxacin. Cells were maintained in this medium for differentiation assays.
For injection assays, cells were infected as described. Immediately following infection, cells were washed, collected in 0.25% trypsin, and centrifuged at 500×g. The cell pellets were lysed in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, and boiled for 10 min. Following separation on 12% SDS-PAGE and polyvinylidene fluoride (PVDF) transfer, the blots were probed with antibodies against MyoD (Santa Cruz sc-304; 1:1000).
Immunocyotchemistry
Following infection or ExoS54–MyoD-mediated transdifferentiation, cells were fixed with 1% paraformaldehyde for 20 min at room temperature. The cells were then washed in PBS, permeablized with 0.1% Triton-X100, and blocked in PBS containing 0.1% Triton-X100 and normal goat serum. The cells were incubated with the primary antibody for 2 h, washed, and incubated with the secondary antibody for 1 h. The cells were then washed, stained with 4′,6-diamidino-2-phenylindole (DAPI), and examined by fluorescence microscopy. Anti-MyoD (Santa Cruz sc-304, 1:400), anti-MyoG (Santa Cruz sc-12732; 1:400), anti-Desmin (Sigma D1033; 1:400), and anti-MHC (Sigma M4276, 1:400) were used as primary antibodies while Alexafluor488 anti-rabbit, Alexafluor594 anti-rabbit, and Alexafluor488 anti-mouse were used as secondary antibodies (Invitrogen).
Flow cytometry
Prior to infection, cells were stained with 10 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; CellTrace CFSE Cell Proliferation Kit; Molecular Probes C34554) at 37°C for 15 min, washed, and retained in DMEM containing 5% FBS until infection. Stained cells were collected by 0.25% trypsin treatment 5 days after P. aeruginosa infection. Cells were centrifuged at 500×g for 5 min, collected, and suspended in 0.5 mL of PBS containing 2% FBS on ice. Cells were analyzed for CFSE content using a Diva v6.2 on LSR-II (BD-Biosciences) flow cytometer.
Reverse transcriptase PCR
Cellular RNA was extracted 18 days postinfection using RNeasy (Qiagen cat. no. #74104). cDNA was generated and amplified using primers shown in Table 2. PCR products were subject to electrophoresis on a 1% agarose gel.
Results
Bacterial production and injection of MyoD fusion protein
In our previous characterization of the T3SS for delivery of Cre recombinase, it was determined that the amino-terminal 54 amino acids of P. aeruginosa exotoxin ExoS, which contain both the secretion signal sequence and chaperone binding domain, were sufficient and optimal to direct Cre for type III-dependent secretion and injection into various types of cell lines (Bichsel et al., 2011). To use this system to incite myogenic conversion, it was necessary to confirm that the strain was capable of producing the fusion protein and directing it for injection. The genetically attenuated P. aeruginosa strain PAK-JΔSTY (ΔSTY) displays hypersecretion of type III effectors and reduced toxicity due to the chromosomal deletion of the exotoxins exoS, exoT, and exoY (Bichsel et al., 2011). PAK-JΔpopD contains a functional T3SS that is capable of protein secretion into culture medium, but it is defective in protein injection into the host cell due to the deletion of ΔpopD, which encodes a protein required for the formation of the pore on host membrane through which the needle injects effectors (Schoehn et al., 2003). The deletion of exsA, a transcriptional activator of the T3SS, results in a strain that is incapable of transcribing genes from type III-specific promoters, and is thus defective in production and secretion of the fusion proteins (Yahr and Wolfgang, 2006). These strains were electroporated with a selectable plasmid expressing the ExoS54-MyoD fusion protein (Fig. 1A) under the control of the endogenous exoS promoter and cultured in the presence of 5 mM EGTA to induce type III secretion. The bacterial cultures were collected 3 h postinduction, separated into pellet and supernatant after centrifugation, and assessed for production and secretion of the fusion protein, respectively. Immunoblot analysis of the lysed bacteria and secreted proteins in supernatants (Fig. 1B) demonstrated that each strain that possessed both a functional T3SS and the ExoS54–MyoD fusion construct was capable of producing the protein and that the ExoS54 signal sequence is sufficient to direct this protein for secretion into culture medium.
FIG. 1.

Bacterial production and injection of ExoS54–MyoD. (A) PAK-JδSTY, PAK-JδpopD, and PAK-JδexsA were electroporated with the vector (V) or a plasmid containing the ExoS54–Cre fusion. (B) Each strain was examined for the ability to produce and secrete the fusion protein by anti-MyoD immunoblot of the bacterial pellet and secretion supernatant. (C) MEFs were infected with each strain at an MOI of 100, unless otherwise noted, lysed and examined for protein injection by anti-MyoD immunoblot.
To assess the capacity for injection, each strain was co-incubated with MEFs for 3 h. Free bacterial cells were subsequently removed by successive washing of the adhered cells. The MEFs were then collected, lysed selectively, and examined for intracellular ExoS54–MyoD by immunoblot (Fig. 1C). Indeed, the injection of the fusion protein occurred in a MyoD and type III secretion-dependent manner, because the protein is not detected without infection, without a functional injection system, or with vector controls. Congruent with our previous findings, ExoS54–MyoD injection occurred in a dose-dependent manner, as protein injection increased with increased MOI.
The ExoS54–MyoD fusion protein retains biological function
To activate myogenesis, MyoD must retain the ability to bind DNA and recruit transcriptional co-activators. Given the structural dependence of MyoD function, it was necessary to determine whether the addition of ExoS54 secretion signal sequence would inhibit myogenic conversion independently of the bacterial infection. To compare the myogenic capacity of MyoD and ExoS54–MyoD, the genes encoding each were cloned into a lentiviral vector under the regulation of the constitutively active ef1α promoter. MEFs were infected with equivalent viral particles (2×107) and monitored over time for myogenic activity. As anticipated, 6 days postinfection, multinucleated myotubes were visible by light microscopy (Fig. 2), and upon immunocytochemical analysis, these cells were found to also express desmin, a muscle-specific intermediate filament and classical marker of myogenesis. Following viral delivery of the genes constitutively expressing MyoD or the ExoS54–MyoD fusion protein, 31.8% and 24.6% of the MEFs express Desmin, respectively, whereas none of the MEFs infected with vector alone resulted in Desmin-positive cells. These results demonstrate that the addition of the amino-terminal secretion signal sequence affects neither the nuclear localization nor the function of MyoD.
FIG. 2.

Lentiviral expression of MyoD and ExoS54–MyoD. MEFs were infected with lentiviral particles expressing, MyoD or ExoS54–MyoD. Cells were fixed and stained 6 dpi for expression of desmin. Nuclei were stained with DAPI.
ExoS54–MyoD induces cell growth arrest
During myogenic conversion, the fibroblasts undergo several distinct changes. Initially the transdifferentiating cells arrest proliferation and then begin to migrate toward one another and ultimately fuse together to form multinucleated myotubes. In myoblasts, MyoD level must be balanced to maintain proliferation and prevent MyoD-meditated differentiation (Wei and Paterson, 2001). Specifically, MyoD is negatively regulated by factors promoting cell cycle progression (Batonnet-Pichon et al., 2006; Kitzmann and Fernandez, 2001; Polesskaya and Rudnicki, 2002; Tapscott, 2005), as such; when the MyoD level rises, p21 expression is activated, which simultaneously abrogates the downregulation of MyoD and promotes differentiation by halting cell proliferation (Tintignac et al., 2004).
The ability of ExoS54–MyoD to arrest the growth of highly proliferative MEFs upon infection would indicate the initiation of early myogenesis. Indeed, we did observe growth arrest specifically in cells injected with ExoS54–MyoD. To quantify this observation, a cell-permeable fluorescent dye was used to monitor proliferation. CFSE can passively diffuse into cells until it is modified by intracellular esterases (Lyons, 2000). The fluorescent dye then remains in the cell cytoplasm and is successively diluted with each cell division. Therefore, nonproliferative cells will retain higher levels of the CFSE dye. MEFs were stained with CFSE prior to infection with P. aeruginosa. Three hours postinfection, the cells were treated with antibiotics and grown for 5 days in DMEM containing 2% horse serum. The fluorescence intensity of the cells, which inversely correlates with proliferation, was assessed by flow cytometry to be either CFSE high (nonproliferative) or CFSE low (proliferative) (Fig. 3A). Mirroring the ExoS54–MyoD injection results, the percentage of nonproliferative cells increased in a MyoD- and dose-dependent manner (Fig. 3B). As is commonly observed with these infections, uninfected cells and those infected with an injection-defective mutant showed little difference in proliferation and often grew over confluent. As the doubling time of MEF under our growth condition (DMEM+2% HS), determined by cell synchronization assay (Fig. 4), is about 28 h, the 10–20% high CFSE cells after 5 days (about 4 doubling time) represent close to 95% original cells that under went growth arrest, consistent with high injection rate by the T3SS.
FIG. 3.

ExoS54–MyoD injection halts cell proliferation. CFSE-stained MEFs were infected with PAK-JΔSTY (pExoS54–MyoD) at MOIs of 25, 50, 100, 150, 200, PAK-JΔpopD and vector controls at an MOI of 100. Following a 3-h infection, bacteria were cleared and cells were allowed to proliferate for 5 days. Cells were then collected for CFSE detection by flow cytometry (A). Efficiency of ExoS54–MyoD-mediated growth arrest was compared for each condition as percent of CFSE retaining cells in the total cell population (B). n=3.
FIG. 4.

MEF cell cycle synchronization. MEFs were either left unsynchronized (U) or synchronized through double thymidine blocking and released every 4 h to determine the duration of the cell replication cycle.
Bacterial injection of ExoS54–MyoD induces myocyte-specific gene expression in MEFs
While the induction of myogenesis, as assessed by growth arrest, may occur rapidly upon ExoS54–MyoD delivery, the changes in morphology and gene expression are progressive. According to the transdifferentiation of fibroblasts expressing the lentiviral myoD transgene, following growth arrest, MEFs destined for myogenic conversion will migrate together, align, and subsequently fuse together to form characteristic multinucleate myotubes. During this process, cells will begin to activate myogenic transcription factors, such as MyoD and myogenin, followed by muscle structural genes, including desmin and myosin heavy chain (MHC) (Wagers and Conboy, 2005; Yokoyama and Asahara, 2011). To determine the ability of ExoS54–MyoD to fully transdifferentiate MEFs upon type III secretion-mediated MyoD protein injection, MEFs were co-incubated with P. aeruginosa cells expressing ExoS54–MyoD for 3 h, followed by antibiotic treatment to eliminate bacterial cells and allowed to differentiate over time. The cells were carefully monitored daily for changes in growth, morphology, and size. Congruent with the CFSE proliferation studies, cells that were uninfected, infected with PAK-JΔSTY containing the vector alone, or PAK-JΔpopD expressing ExoS54–MyoD did not undergo growth arrest. In fact, these cells became over confluent and detached during the time required for ExoS54–MyoD infected cells to transdifferentiate. As such, these cells are not included in the subsequent analysis. In contrast, cells injected with a single dose of ExoS54–MyoD quickly exited the cell cycle, became elongated, aligned, and finally began fusing to generate multinucleate myotubes. Once myotube formation was observed (10–14 dpi), the cells were fixed and stained for the expression of key myogenic proteins. Juxtaposed with uninfected MEFs, which express none of the muscle-specific proteins, infected MEFs are positive for endogenous nuclear MyoD (19.1%) and myogenin (37.6%) as well as cytoplasmic desmin (44.7%) and MHC (15.4%) (Fig. 5), representing on average 29.2% cells expressing the myocyte-specific markers. Cells similarly infected with ExoS54–MyoD were also collected for reverse transcriptase PCR (RT-PCR) analysis for gene expression. Using cDNA derived from mouse tongue muscle and uninfected MEFs as positive and negative controls, respectively, transcripts from the bacterially generated myotubes were examined for nuclear reprogramming (Fig. 6). In accordance with the immunocytochemistry, the RT-PCR demonstrates a lack of muscle-specific gene expression in uninfected MEFs in contrast to the abundant expression in the muscle control and intermediate expression in the bacterially transdifferentiated MEFs. Taken together, these data indicate type III–injected ExoS54–MyoD is able to stimulate myogenic conversion of MEFs, recapitulating the key changes in morphology, behavior, and gene expression to generate terminally differentiated myotubes.
FIG. 5.

ExoS54–MyoD injection causes muscle-specific protein expression in MEFs. Fourteen dpi, infected MEFs and uninfected MEFs were fixed and immunostained for expression of the indicated proteins using protein-specific antibodies and DAPI for nuclear staining.
FIG. 6.
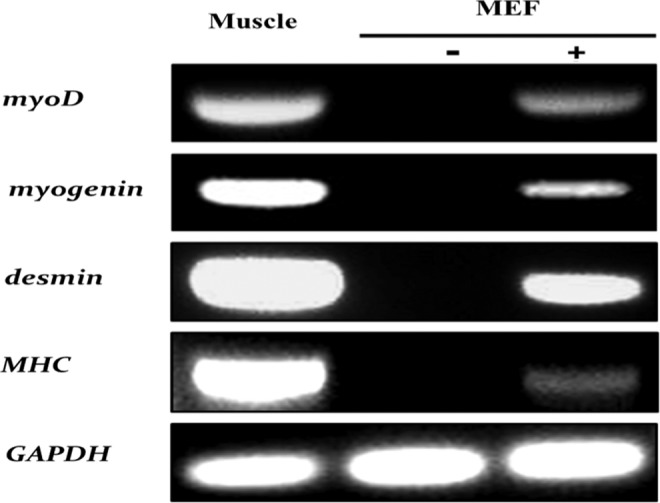
ExoS54–MyoD injection activates muscle-specific gene expression in MEFs. MEFs that had been infected (+) or not infected (−) were collected at 14 dpi for RNA extraction and RT-PCR analysis for the expression of the indicated genes, using cDNA from murine tongue muscle cells as a positive control for expression.
Discussion
P. aeruginosa is one of numerous Gram-negative bacteria that use the type III secretion system to inject toxic proteins into host cells to promote infection (Hauser, 2009). These systems have been optimized evolutionarily to maximize protein injection and conserve bacterial energy. The efficiencies of protein production and injection by this system are not the only advantages it has over currently used transgenic methods. The ease with which bacterial T3SS can be manipulated to control both the quantity and duration of protein activity allow for a unique temporal regulation not found in other inducible systems. As such, multiple proteins can be delivered together or sequentially in varying ratios to incite an optimal effect.
Our previous studies have shown that the endogenous P. aeruginosa effector ExoS is very efficiently injected into cells in vitro, reaching nearly all cells, even when infected at low MOIs (Bichsel et al., 2011). However, with exogenous protein delivery, we do not observe 100% Cre-mediated recombination or myogenic conversion. In this study, almost all cells received ExoS54–MyoD injection as indicated by initial cell cycle arrest in 95% of the cell population, nonetheless on average only 29% cells actually converted to myocytes. Although all of the cells have been injected with the fusion protein, there is surely a threshold level required for induction of myogenesis in MEFs, which may not be reached in each cell. This issue can be overcome by either increasing the MOI or performing multiple rounds of protein injection. Additionally, we have observed a distinct correlation between cell cycle phase and the efficacy of injected proteins. Cells infected during S phase appear to be much more susceptible to ExoS54–MyoD-mediated transdifferentiation (data not shown), similar to the higher rate of chromosomal DNA recombination by injected Cre among S-phase cells (Bichsel et al., 2011). This could occur for a number of reasons, including the increased accessibility of genomic DNA during S phase. As such, synchronizing cells to S phase prior to infection enhances the effectiveness of injected proteins and thus the efficiency of nuclear reprogramming.
Cell proliferation is a major hurdle blocking myogenesis in these MEFs. In the CFSE studies, we demonstrate the ability of ExoS54–MyoD to halt cell proliferation, reaching an efficiency of about 15% CFSE-high cells at 5 days postinfection (Fig. 3). However, cells that do not receive sufficient protein to induce growth arrest will continue to rapidly proliferate, undergoing about four rounds of replication prior to analysis for CFSE content (5 dpi). With each successive division, these proliferative cells will decrease the total percentage of cells undergoing myogenic conversion. By calculating the doubling time of these cells through propidium iodide staining and flow cytometry (Fig. 4), we can determine the number of cell divisions and extrapolate the initial percentage of cells undergoing myogenic conversion to be closer to 95%, which then becomes diluted to around 15% at the time of analysis. When examined from this perspective, the transdifferentiation of these cells is quite efficient. In addition to enriching for myogenesis by fluorescence-activated cell sorting of CFSE-positive cells, it is also possible to select against proliferative cells using chemical agents to increase the myogenic population.
The efficacy of T3SS-mediated protein delivery can also be enhanced by genetically mutating the injected protein to promote superior intracellular stability. Like many transcription factors, the stability of MyoD is regulated by posttranslational modifications, such as ubiquitination, phosphorylation, acetylation, and methylation (Di Padova et al., 2007; Jo et al., 2011; Ling et al., 2012; Sadeh et al., 2008). In the case of MyoD, manipulation of these modified amino acids could not only decrease proteasomal degradation, but also desensitize ExoS54–MyoD to downregulation by cyclin-dependent kinases.
MyoD is a strong transcriptional activator and a key regulator of myogenesis. In addition to activating transcription at many muscle-specific promoters, MyoD is also part of an autoregulatory loop, such that the injected ExoS54–MyoD is capable of activating transcription of the endogenous myoD gene. Given these properties, a single infection is sufficient to activate this cascade and allow the cell to continue the process over a span of several days, long after the majority of injected protein has been degraded, as injected proteins become nondetectable after 24 h. Given the strength of this pathway, delivery of less powerful transcription factors may require multiple rounds of protein injection.
The delivery strain, PAK-JΔSTY, is greatly attenuated in virulence (>70%) due to the deletion of type III secreted exotoxins responsible for the majority of the acute toxicity of the wild-type P. aeruginosa (Bichsel et al., 2011). Although the strain is capable of prolonged and repeated co-incubation with eukaryotic cells in vitro without significant cytotoxicity, we are continuing to optimize this bacterium for use as a protein delivery mechanism. With additional modifications, we are developing a strain with further reduced cytotoxicity, increased protein injection and enhanced sensitivity to antibiotics.
Our recent studies have demonstrated the utility of the type III secretion system in delivery of nuclear proteins to eukaryotic cells (Bichsel et al., 2011). The ability of proteins such as Cre recombinase and MyoD to be injected by the bacterium, localize to the nucleus, and uniquely interact with the eukaryotic DNA to incite physical changes, such as site-directed recombination and alterations of gene expression, as occurs in myogenic conversion, highlights the promise of this system for use with other proteins. Using this modified T3SS, we are also able to deliver well-characterized nuclear reprogramming factors, including Oct4, Sox2, Klf4, and c-Myc, and are currently optimizing the delivery of these proteins to induce pluripotency in a novel manner that is both DNA-free and efficient.
Given the ease with which this system can be applied to transiently deliver diverse proteins and the simplicity of its regulation to control the duration and quantity of delivered protein in a single, repeated, or sequential manner, the use of the bacterial type III secretion system has the potential to replace many current methods of cellular reprogramming.
Acknowledgments
We would like to thank Ranjan Batra for cDNA synthesis, Dr. Vladi Cherepakhin and Neal Benson for flow cytometry, Dr. Jinghua Jia for technical assistance, and Dr. Maury Swanson for helpful discussion. This work was supported by National Institutes of Health (RC1GM091238) and Bankhead-Coley Cancer Research Program (FLDH 09BW-12).
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
- Batonnet-Pichon S. Tintignac L.J. Castro A. Sirri V. Leibovitch M.P. Lorca T. Leibovitch S.A. MyoD undergoes a distinct G2/M-specific regulation in muscle cells. Exp. Cell Res. 2006;312:3999–4010. doi: 10.1016/j.yexcr.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Bichsel C. Neeld D.K. Hamazaki T. Wu D. Chang L.J. Yang L. Terada N. Jin S. Bacterial delivery of nuclear proteins into pluripotent and differentiated cells. PLoS One. 2011;6:e16465. doi: 10.1371/journal.pone.0016465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S.M. Studer L. Cell fate plug and play: Direct reprogramming and induced pluripotency. Cell. 2011;145:827–830. doi: 10.1016/j.cell.2011.05.036. [DOI] [PubMed] [Google Scholar]
- Cherry A.B. Daley G.Q. Reprogramming cellular identity for regenerative medicine. Cell. 2012;148:1110–1122. doi: 10.1016/j.cell.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn B. Sekirov I. Finlay B.B. Type III secretion systems and disease. Clin. Microbiol. Rev. 2007;20:535–549. doi: 10.1128/CMR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis G.R. The type III secretion injectisome. Nat. Rev. Microbiol. 2006;4:811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- Di Padova M. Caretti G. Zhao P. Hoffman E.P. Sartorelli V. MyoD acetylation influences temporal patterns of skeletal muscle gene expression. J. Biol. Chem. 2007;282:37650–37659. doi: 10.1074/jbc.M707309200. [DOI] [PubMed] [Google Scholar]
- Hauser A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009;7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes C.S. Aoki S.K. Low D.A. Bacterial contact-dependent delivery systems. Annu. Genet. 2010;44:71–90. doi: 10.1146/annurev.genet.42.110807.091449. [DOI] [PubMed] [Google Scholar]
- Jo C. Cho S.J. Jo S.A. Mitogen-activated protein kinase kinase 1 (MEK1) stabilizes MyoD through direct phosphorylation at tyrosine 156 during myogenic differentiation. J. Biol. Chem. 2011;286:18903–18913. doi: 10.1074/jbc.M111.225128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzmann M. Fernandez A. Crosstalk between cell cycle regulators and the myogenic factor MyoD in skeletal myoblasts. Cell. Mol. Life Sci. 2001;58:571–579. doi: 10.1007/PL00000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling B.M. Bharathy N. Chung T.K. Kok W.K. Li S. Tan Y.H. Rao V.K. Gopinadhan S. Sartorelli V. Walsh M.J. Taneja R. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA. 2012;109:841–846. doi: 10.1073/pnas.1111628109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A.B. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J. Immunol. Methods. 2000;243:147–154. doi: 10.1016/s0022-1759(00)00231-3. [DOI] [PubMed] [Google Scholar]
- Polesskaya A. Rudnicki M.A. A MyoD-dependent differentiation checkpoint: Ensuring genome integrity. Dev. Cell. 2002;3:757–758. doi: 10.1016/s1534-5807(02)00372-6. [DOI] [PubMed] [Google Scholar]
- Sadeh R. Breitschopf K. Bercovich B. Zoabi M. Kravtsova-Ivantsiv Y. Kornitzer D. Schwartz A. Ciechanover A. The N-terminal domain of MyoD is necessary and sufficient for its nuclear localization-dependent degradation by the ubiquitin system. Proc. Natl. Acad. Sci. USA. 2008;105:15690–15695. doi: 10.1073/pnas.0808373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoehn G. Di Guilmi A.M. Lemaire D. Attree I. Weissenhorn W. Dessen A. Oligomerization of type III secretion proteins PopB and PopD precedes pore formation in Pseudomonas. EMBO J. 2003;22:4957–4967. doi: 10.1093/emboj/cdg499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott S.J. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development. 2005;132:2685–2695. doi: 10.1242/dev.01874. [DOI] [PubMed] [Google Scholar]
- Tapscott S.J. Davis R.L. Thayer M.J. Cheng P.F. Weintraub H. Lassar A.B. MyoD1: A nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science. 1988;242:405–411. doi: 10.1126/science.3175662. [DOI] [PubMed] [Google Scholar]
- Tintignac L.A. Sirri V. Leibovitch M.P. Lecluse Y. Castedo M. Metivier D. Kroemer G. Leibovitch S.A. Mutant MyoD lacking Cdc2 phosphorylation sites delays M-phase entry. Mol. Cell. Biol. 2004;24:1809–1821. doi: 10.1128/MCB.24.4.1809-1821.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagers A.J. Conboy I.M. Cellular and molecular signatures of muscle regeneration: Current concepts and controversies in adult myogenesis. Cell. 2005;122:659–667. doi: 10.1016/j.cell.2005.08.021. [DOI] [PubMed] [Google Scholar]
- Wei Q. Paterson B.M. Regulation of MyoD function in the dividing myoblast. FEBS Lett. 2001;490:171–178. doi: 10.1016/s0014-5793(01)02120-2. [DOI] [PubMed] [Google Scholar]
- Weintraub H. Tapscott S.J. Davis R.L. Thayer M.J. Adam M.A. Lassar A.B. Miller A.D. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl. Acad. Sci. USA. 1989;86:5434–5438. doi: 10.1073/pnas.86.14.5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S.E. Schweizer H.P. Dall C. Sample A.K. Runyen-Janecky L.J. Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa. Gene. 1994;148:81–86. doi: 10.1016/0378-1119(94)90237-2. [DOI] [PubMed] [Google Scholar]
- Yahr T.L. Wolfgang M.C. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 2006;62:631. doi: 10.1111/j.1365-2958.2006.05412.x. [DOI] [PubMed] [Google Scholar]
- Yamanaka S. Blau H.M. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465:704–712. doi: 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama S. Asahara H. The myogenic transcriptional network. Cell. Mol. Life Sci. 2011;68:1843–1849. doi: 10.1007/s00018-011-0629-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J. Rossi R. Hale C. Goulding D. Dougan G. Interaction of enteric bacterial pathogens with murine embryonic stem cells. Infect. Immun. 2009;77:585–597. doi: 10.1128/IAI.01003-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss A.K. Son S. Chang L.J. RNA 3′ readthrough of oncoretrovirus and lentivirus: Implications for vector safety and efficacy. J. Virol. 2002;76:7209–7219. doi: 10.1128/JVI.76.14.7209-7219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]