

Abstract
Dystrophia myotonica type 1 (DM1) is an autosomal dominant multisystem disorder. The pathogenesis of central nervous system (CNS) involvement is poorly understood. Disease-specific induced pluripotent stem cell (iPSC) lines would provide an alternative model. In this study, we generated two DM1 lines and a normal iPSC line from dermal fibroblasts by retroviral transduction of Yamanaka's four factors (hOct4, hSox2, hKlf4, and hc-Myc). Both DM1 and control iPSC clones showed typical human embryonic stem cell (hESC) growth patterns with a high nuclear-to-cytoplasm ratio. The iPSC colonies maintained the same growth pattern through subsequent passages. All iPSC lines expressed stem cell markers and differentiated into cells derived from three embryonic germ layers. All iPSC lines underwent normal neural differentiation. Intranuclear RNA foci, a hallmark of DM1, were detected in DM1 iPSCs, neural stem cells (NSCs), and terminally differentiated neurons and astrocytes. In conclusion, we have successfully established disease-specific human DM1 iPSC lines, NSCs, and neuronal lineages with pathognomonic intranuclear RNA foci, which offer an unlimited cell resource for CNS mechanistic studies and a translational platform for therapeutic development.
Introduction
Myotonic dystrophy type 1 (DM1) is a dominantly inherited multisystemic disorder and is the most prevalent muscular dystrophy in adults, affecting 1 in 8500 individuals worldwide (Harper, 2001). The disease is caused by an unstable CTG nucleotide repeat expansion within the 3′-untranslated region of the dystrophia myotonica protein kinase (DMPK) gene on chromosome 19q13.3 (Fu et al., 1992). In the classic form, the major features include myotonia, muscle weakness and wasting, cardiomyopathy with conduction defects, insulin resistance, frontal balding, and early-onset cataracts. The disease mechanism in skeletal muscle has been mostly deciphered through studies of mouse models and fibroblast/myoblast cell cultures (for review, see Ashizawa and Sarkar, 2011; Gomes-Pereira et al., 2011; Lee and Cooper, 2009; Ranum and Cooper, 2006; Romeo, 2012). However, the effects of the CTG repeat expansion on the central nervous system (CNS) are less defined. A major obstacle is difficulty in obtaining viable human DM1 CNS tissues. DM1 CNS studies in humans have been largely restricted to investigations of clinical findings, neuropsychiatry, neuroradiology, and neuropathology. The CNS involvement in DM1 has a dramatic spectrum in presentation and the degree of impairment, which generally correlates with CTG expansion size (Ekstrom et al., 2008; Sistiaga et al., 2010). Reported CNS symptoms range from memory impairment, executive dysfunction, psychiatric disorders, and personality abnormalities in adult-onset cases and mental retardation, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD) in congenital and juvenile forms (Bird et al., 1983; Douniol et al., 2009; Ekstrom et al., 2008; Portwood et al., 1986; Roig et al., 1994; Rubinsztein et al., 1997; Sistiaga et al., 2010; Steyaert et al., 1997). Additionally, excessive daytime somnolence with abnormal rapid eye movement (REM) sleep patterns and abnormal respiratory center function, including central sleep apnea and excessive sensitivity to anesthetics and narcotics with serious respiratory suppression, are attributable to CNS pathology in this disease. Imaging studies suggest that DM1 is likely a white matter disease (Ogata et al., 1998; Romeo et al., 2010) and larger expansion sizes are associated with more extensive brain involvement (Romeo, et al., 2010). In contrast, neuropathological studies of DM1 brains identified characteristic abnormalities in the gray matter, including neurofibrillary tangles (NFTs)(Kiuchi et al., 1991; Maurage et al., 2005; Mitake et al., 1989; Yoshimura et al., 1990) and intranuclear RNA foci of expanded CUG repeats (Jiang et al., 2004).
The underlying molecular mechanism for CNS involvement in DM1 has been explained as a “spliceopathy” where aberrant splicing of critical RNA transcripts causes cellular dysfunction. For example, abnormal splicing of the microtubule-associated protein tau (MAPT) gene has been identified in DM1 brain with corresponding pathological findings of NFTs (Dhaenens et al., 2008; Dhaenens et al., 2011; Jiang et al., 2004; Leroy et al., 2006a; Leroy et al., 2006b; Sergeant et al., 2001; Vermersch et al., 1996). Additionally, the requirement of specific splicing factors differs between muscle and brain tissues. Studies of mouse genetic models suggested that sequestration of mbnl1 appears to play a major pathogenic role in the DM1 muscle (Dhaenens et al., 2011; Suenaga et al., 2012), whereas a loss of function of mbnl 2 may be more critical than mbnl1 in the DM1 brain pathology (Charizanis et al., 2012).
These studies have shortcomings. Although mouse models are powerful tools to study the disease mechanism of genetic disorders such as DM1, interspecies differences between human and mouse exist. Unfortunately, fresh CNS tissues from human DM1 patients are not readily available, and tissues derived from autopsy of patients in advanced stages of the disease often suffer from many confounding factors, including age and co-morbidities prior to death. Autopsy tissues are not suitable for electrophysiological studies or interventional mechanistic experiments. Viable CNS neurons and glia are exceedingly difficult to obtain, and primary cultures of these CNS cells are short-lived. Although human fibroblasts and myoblasts from DM1 patients may be relatively easy to obtain, they differ from CNS cells in their morphology and cellular functions. Thus, there is a gap that needs to be filled in the experimental system for mechanistic studies of DM1 brain disorders. Induced pluripotent stem cells (iPSCs) can provide an unlimited resource for DM1 studies, and they can be differentiated in vitro to multiple cell types, including neuronal and glial lineages.
In this study, we established two DM1 iPSC lines and one control iPSC line. The human DM1 iPSCs and their neural cell lineages showed intranuclear RNA foci, which are pathognomonic of DM1.
Materials and Methods
Reagents and cells
Culture medium
Media for iPSCs culture [Dulbecco's modified Eagle medium (DMEM)/F12, 20% knockout serum replacement (KSR), Glutamax, 2-mercaptoethanol, sodium pyruvate, minimal essential medium and nonessential amino acids (MEM NEAA), penicillin/streptomycin], recombinant basic human fibroblast growth factor (bFGF) (#PHG0021), recombinant human epidermal growth factor (hEGF) (#PHG0311L), collagenase IV (# 17104-019), and SuperScript III Reverse Transcriptase (#18080)] were all from Life Technologies (Grand Island, NY, USA). Defined Cryopreservation Medium for hESC and hiPSCs (#05854), Accutase™ (#07920), Anti-Oct-3/4 (#01550), Anti-SSEA-4 (#01554), fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse immunoglobulin G (IgG; #10210), heparin (#07980), Y-27632 ROCK inhibitor (#07171), AggreWell™ 800 (#27865), STEMdiff™ Neural Induction Medium (#05831), STEMdiff™ Neural Rosette Selection Reagent (# 05832), NeuroCult™ NS-A proliferation kit (#05751), and NeuroCult™ NS-A Differentiation kit (# 05752) were from STEMCELL Technologies (Vancouver, Canada). Poly-L-Ornithine (#P4957), Laminin (#L2020), and Mitomycin (M4287) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The following antibodies are from companies in parentheses: Nestin (#MAB 1259) (R&D), alpha-fetoprotein (AFP) (R&D), Neurofilament H (Cell Signaling #2836), glial fibrillary acidic protein (GFAP; Novus #NB300-41A), β-Tubulin III (Millipore # CBL 412 X), Desmin (Lab-Vision). IbidiTreat μ-Slides are from ibidi GmbH (Martinsried, Germany).
Clinical information of study subjects
This study was approved by the University of Florida Institutional Review Board. All subjects consented to the study. DM1-03 is a 46-year-old Caucasian male with onset of symptoms at age 25. At the time of biopsy, he had severe myotonia, dysphagia (percutaneous endoscopic gastrostomy [PEG] placed at age 43), conduction block (pacemaker placed at age of 45), cataracts (removal surgery at the age of 30), hypersomnia, and cognitive impairment, but still functioned independently with his activities of daily living. DM1-05 is a 45-year-old Caucasian female with symptom onset at the age of 26. She suffered from myotonia, mild dysphagia, no conduction block, no cataracts, no diabetes, some cognitive impairment, but functioned well both socially and occupationally. A 51-year-old Caucasian male with no medical problems was recruited as a normal control.
Skin biopsy and culture of human dermal fibroblasts
Skin biopsies were performed by punch biopsy (6 mm in diameter) in the lateral thigh at the middle level between gluteal fold and popliteal fossa under local anesthesia. Biopsy specimens were processed into 0.5-mm cubes and placed into duplicate 25-cm2 flask and cultured in primary culture medium (DMEM with 20% fetal bovine serum [FBS]). When fibroblast cells from adjacent explants started merging, the flasks were treated with 0.05% trypsin/EDTA and passed to a 75-cm2 flask. Cells at passage 3 were used for reprogramming.
Southern blot
High-molecular-weight DNA was extracted from DM1 fibroblasts cells using conventional methods. Five micrograms of DNA was digested by NcoI and separated by 0.5% agarose gel before transferring to a Nitran supercharged membrane, which was hybridized overnight at 74°C with a DM1 probe. This probe is a 777-bp long PCR product amplified from a region of the DMPK gene located upstream of CTG repeat (forward primer, 5′-TGCCTCAGACCTGCTGCCCA-3′; reverse primer, 5′-AACCCAATGCAGCCCAGGGC-3′).
Generation of iPSC lines
Reprogramming was performed with the traditional Yamanaka four factors using retroviral vectors. Retroviruses, one for each of the four reprogramming factors (i.e., Oct4, Sox2, Klf4, and c-Myc) and enhanced green fluorescent protein (eGFP), was individually packaged using 293FT human embryonic kidney cell line (Invitrogen). After 72 h of virus production, cell supernatant was filtered through a 0.45-μm pore size filter, and the virus was concentrated by centrifugation for 16 h at 8000×g at 4°C. Oct4, Sox2, Klf4, and c-Myc viruses were combined and used for transduction. Transduction efficiency was monitored using additional fibroblasts retrovirally transduced with eGFP virus alone. One day prior to transduction, fibroblasts were plated at a density of 1×105 cells per 9.5-cm2 cell culture dish in DMEM containing 15% FBS. On day 5 after transduction, each 9.5-cm2 plate was passaged to a 56-cm2 dish with feeders (6.7×105 mitomycin C mitotically inactivated mouse embryonic fibroblasts [MEFs]). On day 6, the medium was changed from DMEM containing 15% FBS to iPSC medium. Half of the medium was removed and replaced with fresh iPSC medium every other day (day 8, 10, 12, etc.) until colonies were ready to pick up. Propagation and freezing of iPSCs was performed according to published protocols (Baharvand et al.; Mollamohammadi et al., 2009). mFreSR freezing medium (STEMCELL Technologies) was used to prepare iPSC frozen stocks.
Characterization of iPSCs
Morphology
iPSCs were cultured with MEF feeders. The morphology of iPSC clones was examined and compared to that of human embryonic stem cells (hESCs).
RT-PCR of stem cell markers
Total RNA was extracted from fibroblasts (passage 4) and iPSCs (passage 13 of DM1-03 and DM1-05, passage 15 of normal control) using RNeasy Micro Kit (Qiagen, Valencia, CA). cDNA was synthesized using SuperScript III Reverse Transcriptase (Invitrogen) from 1μg total RNA in a final volume of 20 μL. A 1-μL amount was used for subsequent PCR. Stem cell marker RT-PCR was performed using published primers (Takahashi et al., 2007) targeting endogenous human OCT4, SOX2, NANOG, and MYC. Electrophoresis of the PCR products was carried out on a 1.6% agarose gel.
Alkaline phosphatase activity assay and immunocytofluorescence staining of stem cell markers
The assays were performed on ibidiTreat μ-Slides. 3-5 iPSC clumps (around 100 cells) were seeded into chambers with feeders and cultured for 2–3 days to allow the colonies to expand. For the alkaline phosphatase activity assay, the chamber was incubated with Liquid Fast-Red Substrate System (Thermo Scientific, #TA-060-AC) overnight at 4°C. Nuclear transcription factor Oct4 and cell-surface marker SSEA4 were examined by immunocytofluorescence staining. The slides were washed with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 5 min, permeabilized with 0.3% Triton X-100 for 15 min, blocked with 10% normal goat serum (Vector Laboratories) for 30 min, and incubated with Oct 4 or SSEA4 (1:100) overnight at 4°C. The slides were washed and incubated with goat anti-mouse FITC-conjugated secondary antibody (1:500) (StemCell Technologies) for 30 min. Slides were washed and mounted with Vectashield mounting medium with DAPI (Vector Laboratories). Pictures were taken using an Olympus IX81-DSU Spinning Disk confocal microscope.
Prolonged self-renewal
iPSCs were passaged at a 1:4 ratio approximately every 4–5 days and were maintained to passage 20 for DM1-03 and DM1-05 and passage 21 for normal control iPSCs cells at the time of this submission. Over this time, iPSC growth pattern and morphology have not changed.
Embryoid body formation
iPSCs growing on mitomycin-inactivated MEF feeders (passage 17 of DM1-03, passage 16 of DM1-05, and passage 17 of normal control iPSCs) were first detached with collagenase IV (1 mg/mL) for 7–10 min at 37°C until feeder cells and differentiated cells were detached while iPSC colonies still remained attached (the edge may be off the surface). The supernatant and floating cells were removed and the dish was flushed with iPSC medium with 1-mL big bore tips. The suspension was then passed through a 70-μ m cell strainer to remove the feeder cells. The strainer was then reversed and iPSC clumps were collected with DMEM/F-12 medium. After a brief centrifugation, the colonies were resuspended in 3 mL of Accutase and incubated at 37°C with intermittent pipetting (every 5 min) for as long as 25 min to generate a single-cell suspension. The suspension was filtered through a 37-μ m cell strainer. The cell strainer was washed with an additional 7 mL of DMEM/F-12 medium. Cells were then centrifuged at 300×g for 5 min. The cell pellet was resuspended in EB medium to generate 2.5×105 cells/mL and 2 mL was added to each well of an AggreWell 400 plate (STEMCELL Technologies). The plate was centrifuged at 100×g for 3 min to capture the cells in the microwells. This generated roughly 200 cells per EB. At 24 h later, EBs were detached by flushing the microwells with induction medium and transferred to a low-attachment six-well plate for further culture of 5 days. The EBs were transferred to ibidiTreat μ-Slides (coated with Poly-Orthnine-Laminin) for further differentiation.
Neural differentiation
Neural differentiation using the AggreWell 800 (Stemcell Technologies) was performed following manufacturer's protocol. Briefly, a single-cell suspension was prepared, and 1.2–2.5×106 cells were seeded to each well of the AggreWell 800. The cells were captured in the microwells by centrifugation and cultured for 5 days. Over this time, the iPSCs collected at the bottom of the well aggregated to form neurospheres. The spheres were flushed from the microwells using neural induction medium and were transferred to a polyornithine laminin-coated six-well plate. On day 7, neural rosettes were detached using STEMdiff Neural Rosette Selection Reagent (STEMCELL Technologies) and transferred to a six-well plate coated with polyornithine laminin. Neural stem cells (NSCs) started growing out of neural rosettes after 12 h. Cells were cultured for 7 days. NSCs were detached with Accutase to make a single-cell suspension, and 4×104 cells were seeded to ibidiTreat μSlides and cultured in Neural Induction medium (STEMCELL Technologies) for observation and staining of neural markers. For further propagation and differentiation, NSCs were detached using Accutase and grown either in suspension or as attached cells using NeuroCult NS-A proliferation medium (STEMCELL Technologies).
RNA fluorescence in situ hybridization
Intranuclear foci containing (CUG)exp RNA were detected in DM1 fibroblasts, iPSCs, NSCs, and neurons via RNA fluorescence in situ hybridization (FISH) using a Cy5-labeled (CAG)10 DNA probe. Cells plated in ibidiTreat μ-slides were washed twice in sterile PBS (pH 7.4), fixed in 10% buffered formalin phosphate (Fisher Scientific) for 10 min at room temperature, washed three times in sterile PBS (pH 7.4), and dehydrated in prechilled 70% ethanol for 3 h at 4°C. The cells were washed in 40% formamide (EMD Chemicals) in 2× saline-sodium citrate (SCC) buffer (300 mM sodium chloride, 30 mM sodium citrate, pH 7.0) for 10 min at room temperature, then blocked in hybridization buffer (40% formamide, 2× SCC buffer, 200μg/mL bovine serum albumin [BSA], 100 mg/mL dextran sulfate, 2 mM vanadyl sulfate, 1 mg/mL yeast tRNA [Invitrogen]) for 15 min at 37°C. The Cy5-labeled (CAG)10 DNA probe was denatured for 10 min at 100°C, chilled on ice for 10 min, then added to prechilled hybridization buffer for a final concentration of 500 pg/μL probe. Hybridization buffer containing the probe was added to the cells, and hybridization was performed in a humidified chamber for 2 h at 37°C. As a negative control, RNA FISH for (CUG)exp RNA was performed on control patient cells. As an additional negative control, the experiment was performed in parallel on all cells using hybridization buffer lacking the (CAG)10 DNA probe. After hybridization, cells were washed three times in prewarmed 40% formamide/2× SCC buffer for 30 min at 37°C and once in sterile PBS (pH 7.4), followed by counterstaining with Vectashield containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories).
Immunocytofluorescence staining of markers of three embryonic germ layers and neural cells
The studies were carried out on ibidiTreat μ-Slides. In all, 40 μL of NSCs at a density of 1×106/mL were seeded to ibidiTreat μ-Slides and cultured in Neural Induction medium for observation and staining of neural markers. Cells were cultured for 3–5 days to allow for spontaneous differentiation. The slides were then fixed in 4% paraformaldehyde and processed for immunocytofluorescence staining. Primary antibody was incubated overnight at 4°C with AFP (1:100), Desmin (1:100), Nestin (1:100), Neurofilament H (1:50), β-tubulin III (1:250), or GFAP (1:500). The following day, slides were washed three times with PBS, incubated with appropriate secondary antibody conjugated with either Alexa Fluor 555 or Alexa Fluor 488 (Invitrogen) (1:500) for 30 min, washed with PBS, and mounted with Vectashield Mounting Medium with DAPI. Pictures were taken using Olympus IX81-DSU Spinning Disk confocal microscope.
RNA FISH plus immunocytofluoroscence of stem cell marker for iPSCs, three germ-line layer markers for EB in vitro differentiation and neural differentiation
These assays were performed in the same ibidiTreat μ-Slides. We found combining the two studies in one slide is feasible and can better illustrate the relationship. Briefly, the slides were first processed for intranuclear foci as described above and then were incubated with primary antibody (Oct4, both AFP and Desmin simultaneously, or neural markers) and staining was completed as described above.
Results
Confirmation of expansion of CTG repeats in DM1 fibroblast cells
The Southern blot analysis of genomic DNA isolated from cultured fibroblast cells showed expanded alleles as a smear in a range of approximately 2829–3575 repeats for DM1-03 and 1933–3152 repeats for DM1-05, suggesting repeat size mosaicism. In the normal control, the two normal alleles were detected as a single NcoI restriction fragment of 8.1 kb (Fig. 1).
FIG. 1.

Southern blot for CTG repeats in DM1 and normal control fibroblasts. Both DM1-03 and DM1-05 contain a normal allele and a smear of expanded allele. Control cell has no expanded allele.
Reprogramming efficiency is similar for DM1 and control iPSCs
The traditional retroviral method had similar transduction efficiencies between DM1 and control cells as demonstrated by GFP fluorescence (Fig. 2). Each 10-cm dish generated approximately 10 clones with a reprogramming efficiency of roughly 0.01% for both DM1 and control fibroblast cells. Colonies with typical hESC-like morphology were ready to be isolated around day 30 for both DM1 and control iPSCs. These cells were allowed to expand in culture, and 4–10 individual clones were further propagated.
FIG. 2.

Retroviral infection was conducted on a six-well plate. eGFP was used as a control to monitor infection efficiency. The infection efficiency is higher than 80%. There is no difference between DM1 and normal control.
DM1 and control iPSCs cells have stem cell characteristics
Morphology
DM1 and control iPSCs grew as flat, well-circumscribed colonies with cells having a high nuclear-to-cytoplasm ratio (Fig. 3). There was no change in their morphology or growth through subsequent passages (all beyond passage 20).
FIG. 3.

Typical DM1 and control iPSC colonies growing on MEFs are shown to be flat and well-circumscribed (A–C). High magnification shows tightly compacted cells with high nuclear/cytoplasm (D–F).
Stem cell markers
DM1 and control iPSCs, but not the original fibroblasts, were shown by RT-PCR to express stem cell markers (OCT4, NANOG, and SOX2) (Fig. 4). Immunocytofluorescence analysis revealed positive staining of these stem cell markers, alkaline phosphatase, OCT4, and SSEA4 in DM1 and control iPSCs (Fig. 5).
FIG. 4.
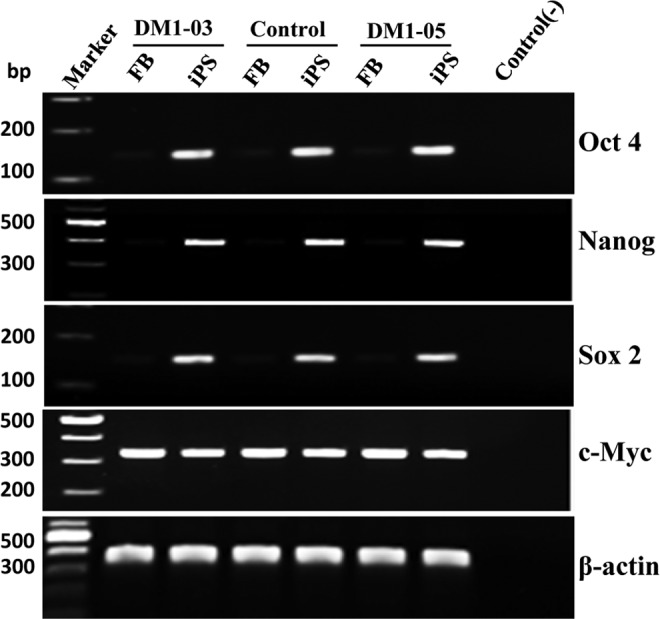
RT-PCR products of stem cell markers show that DM1 and normal control iPSCs, but not their progenitor fibroblasts (FB), express stem cell markers (Oct4, Nanog, and Sox2). Control (−), no RNA control. β-actin, loading control.
FIG. 5.
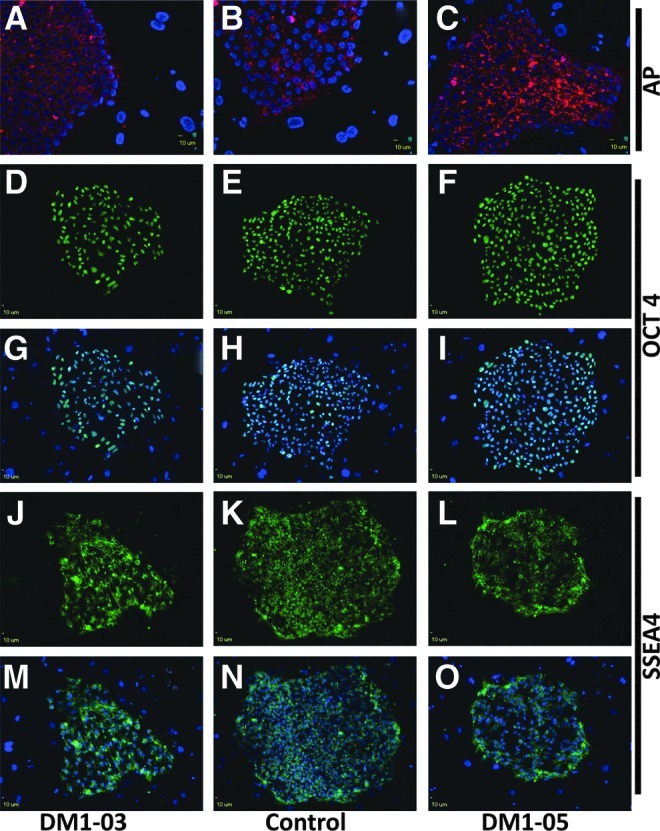
Immunofluorescence staining for stem cell markers. All iPSC colonies express alkaline phosphatase (AP) (A–C), Oct 4 (D–I), and SSEA 4 (J–O). G–I and M–O also show DAPI stain (blue). The surrounding feeder cells have no fluorescence for stem cell markers.
DM1 iPSCs are pluripotent
To examine the pluripotency of established iPSC lines, EBs were generated. All EBs showed a normal pattern of differentiation in culture chambers and differentiated into cells with various types of morphology (Fig. 6). Endodermal and mesodermal cell differentiation was confirmed by immunohistochemical staining of AFP and Desmin (Fig. 7A–C), respectively. Less endoderm differentiation appears under this experimental condition. Ectodermal differentiation was demonstrated by neural differentiation as described below.
FIG. 6.

DM1-03 EBs undergo differentiation in cultured chamber into cells with different morphology. Spontaneous neuronal differentiation can be seen with extending axons.
FIG. 7.

Intranuclear CUG repeats foci in EBs and fibroblasts. The endodermal and mesodermal cell differentiation of EB is confirmed by AFP (green) and Desmin (red) stains. Intranuclear foci are found in DM1 cells of three embryonic germ layers (A, B), as well as in their progenitor fibroblasts (D, E), but not normal control (C, F). Insets indicate areas of the image that are enlarged for greater detail. Blue indicates DAPI staining.
Intranuclear RNA foci are present within stem cells and terminally differentiated cells
Intranuclear RNA foci were detected in original DM1 fibroblasts, iPSCs, and all three embryonic germ layer cells derived from DM1 iPSCs, whereas no foci were found in the normal control at any stage (enlarged areas in Fig. 7A–F and Fig. 8).
FIG. 8.

Intranuclear CUG repeat foci in DM1-03 iPSCs. OCT 4 (green) and CUG RNA foci (red) were detected in DM1 iPSCs but not in feeder cells (arrows). (A) DAPI (blue) and RNA foci (red). (B) OCT4 (green) and RNA foci (red). (C) DAPI (blue), RNA foci (red), and Oct4 (green) merge.
DM1 and control iPSCs undergo lineage-directed neural differentiation
Neural differentiation of DM1 iPSCs was compared to control iPSCs. Both DM1 and control iPSCs underwent normal neural differentiation. iPSCs aggregated and formed neurospheres in the microwells (Fig. 9A). Neural rosettes started forming 3 days following attachment of neurospheres (Fig. 9B). Following replating of neural rosettes, NSCs began egressing from the edges and had the tendency to form loosely arranged rosettes (Fig. 9C) from which spontaneous differentiation occurred (Fig. 9D). Eventually, the entire plate was dominated by NSCs. The NSCs have been passed up to passage 6 without losing neural differentiation potential.
FIG. 9.

Neural differentiation of DM1-03 iPSCs. (A) Aggregates and neurosphere formation. (B) Neural rosettes 3 days after neurosphere attachment. NSCs egressing replated neural rosettes and reforming into small loosely arranged rosettes (C), from which spontaneous differentiation occurs (D).
Intranuclear RNA foci are formed in NSCs and terminally differentiated neurons and astrocytes
DM1 and control NSCs express Nestin and can further differentiate into neurons and astrocytes (confirmed by neuron and astrocyte markers). Intranuclear foci, a pathognomonic finding in DM1, are present in NSCs as well as terminally differentiated neurons and astrocytes (Fig. 10). No intranuclear foci were observed in control cells (data not shown).
FIG. 10.

Intranuclear CUG repeats foci in neural cells of DM1-03. DM1 NSCs express Nestin (A, green) and can further differentiate into astrocytes positive for GFAP (F, green) and neurons positive for Neurofilament H (B, green) and β-tubulin III (C–E, green). Intranuclear foci (red) are formed in NSCs, neurons, and astrocytes (see insets). Blue indicates DAPI staining.
Discussion
The main results of the present study were successful generation of disease-specific DM1 iPSC lines, neural differentiation, and demonstration of pathognomonic intranuclear CUG repeat foci in these cells. Once established, DM1 and control iPSCs grew in a similar pattern to hESCs with a high nuclear-to-cytoplasm ratio, expressed stem cell markers, and had stem cell features of self-renewal and pluripotency. These cells harbor the naturally mutated gene in the human genomic background and provide an unlimited resource of cells for repeated studies and comparisons, making them an excellent isogenic cellular model for DM1 pathology. The generated iPSCs undergo successfully neural differentiation into viable neural cells—cells that are otherwise difficult to obtain. Through lineage-specific differentiation and cell sorting, human DM1 iPSCs can be used to analyze the expression of DMPK, MBNL, CELF, and alternative splicing of their target genes in a cell-type specific manner.
Intranuclear CUG RNA foci are pathognomonic findings in DM1. These RNA foci are found in cultured human fibroblasts, myoblasts, biopsied muscle tissues (Taneja et al., 1995), and neuronal cells in postmortem brains from DM1 patients (Jiang et al., 2004) and transgenic mouse models (Lin et al., 2006; Mahadevan et al., 2006; Mankodi et al., 2000; Orengo et al., 2008; Seznec et al., 2001; Wang et al., 2007). The foci contain mutant DM1 mRNA and sequester splicing factors. Among them, MBNL 1, 2, and 3 are factors that have been extensively studied for their roles in DM1 pathogenesis (Ashizawa and Sarkar, 2011; de Leon and Cisneros, 2008; Lee and Cooper, 2009; Llamusi and Artero, 2008; Ranum and Cooper, 2006; Schara and Schoser, 2006). In this study, we found these foci not only in terminally differentiated cells but also in cells in pluripotent and multipotent cell stages. To our knowledge, this is the first report to track the foci formation during the developmental course from stem cell to terminally differentiated neuron. DM1 hESCs have been previously established from discarded embryos but without mention of intranuclear foci (Marteyn et al., 2011; Mateizel et al., 2006; Turetsky et al., 2008). As early as 10.5 days postconception, Dmpk in wild-type mouse is expressed at high levels in the developing nervous system (Kanadia et al., 2003), but nuclear foci have not been reported in the developing brain of early-stage embryos of DM1 transgenic mouse models. If the formation of intranuclear foci can be considered to represent the pathogenic process, the presence of foci in these iPSCs would suggest that adult DM1 patients may have a subclinical developmental disorder. Because sequestration of MBNL proteins into the nuclear foci would drive the alternative splicing of target transcripts toward their fetal isoforms, the pathogenic implication of the foci during embryonic development may be limited in DM1, particularly in adult-onset patients. However, it may be of interest to investigate subclinical phenotypes in presymptomatic DM1 mutation carriers. Thus, the presence of nuclear foci in iPSCs derived from adult-onset DM1 patients is relevant to the controversy of whether DM1 is a developmental or degenerative disorder.
Multiple approaches, including antisense oligonucleotides, small interfering RNA (siRNA), ribozymes, and chemicals, have been investigated for development of future treatments of DM1 (Furling et al., 2003; Langlois et al., 2003; Larsen et al., 2011; Lee et al., 2012; Mulders et al., 2009; Warf et al., 2009; Wheeler et al., 2009;Wheeler et al., 2012). However, all are based on animal models and cultured fibroblasts and myoblasts. The early results of these studies can be further validated in human disease-specific DM1 iPSCs and various cell lineages differentiated from the iPSCs. The in vivo efficacy can also be analyzed in transplanted teratoma tissues derived from DM1 iPSCs in nude mice. These cell lines represent an additional, more applicable model to further translate results from basic studies into clinical applications. Furthermore, recent technological developments involving genomic editing with either zinc-finger nucleases (ZFNs) or transcription activator-like effector nucleases (TALENs) could correct the genetic mutation in iPSCs (Hockemeyer and Jaenisch, 2010; Hockemeyer et al., 2009; Hockemeyer et al., 2011; Sebastiano et al., 2011; Zou et al., 2009; Zou et al., 2011), which can be transplanted for replacement of damaged cells with or without in vitro differentiation. Although there are issues such as tumorigenicity, imprinting, and immunogenicity (Blum and Benvenisty, 2009; Izpisua Belmonte et al., 2009; Kiuru et al., 2009; Pick et al., 2009; Stadtfeld et al., 2010; Zhao et al., 2011), we believe that DM1-specific human iPSCs are useful for testing these technologies and establishing the basis for future cell-based therapy.
In conclusion, we have established disease-specific human DM1 iPSC lines. These mutant iPSCs harbor mutations in the native genomic background of DM1 patients and have the potential of differentiation into cells of the three embryonic germ layers. Neural cells differentiated form these iPSCs contain pathognomonic intranuclear RNA foci. These cell lines represent an invaluable tool for the study of DM1 CNS neuropathogenesis and can readily be exploited as a translational platform for therapeutic drug development.
Acknowledgments
Dr. Guangbin Xia was a trainee of the Clinical Research Consortium for Spinocerebellar Ataxias (CRC-SCA), which was supported by NIH NS068897 (T.A.). Part of the study was supported by NIH AR046799 (M.S.S.) and Marigold Foundation (T.A.).
Author Disclosure Statement
All authors declare that they have nothing to disclose.
Author contributions are as follows: Study concept and design: Ashizawa, Terada, Xia, Swanson. Acquisition of data: Xia, Santostefano, Goodwin, Liu. Analysis and interpretation of data: Xia, Santostefano, Goodwin, Swanson, Terada, Ashizawa. Drafting of the manuscript: Xia, Ashizawa, and Terada. Critical revision of the manuscript for important intellectual content: Subramony, Santostefano, Goodwin, Swanson. Administrative, technical, and material support: Liu. Study supervision: Ashizawa, Terada.
References
- Ashizawa T. Sarkar P.S. Myotonic dystrophy types 1 and 2. Handb. Clin. Neurol. 2011;101:193–237. doi: 10.1016/B978-0-08-045031-5.00015-3. [DOI] [PubMed] [Google Scholar]
- Baharvand H. Totonchi M. Taei A. Seifinejad A. Aghdami N. Salekdeh G.H. Human-induced pluripotent stem cells: Derivation, propagation, and freezing in serum- and feeder layer-free culture conditions. Methods Mol. Biol. 2010;584:425–443. doi: 10.1007/978-1-60761-369-5_23. [DOI] [PubMed] [Google Scholar]
- Bird T.D. Follett C. Griep E. Cognitive and personality function in myotonic muscular dystrophy. J. Neurol. Neurosurg. Psychiatry. 1983;46:971–980. doi: 10.1136/jnnp.46.11.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum B. Benvenisty N. The tumorigenicity of diploid and aneuploid human pluripotent stem cells. Cell Cycle. 2009;8:3822–3830. doi: 10.4161/cc.8.23.10067. [DOI] [PubMed] [Google Scholar]
- Charizanis K. Lee K.Y. Batra R. Goodwin M. Zhang C. Yuan Y. Shiue L. Cline M. Scotti M.M. Xia G. Kumar A. Ashizawa T. Clark H.B. Kimura T. Takahashi M. P. Fujimura H. Jinnai K. Yoshikawa H. Gomes-Pereira M. Gourdon G. Sakai N. Nishino S. Foster T. C. Ares M., Jr. Darnell R.B. Swanson M.S. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75:437–450. doi: 10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon M.B. Cisneros B. Myotonic dystrophy 1 in the nervous system: from the clinic to molecular mechanisms. J. Neurosci. Res. 2008;86:18–26. doi: 10.1002/jnr.21377. [DOI] [PubMed] [Google Scholar]
- Dhaenens C.M. Schraen-Maschke S. Tran H. Vingtdeux V. Ghanem D. Leroy O. Delplanque J. Vanbrussel E. Delacourte A. Vermersch P. Maurage C.A. Gruffat H. Sergeant A. Mahadevan M.S. Ishiura S. Buee L. Cooper T.A. Caillet-Boudin M.L. Charlet-Berguerand N. Sablonniere B. Sergeant N. Overexpression of MBNL1 fetal isoforms and modified splicing of Tau in the DM1 brain: Two individual consequences of CUG trinucleotide repeats. Exp. Neurol. 2008;210:467–478. doi: 10.1016/j.expneurol.2007.11.020. [DOI] [PubMed] [Google Scholar]
- Dhaenens C.M. Tran H. Frandemiche M.L. Carpentier C. Schraen-Maschke S. Sistiaga A. Goicoechea M. Eddarkaoui S. Van Brussels E. Obriot H. Labudeck A. Gevaert M. H. Fernandez-Gomez F. Charlet-Berguerand N. Deramecourt V. Maurage C.A. Buee L. de Munain A.L. Sablonniere B. Caillet-Boudin M.L. Sergeant N. Mis-splicing of Tau exon 10 in myotonic dystrophy type 1 is reproduced by overexpression of CELF2 but not by MBNL1 silencing. Biochim. Biophys. Acta. 2011;1812:732–742. doi: 10.1016/j.bbadis.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Douniol M. Jacquette A. Guile J.M. Tanguy M.L. Angeard N. Heron D. Plaza M. Cohen D. Psychiatric and cognitive phenotype in children and adolescents with myotonic dystrophy. Eur. Child Adolesc. Psychiatry. 2009;18:705–715. doi: 10.1007/s00787-009-0037-4. [DOI] [PubMed] [Google Scholar]
- Ekstrom A.B. Hakenas-Plate L. Samuelsson L. Tulinius M. Wentz E. Autism spectrum conditions in myotonic dystrophy type 1: A study on 57 individuals with congenital and childhood forms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008;147B:918–926. doi: 10.1002/ajmg.b.30698. [DOI] [PubMed] [Google Scholar]
- Fu Y.H. Pizzuti A. Fenwick R.G., Jr. King J. Rajnarayan S. Dunne P.W. Dubel J. Nasser G.A. Ashizawa T. de Jong P., et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- Furling D. Doucet G. Langlois M.A. Timchenko L. Belanger E. Cossette L. Puymirat J. Viral vector producing antisense RNA restores myotonic dystrophy myoblast functions. Gene Ther. 2003;10:795–802. doi: 10.1038/sj.gt.3301955. [DOI] [PubMed] [Google Scholar]
- Gomes-Pereira M. Cooper T.A. Gourdon G. Myotonic dystrophy mouse models: Towards rational therapy development. Trends Mol. Med. 2011;17:506–517. doi: 10.1016/j.molmed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper P. Myotonic Dystrophy. 3rd. London: WB Saunders; 2001. [Google Scholar]
- Hockemeyer D. Jaenisch R. Gene targeting in human pluripotent cells. Cold Spring Harb. Symp. Quant. Biol. 2010;75:201–209. doi: 10.1101/sqb.2010.75.021. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D. Soldner F. Beard C. Gao Q. Mitalipova M. DeKelver R.C. Katibah G.E. Amora R. Boydston E.A. Zeitler B. Meng X. Miller J.C. Zhang L. Rebar E.J. Gregory P.D. Urnov F.D. Jaenisch R. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D. Wang H. Kiani S. Lai C.S. Gao Q. Cassady J.P. Cost G.J. Zhang L. Santiago Y. Miller J.C. Zeitler B. Cherone J.M. Meng X. Hinkley S.J. Rebar E. J. Gregory P.D. Urnov F.D. Jaenisch R. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izpisua Belmonte J.C. Ellis J. Hochedlinger K. Yamanaka S. Induced pluripotent stem cells and reprogramming: Seeing the science through the hype. Nat. Rev. Genet. 2009;10:878–883. doi: 10.1038/nrg2700. [DOI] [PubMed] [Google Scholar]
- Jiang H. Mankodi A. Swanson M.S. Moxley R.T. Thornton C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- Kanadia R.N. Urbinati C.R. Crusselle V.J. Luo D. Lee Y.J. Harrison J.K. Oh S.P. Swanson M.S. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr. Patterns. 2003;3:459–462. doi: 10.1016/s1567-133x(03)00064-4. [DOI] [PubMed] [Google Scholar]
- Kiuchi A. Otsuka N. Namba Y. Nakano I. Tomonaga M. Presenile appearance of abundant Alzheimer's neurofibrillary tangles without senile plaques in the brain in myotonic dystrophy. Acta Neuropathol. 1991;82:1–5. doi: 10.1007/BF00310916. [DOI] [PubMed] [Google Scholar]
- Kiuru M. Boyer J.L. O'Connor T.P. Crystal R.G. Genetic control of wayward pluripotent stem cells and their progeny after transplantation. Cell Stem Cell. 2009;4:289–300. doi: 10.1016/j.stem.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois M.A. Lee N.S. Rossi J.J. Puymirat J. Hammerhead ribozyme-mediated destruction of nuclear foci in myotonic dystrophy myoblasts. Mol. Ther. 2003;7:670–680. doi: 10.1016/s1525-0016(03)00068-6. [DOI] [PubMed] [Google Scholar]
- Larsen J. Pettersson O.J. Jakobsen M. Thomsen R. Pedersen C.B. Hertz J.M. Gregersen N. Corydon T.J. Jensen T.G. Myoblasts generated by lentiviral mediated MyoD transduction of myotonic dystrophy type 1 (DM1) fibroblasts can be used for assays of therapeutic molecules. BMC Res. Notes. 2011;4:490. doi: 10.1186/1756-0500-4-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.E. Cooper T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009;37:1281–1286. doi: 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.E. Bennett C.F. Cooper T.A. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA. 2012;109:4221–4226. doi: 10.1073/pnas.1117019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy O. Dhaenens C.M. Schraen-Maschke S. Belarbi K. Delacourte A. Andreadis A. Sablonniere B. Buee L. Sergeant N. Caillet-Boudin M.L. ETR-3 represses Tau exons 2/3 inclusion, a splicing event abnormally enhanced in myotonic dystrophy type I. J. Neurosci. Res. 2006a;84:852–859. doi: 10.1002/jnr.20980. [DOI] [PubMed] [Google Scholar]
- Leroy O. Wang J. Maurage C.A. Parent M. Cooper T. Buee L. Sergeant N. Andreadis A. Caillet-Boudin M.L. Brain-specific change in alternative splicing of Tau exon 6 in myotonic dystrophy type 1. Biochim. Biophys. Acta. 2006b;1762:460–467. doi: 10.1016/j.bbadis.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Lin X. Miller J.W. Mankodi A. Kanadia R.N. Yuan Y. Moxley R.T. Swanson M.S. Thornton C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- Llamusi B. Artero R. Molecular effects of the CTG repeats in mutant dystrophia myotonica protein kinase gene. Curr. Genomics. 2008;9:509–516. doi: 10.2174/138920208786847944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan M.S. Yadava R.S. Yu Q. Balijepalli S. Frenzel-McCardell C.D. Bourne T.D. Phillips L.H. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 2006;38:1066–1070. doi: 10.1038/ng1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A. Logigian E. Callahan L. McClain C. White R. Henderson D. Krym M. Thornton C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- Marteyn A. Maury Y. Gauthier M.M. Lecuyer C. Vernet R. Denis J.A. Pietu G. Peschanski M. Martinat C. Mutant human embryonic stem cells reveal neurite and synapse formation defects in type 1 myotonic dystrophy. Cell Stem Cell. 2011;8:434–444. doi: 10.1016/j.stem.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Mateizel I. De Temmerman N. Ullmann U. Cauffman G. Sermon K. Van de Velde H. De Rycke M. Degreef E. Devroey P. Liebaers I. Van Steirteghem A. Derivation of human embryonic stem cell lines from embryos obtained after IVF and after PGD for monogenic disorders. Hum. Reprod. 2006;21:503–511. doi: 10.1093/humrep/dei345. [DOI] [PubMed] [Google Scholar]
- Maurage C.A. Udd B. Ruchoux M.M. Vermersch P. Kalimo H. Krahe R. Delacourte A. Sergeant N. Similar brain tau pathology in DM2/PROMM and DM1/Steinert disease. Neurology. 2005;65:1636–1638. doi: 10.1212/01.wnl.0000184585.93864.4e. [DOI] [PubMed] [Google Scholar]
- Mitake S. Inagaki T. Niimi T. Shirai T. Yamamoto M. [Development of Alzheimer neurofibrillary changes in two autopsy cases of myotonic dystrophy] Rinsho Shinkeigaku. 1989;29:488–492. [PubMed] [Google Scholar]
- Mollamohammadi S. Taei A. Pakzad M. Totonchi M. Seifinejad A. Masoudi N. Baharvand H. A simple and efficient cryopreservation method for feeder-free dissociated human induced pluripotent stem cells and human embryonic stem cells. Hum. Reprod. 2009;24:2468–2476. doi: 10.1093/humrep/dep244. [DOI] [PubMed] [Google Scholar]
- Mulders S.A. van den Broek W.J. Wheeler T.M. Croes H.J. van Kuik-Romeijn P. de Kimpe S.J. Furling D. Platenburg G.J. Gourdon G. Thornton C.A. Wieringa B. Wansink D.G. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA. 2009;106:13915–13920. doi: 10.1073/pnas.0905780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata A. Terae S. Fujita M. Tashiro K. Anterior temporal white matter lesions in myotonic dystrophy with intellectual impairment: An MRI and neuropathological study. Neuroradiology. 1998;40:411–415. doi: 10.1007/s002340050613. [DOI] [PubMed] [Google Scholar]
- Orengo J.P. Chambon P. Metzger D. Mosier D.R. Snipes G.J. Cooper T.A. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA. 2008;105:2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick M. Stelzer Y. Bar-Nur O. Mayshar Y. Eden A. Benvenisty N. Clone- and gene-specific aberrations of parental imprinting in human induced pluripotent stem cells. Stem Cells. 2009;27:2686–2690. doi: 10.1002/stem.205. [DOI] [PubMed] [Google Scholar]
- Portwood M.M. Wicks J.J. Lieberman J.S. Duveneck M.J. Intellectual and cognitive function in adults with myotonic muscular dystrophy. Arch. Phys. Med. Rehabil. 1986;67:299–303. [PubMed] [Google Scholar]
- Ranum L.P. Cooper T.A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- Roig M. Balliu P.R. Navarro C. Brugera R. Losada M. Presentation, clinical course, and outcome of the congenital form of myotonic dystrophy. Pediatr. Neurol. 1994;11:208–213. doi: 10.1016/0887-8994(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Romeo V. Myotonic dystrophy type 1 or Steinert's disease. Adv. Exp. Med. Biol. 2012;724:239–257. doi: 10.1007/978-1-4614-0653-2_18. [DOI] [PubMed] [Google Scholar]
- Romeo V. Pegoraro E. Ferrati C. Squarzanti F. Soraru G. Palmieri A. Zucchetta P. Antunovic L. Bonifazi E. Novelli G. Trevisan C.P. Ermani M. Manara R. Angelini C. Brain involvement in myotonic dystrophies: Neuroimaging and neuropsychological comparative study in DM1 and DM2. J. Neurol. 2010;257:1246–1255. doi: 10.1007/s00415-010-5498-3. [DOI] [PubMed] [Google Scholar]
- Rubinsztein J.S. Rubinsztein D.C. McKenna P.J. Goodburn S. Holland A.J. Mild myotonic dystrophy is associated with memory impairment in the context of normal general intelligence. J. Med. Genet. 1997;34:229–233. doi: 10.1136/jmg.34.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schara U. Schoser B.G. Myotonic dystrophies type 1 and 2: A summary on current aspects. Semin. Pediatr. Neurol. 2006;13:71–79. doi: 10.1016/j.spen.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Sebastiano V. Maeder M.L. Angstman J.F. Haddad B. Khayter C. Yeo D.T. Goodwin M.J. Hawkins J.S. Ramirez C.L. Batista L.F. Artandi S.E. Wernig M. Joung J.K. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells. 2011;29:1717–1726. doi: 10.1002/stem.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N. Sablonniere B. Schraen-Maschke S. Ghestem A. Maurage C.A. Wattez A. Vermersch P. Delacourte A. Dysregulation of human brain microtubule-associated tau mRNA maturation in myotonic dystrophy type 1. Hum. Mol. Genet. 2001;10:2143–2155. doi: 10.1093/hmg/10.19.2143. [DOI] [PubMed] [Google Scholar]
- Seznec H. Agbulut O. Sergeant N. Savouret C. Ghestem A. Tabti N. Willer J.C. Ourth L. Duros C. Brisson E. Fouquet C. Butler-Browne G. Delacourte A. Junien C. Gourdon G. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum. Mol. Genet. 2001;10:2717–2726. doi: 10.1093/hmg/10.23.2717. [DOI] [PubMed] [Google Scholar]
- Sistiaga A. Urreta I. Jodar M. Cobo A.M. Emparanza J. Otaegui D. Poza J.J. Merino J.J. Imaz H. Marti-Masso J.F. Lopez de Munain A. Cognitive/personality pattern and triplet expansion size in adult myotonic dystrophy type 1 (DM1): CTG repeats, cognition and personality in DM1. Psychol. Med. 2010;40:487–495. doi: 10.1017/S0033291709990602. [DOI] [PubMed] [Google Scholar]
- Stadtfeld M. Apostolou E. Akutsu H. Fukuda A. Follett P. Natesan S. Kono T. Shioda T. Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyaert J. Umans S. Willekens D. Legius E. Pijkels E. de Die-Smulders C. Van den Berghe H. Fryns J.P. A study of the cognitive and psychological profile in 16 children with congenital or juvenile myotonic dystrophy. Clin. Genet. 1997;52:135–141. doi: 10.1111/j.1399-0004.1997.tb02533.x. [DOI] [PubMed] [Google Scholar]
- Suenaga K. Lee K.Y. Nakamori M. Tatsumi Y. Takahashi M.P. Fujimura H. Jinnai K. Yoshikawa H. Du H. Ares M., Jr. Swanson M.S. Kimura T. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PLoS One. 2012;7:e33218. doi: 10.1371/journal.pone.0033218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K. Tanabe K. Ohnuki M. Narita M. Ichisaka T. Tomoda K. Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Taneja K.L. McCurrach M. Schalling M. Housman D. Singer R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995;128:995–1002. doi: 10.1083/jcb.128.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turetsky T. Aizenman E. Gil Y. Weinberg N. Shufaro Y. Revel A. Laufer N. Simon A. Abeliovich D. Reubinoff B.E. Laser-assisted derivation of human embryonic stem cell lines from IVF embryos after preimplantation genetic diagnosis. Hum. Reprod. 2008;23:46–53. doi: 10.1093/humrep/dem351. [DOI] [PubMed] [Google Scholar]
- Vermersch P. Sergeant N. Ruchoux M.M. Hofmann-Radvanyi H. Wattez A. Petit H. Dwailly P. Delacourte A. Specific tau variants in the brains of patients with myotonic dystrophy. Neurology. 1996;47:711–717. doi: 10.1212/wnl.47.3.711. [DOI] [PubMed] [Google Scholar]
- Wang G.S. Kearney D.L. De Biasi M. Taffet G. Cooper T.A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Invest. 2007;117:2802–2811. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf M.B. Nakamori M. Matthys C.M. Thornton C.A. Berglund J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. USA. 2009;106:18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T.M. Sobczak K. Lueck J.D. Osborne R.J. Lin X. Dirksen R.T. Thornton C. A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325:336–339. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T.M. Leger A.J. Pandey S.K. MacLeod A.R. Nakamori M. Cheng S.H. Wentworth B.M. Bennett C.F. Thornton C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488:111–115. doi: 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura N. Otake M. Igarashi K. Matsunaga M. Takebe K. Kudo H. Topography of Alzheimer's neurofibrillary change distribution in myotonic dystrophy. Clin. Neuropathol. 1990;9:234–239. [PubMed] [Google Scholar]
- Zhao T. Zhang Z.N. Rong Z. Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–215. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
- Zou J. Maeder M.L. Mali P. Pruett-Miller S.M. Thibodeau-Beganny S. Chou B.K. Chen G. Ye Z. Park I.H. Daley G.Q. Porteus M.H. Joung J.K. Cheng L. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J. Mali P. Huang X. Dowey S.N. Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011;118:4599–4608. doi: 10.1182/blood-2011-02-335554. [DOI] [PMC free article] [PubMed] [Google Scholar]