

Abstract
Human cytomegalovirus (HCMV) is a prevalent virus that infects up to 90% of the population. The goal of this research is to determine if small molecular prodrug substrates can be developed for a specific HCMV encoded protease and thus achieve site-specific activation. HCMV encodes a 256 amino acid serine protease that is responsible for capsid assembly, an essential process for herpes virus production. The esterase activity of the more stable HCMV A143T/A144T protease mutant was evaluated with model p-nitrophenol (ONp) esters, Boc-Xaa-ONp (Ala, Leu, Ile, Val, Gln, Phe at the Xaa position). We demonstrate that the A143T/A144T mutant has esterase activity toward specific small ester compounds, e.g., Boc-l-Ala-ONp. Mono amino acid and dipeptide prodrugs of ganciclovir (GCV) were also synthesized and evaluated for hydrolysis by the A143T/A144T protease mutant in solution. Hydrolysis of these prodrugs was also evaluated in Caco-2 cell homogenates, human liver microsomes (HLMs), and rat and human plasma. For the selectivity potential of the prodrugs, the hydrolysis ratio was evaluated as a percentage of prodrug hydrolyzed by the HCMV protease over the percentages of prodrug hydrolyses by Caco-2 cell homogenates, HLMs, and human/rat plasma. A dipeptide prodrug of ganciclovir, Ac-l-Gln-l-Ala-GCV, emerged as a potential selective prodrug candidate. The results of this research demonstrate that targeting prodrugs for activation by a specific protease encoded by the infectious HCMV pathogen may be achievable.
Keywords: prodrug, human cytomegalovirus protease, ganciclovir, prodrug activation, prodrug targeting, site-specific, drug delivery
Introduction
The prodrug approach has often been used to overcome pharmaceutical, pharmacokinetic, and pharmacodynamic barriers that limit parent drug efficacy. Maintaining the efficacy of the parent drug requires that the prodrug be transformed back to its pharmacologically active parent form in order to exert its therapeutic effects.1 The molecular insights of the past decade can now be used to develop mechanistic approaches to prodrug design specifically targeting drug transporters and activating enzymes expressed in infected cells or overexpressed in diseased tissue.2,3
The human cytomegalovirus (HCMV) is a ubiquitous betaherpesvirinae, a highly prevalent pathogen.4 Infection in normal individuals is generally asymptomatic; however, HCMV is associated with important morbidity and mortality in immune compromised patients.5 One of the most potent anti-HCMV therapeutic agents is the nucleoside analogue 9-(1,3-dihydroxy-2-propoxymethyl)guanine (ganciclovir; GCV).6,7 An l-valine ester prodrug of GCV (valganciclovir) has been developed and approved which provides improved systemic availability of GCV.8
Common to other herpes viruses, HCMV has the capacity for lifelong persistence within the host by establishment of cellular sites of viral latency.9 Occasionally the virus is reactivated but is suppressed mainly by cell-mediated immune surveillance in normal individuals. However, in immunocompromised patients, reactivation may result in relapse of infection.10 The latent nature of herpes viruses and the frequent relapses, especially in immunocompromised patients, require long-term drug treatment. The major adverse effects with GCV treatment are hematological in nature, including neutropenia, anemia, and thrombocytopenia.11
HCMV encodes a unique protease that is responsible for capsid assembly, a crucial process for herpes virion production.12 A 708 amino acid precursor protein of HCMV protease is encoded by the UL80 gene.13 Studies have demonstrated that this 80 kDa precursor protein undergoes autoproteolysis at several sites, resulting in a 30 kDa active serine protease.13,14
HCMV protease is a serine protease. It has little sequence homology with chymotrypsin-like or subtilisin-like proteases and shares 30% homology with different herpes virus subfamilies, with 90% similarity within the subfamily. It has been reported that dimerization of this 30 kDa protease is important in its catalytic activity.15 The unique fold and active site of HCMV protease have been revealed in its 2.5 Å crystal structure.16,17 The catalytic triad in the active site of HCMV protease consists of Ser132, His63, and His157. This is unique in the serine hydrolase superfamily; the general hydrolase catalytic triad contains aspartic/glutamic acid as the third member. The HCMV protease cleaves its natural substrates between alanine and serine.18 In terms of small molecular weight substrates, there are a few 9-amino-acid peptide substrates, with sequences derived from the HCMV protease endogenous peptide substrates.19 The shortest substrate tested to date is a fluorogenic tetrapeptide, N-Ac-Tag-Asn(NMe2)-Ala-AMC.20
The structural and functional uniqueness of HCMV protease and the fact that it is virally encoded suggest that it is a potential target for site-specific prodrug activation at the site of infection. In this study, we demonstrate that HCMV protease is able to hydrolyze an ester bond with substrate specificity. First, we generated a stable mutant of HCMV protease for prodrug evaluation, and determined its esterase activity against p-nitrophenol (ONp) esters, with Boc-l-Ala-ONp being preferred. To evaluate the prodrug substrate specificity of HCMV protease, we used GCV as the model parent drug with blocked amino acid and dipeptides as potential prodrug substrates. The goal of this strategy was to determine if amino acid/dipeptide prodrugs of GCV could be readily and selectively activated by HCMV protease. Finally, the stabilities of these prodrugs in Caco-2 cell homogenates, human liver microsomes, and rat and human plasma were evaluated to determine their potential for selective activation by the HCMV protease. The results of this study demonstrate that site-specific targeting is possible via targeting the virally encoded CMV protease.
Materials and Methods
Materials
HCMV protease cDNA was kindly provided by Professor John C. Drach of the University of Michigan. All chemicals were either analytical or HPLC grade. tert-Butyloxycarbonyl (Boc) protected l-amino acid p-nitrophenol (ONp) esters (Boc-l-Ala-ONp, Boc-l-Ile-ONp, Boc-l-Leu-ONp, Boc-l-Val-ONp, Boc-l-Gly-ONp and Boc-l-Phe-ONp), Nα-acetyl-l-alanine (Ac-l-Ala-OH), N-(tert-butoxycarbonyl)-l-alanine (Boc-l-Ala-OH), N-carbobenzyloxy-l-alanine (CBz-l-Ala-OH), N-(benzyloxycarbonyl)-l-alanine (Bz-l-Ala-OH), Nα-acetyl-l-glutamine (Ac-l-Gln-OH), Nα-acetyl-l-asparagine (Ac-l-Asn-OH), and l-alanine ethyl ester (NH2-l-Ala-OEt) hydrochloride were purchased from Chem-Impex International,Inc. (Wood Dale, IL). Trifluoroacetic acid (TFA), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC HCl), 4-(dimethylamino)pyridine (DMAP), N,N-dimethylformamide (DMF), Hydroxybenzotriazole (HOBt), O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU), N,N-diisopropylethylamine (DIPEA) triethylamine (TEA), trifluoroacetic acid (TFA), dithiothreitol (DTT), and ganciclovir (GCV) were purchased from Sigma-Aldrich Co. (St. Louis, MO). PD-10 columns were purchased from Amersham Pharmacia Biotech (Piscataway, NJ). DNA extraction and purification kits and Ni-NTA superflow cartridges were obtained from Qiagen Inc. (Valencia, CA). Enzymes for molecular cloning were acquired from New England Biolabs (Ipswich, MA) and Roche Applied Science (Indianapolis, IN). Plasmid pBluescript II SK(+) was obtained from Stratagene (La Jolla, CA).
Ganciclovir Prodrug Synthesis
1. Ac-l-Ala-GCV Mono Ester (3a)
As described in Scheme 1, 150 mg (0.588 mmol) of GCV was dissolved in 10 mL of anhydrous DMF by heating at 110–130 °C (oil bath). 107.8 mg (0.882 mmol) of Ac-l-Ala-OH, 281.0 mg of EDC (1.47 mmol), and 154.2 mg (1.176 mmol) of DMAP were dissolved in 5 mL of DMF. This solution was stirred at room temperature for 1 h, and the GCV in DMF was added to it dropwise. After the reaction mixture was stirred at room temperature under argon gas for 48 h, the solvent was removed in vacuo. The resulting reaction mixture was dissolved in ethyl acetate (EA) and water. The aqueous phase was collected and run in preparative HPLC using an Xterra Prep MS C18 column (Waters) for purification. A water/methanol gradient, both containing 0.1% TFA, was used as the mobile phase. Fractions containing Ac-l-Ala-GCV monoester were pooled, and the solvent was removed in vacuo. Freeze-drying overnight of this final sample resulted in 31.2 mg (0.0847 mmol, yield 14.4%) of monoester product: 1H NMR (CD3OD) δ 8.37 (1H, br, H-8), 5.64 (2H, m, H-1′), 4.28 (2H, m, COOCH2), 4.07 (1H, m, H-α), 4.00 (1H, m, H-4′), 3.57 (2H, m, CH2OH), 1.96 (3H, s, COCH3), 1.31 (3H, m, CH3-β); MS m/z 369.1 [M + H]+.
Scheme 1. Synthesis of Ac-l-Ala-GCV (3a), Boc-l-Ala-GCV (3b), and CBz-l-Ala-GCV (3c).

2. Boc-l-Ala-GCV Mono Ester (3b)
3b was synthesized using a procedure similar to that used for the synthesis of Ac-l-Ala-GCV (3a) (yield 46.4%): 1H NMR (CD3OD) δ 8.02 (1H, s, H-8), 5.59 (2H, m, H-1′), 4.28 (1H, m, COOCH2), 4.08 (1H, m, COOCH2, H-α), 3.97 (1H, m, H-4′), 3.57 (2H, m, CH2OH), 1.42 (9H, s, Boc), 1.27 (3H, m, CH3-β); MS m/z 427.1 [M + H]+.
3. CBz-l-Ala-GCV Mono Ester (3c)
3c was synthesized using a procedure similar to that used for the synthesis of Ac-l-Ala-GCV (3a) (yield 47.6%): 1H NMR (CD3OD) δ 8.61(1H, br, H-8), 7.30(5H, m, aromatic protons), 5.64 (2H, s, H-1′), 5.08 (2H, m, CH2Ph), 4.27 (1H, m, COOCH2), 4.05–4.17 (2H, m, COOCH2, H-α), 4.00 (1H, m, H-4′), 3.57 (2H, m, CH2OH), 1.33 (3H, m, CH3-β); MS m/z 369.1 [M + H]+.
4. l-Ala-GCV Mono Ester (4)
As shown in Scheme 2, 60 mg (0.141 mmol) of Boc-l-Ala-GCV (3b) was dissolved in TFA:CH2Cl2 (1:1) under argon for 4 h. After removal of the solvent, the residue was dissolved in 0.1% TFA, filtered, and lyophilized to obtain 40.47 mg (0.124 mmol) of l-Ala-GCV (4) (yield 87.9%): 1H NMR (CD3OD) δ 8.42 (1H, br, H-8), 5.66, 5.65 (2H, s, H-1′), 4.38 (1H, m, COOCH2), 4.21 (1H, m, COOCH2), 4.02 (2H, m, H-α, H-4′), 3.62 (2H, m, CH2OH), 1.45 (3H, m, CH3-β); MS m/z 327.0 [M + H]+.
Scheme 2. Synthesis of l-Ala-GCV (4) and Bz-l-Ala-GCV (5).

5. Bz-l-Ala-GCV Mono Ester (5)
As shown in Scheme 2, 64 mg (0.196 mmol) of l-Ala-GCV (4) and 44.4 mg (0.196 mmol) of benzoic anhydride were dissolved in 6 mL of dichloromethane (DCM), and the reaction mixture was stirred under argon at room temperature for 4 h. After removal of the solvent, the residue was dissolved in methanol and purified using preparative HPLC. Fractions containing Bz-l-Ala-GCV (5) were pooled, and the solvent was removed in vacuo and lyophilized (yield 17.2%): 1H NMR (CD3OD) δ 8.45(1H, br, H-8), 7.44–7.85 (5H, m, aromatic protons) 5.64 (2H, m, H-1′), 4.51 (1H, m, COOCH2), 4.32 (1H, m, COOCH2), 4.14 (1H, m, H-α), 4.03(1H, m, H-4′), 3.57 (2H, m, CH2OH), 1.46 (3H, m, CH3-β); MS m/z 431.1 [M + H]+.
6. Ac-l-Asn-l-Ala-GCV (9a)
As shown in Scheme 3, to a stirred solution of 200 mg (1.148 mmol) of Ac-l-Asn-OH (6a) in DMF were added HOBt (155 mg, 1.148 mmol), HBTU (435.4 mg, 1.148 mmol), and 399 μL (2.296 mmol) of DIPEA at 0 °C, and the mixture was stirred at room temperature for 30 min. NH2-Ala-OEt (7) (176.3 mg, 1.148 mmol) in DMF was added to the above reaction mixture. The reaction mixture was stirred under argon for 48 h. The solvent was removed in vacuo, and the product was extracted using water/EA. The product remained in the aqueous phase and was separated from reaction impurities using a Shimadzu preparative HPLC with an Xterra Prep MS C18 column for purification. Prep HPLC fractions containing the dipeptide product, Ac-l-Asn-l-Ala-OEt (211 mg, 0.773 mmol), were pooled, and the solvent was removed in vacuo. After freeze-drying (yield 52%), the ethyl group was removed from the carboxylic acid end of the dipeptide by addition of LiOH (9.3 mg, 0.386 mmol) to a solution of 211 mg (0.773 mmol) of Ac-l-Asn-l-Ala-OEt in 25 mL of methanol/H2O (4:1, v/v). This was stirred at room temperature for 4 h, after which the solvent was removed in vacuo. The product was then dissolved in EA and washed with 10% (w/v) citric acid. The solvent was removed in vacuo to give 149.5 mg of Ac-l-Asn-l-Ala-OH (8a) (0.610 mmol, yield 78.9%). GCV (155.6 mg, 0.61 mmol) was dissolved in 8 mL of DMF by heating at 110–130 °C. Ac-l-Asn-l-Ala-OH (8a) (149.5 mg, 0.61 mmol), EDC (174.9 mg, 0.915 mmol), and DMAP (111.8 mg, 0.915 mmol) were dissolved in 5 mL of DMF and stirred at room temperature for 1 h. To this solution, GCV in DMF was added dropwise. The reaction mixture was stirred under argon at room temperature for 48 h. After removing the solvent in vacuo, the solid content was dissolved and extracted with EA/water. The water phase was separated, and 28.0 mg of Ac-l-Asn-l-Ala-GCV (9a) (0.058 mmol, yield 9.5%) was purified using Shimadzu preparative HPLC as described above: 1H NMR (CD3OD) δ 8.48 (1H, br, H-8), 5.66 (2H, s, H-1′), 4.76 (1H, m, H-α of Asn), 4.00–4.35 (4H, m, COOCH2, H-α of Ala, H-4′), 3.56 (2H, m, CH2OH), 2.71 (1H, m, H-β of Asn), 2.62 (1H, m, H-β of Asn), 1.99 (3H, s, COCH3), 1.34 (3H, m, CH3-β of Ala); MS m/z 483.2 [M + H]+.
Scheme 3. Synthesis of Ac-l-Asn-l-Ala-GCV (9a) and Ac-l-Gln-l-Ala-GCV (9b).

7. Ac-l-Gln-l-Ala-GCV (9b)
9b was synthesized using a procedure similar to that used for the synthesis of Ac-l-Asn-l-Ala-GCV (yield 15%): 1H NMR (CD3OD) δ 8.64 (1H, br, H-8), 5.68 (2H, m, H-1′), 4.01–4.39 (5H, m, COOCH2, H-α of Ala and Gln, H-4′), 3.58 (2H, m, CH2OH), 2.32 (2H, m, H-β of Gln), 2.08 (1H, m, H-γ of Gln), 1.99 (3H, m, COCH3), 1.91 (1H, m, H-γ of Gln), 1.34 (3H, m, CH3-β of Ala); MS m/z 497.1 [M + H]+.
Cloning, Expression, and Purification of His-Tagged Wild Type HCMV Protease
The gene encoding the catalytic domain of human cytomegalovirus protease was amplified via PCR using the cDNA as the template. The forward and reverse primers were GCTAGGCTCATATGACGATGGACGAGCAGCAG and GCTAGGCTAGATCTAACGCCTTGACGTATGACTCGC, respectively. Restriction digestion sequences for BglII and NdeI were included in the forward and reverse primers, respectively. The phosphorylated PCR product was blunt-end ligated into the EcoRV site of pBluescript II SK(+), which was subsequently transformed into NovaBlue competent Escherichia coli. pBluescript II SK(+)-UL80.5 miniprepped from NovaBlue cells was digested with BglII and NdeI restriction enzymes and the HCMV cDNA subcloned into pET29b. The resulting plasmid was transformed into competent E. coli BL21 (DE3) cells. The bacteria were grown overnight in LB medium containing ampicillin (100 μg/mL) at 30 °C with 0.3 mM isopropyl-thio-β-d-galactopyranoside for induction of recombinant protein.
Cell pellets were suspended in lysis buffer (50 mM Tris buffer pH 8.0, 100 mM NaCl, 10 mM MgCl2, 2 mM β-mercaptoethanol), and the suspension was alternately frozen (−80 °C) and thawed (4 °C) until cell lysis. Lysozyme powder was added to the suspension (lysozyme final concentration 250 μg/mL) and shaken at 37 °C for 1 h, followed by sonication (5 × 15 s with cooling in between). The resulting suspension was centrifuged at 7500g for 30 min. The resulting pellets were resuspended with lysis buffer, and the previous procedure was repeated one more time. The combined supernatants were stored at −80 °C until purification.
Purification of recombinant HCMV protease was performed using the Biologic HR chromatography system (Bio-Rad, Hercules, CA) with automatic monitoring at A280. The above supernatant was mixed with an equal volume of NPI-20 buffer (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, pH 8.0). Triton X-100 was added to a final concentration of 1%, and the resulting mixture was rocked at 4 °C for 1 h, followed by centrifugation at 10000g for 10 min. The supernatant was desalted using a PD-10 desalting column equilibrated with NPI-10 binding buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0) and then loaded onto a Ni-NTA Superflow cartridge (5 mL) (Qiagen, Valencia, CA). The Ni-NTA cartridge was washed with 200 mL of buffer NPI-20, and the His-tagged protein was eluted with a gradient from 20 mM to 250 mM imidazole at a flow rate of 1 mL/min.
Generation of HCMV Protease Mutants
Point mutations were introduced using site-directed mutagenesis. Briefly, A143S and A143T/A144T mutants were generated by PCR using plasmid containing wild type protease (described above) as the template and the complementary mutagenic primers GACGACGTGGAGTCCGCGACGTCGC (sense and antisense) for A143S and CGACGACGTGGAGACCACGACGTCGCTTTC (sense and antisense) for A143T/A144T. The resulting plasmids were transformed into NovaBlue competent E. coli, and positives were confirmed by sequencing before subcloning into pET29b and transformation into E. coli BL21 (DE3). The expression and purification procedures were the same as for wild type enzyme. The A143T/A144T mutant was used for evaluation of potential prodrugs.
Enzyme Activity toward p-Nitrophenol Substrates
Hydrolysis of Boc-Xaa-ONp and Boc-l-Gly-l-Gly-ONp were carried out in a 96-well plate-based assay. Hydrolysis activity was measured from the rate of p-nitrophenol release at 405 nm using a Synergy HT Multi-Mode Microplate Reader (Biotek, Winooski, VT). 10 μg/mL HCMV A143T/A144T protease mutant was added to 200 μL (final reaction volume) of 100 mM phosphate buffer (pH 7.4) containing 0.5 M Na2SO4 and 10% glycerol and incubated for 3 min, after which Boc-Xaa-ONp (50 μM) was added to the mixture. The absorbance was recorded every 30 s with 10 s of shaking of the plate prior to the reading.
Hydrolysis in Caco-2 Cell Homogenates
For preparation of the S10 fraction, Caco-2 cells (passage 20 to 35) were grown on 100 mm × 150 mm culture plates for 14 to 16 days postconfluence. The cells were washed twice with ice-cold phosphate-buffered saline (PBS) and collected in PBS using a cell scraper. The cells were centrifuged at 6000g for 10 min, and the pellet was kept at −80 °C until use.
Caco-2 cell pellets were resuspended in 50 mM Tris buffer (pH 7.4) containing 0.25 M sucrose and homogenized using a Dounce tissue grinder (Bellco Glass, Inc. Vineland, NJ) 100 times on ice. The cell lysate was sonicated 3 × 10 s and centrifuged at 10000g for 20 min. The resulting pellet was resuspended in 50 mM Tris buffer (pH 7.4) containing 1% Triton X-100, vigorously vortexed and rocked at 4 °C for 1 h, and centrifuged at 10000g for 20 min. The supernatants (S10 fraction) were combined and stored at −80 °C until use. The protein concentration was measured with the Bio-Rad DC assay kit (Hercules, CA).
For measuring the percentage of prodrug hydrolysis by the S10 fraction, the prodrug (400 μM final) was incubated with S10 (1 mg/mL) at 37 °C, and samples were taken at various time points. The reaction was quenched with a 2 vol equivalent of ice-cold acetonitrile/water (1:1, v/v). After brief centrifugation at 10000g, the supernatant was filtered through a PVDF filter microplate (Corning, Tewksbury, MA). The filtrates were subjected to analysis by HPLC with ZORBAX SB-Aq column on Agilent HPLC system.
Hydrolysis in Human Liver Microsomes
The prodrugs of ganciclovir (3a–c, 4, 5 and 9a,b) (400 μM) were incubated in Tris buffer containing 400 μg/mL of HLMs at 37 °C, and samples were taken at various time points. The reaction was quenched by adding the samples to 50% acetonitrile containing 0.1% TFA. After evaporating the solvent, the pellet was resuspended in 50% acetonitrile/0.1% TFA. After 30 min incubation at 37 °C, the samples were centrifuged at 15000g for 5 min, and the supernatants were analyzed by HPLC with ZORBAX SB-Aq column on Agilent HPLC system.
Hydrolysis of Prodrugs by HCMV Protease A143T/A144T Mutant
HCMV A143T/A144T protease mutant protein was cloned, expressed, and purified as described. HCMV protease (80 μg/mL) was added to 50 mM Tris buffer (pH 7.4) containing 0.5 mM Na2SO4, 10% glycerol, and 5 mM DTT in a 100 μL reaction mixture, followed by the addition of internal standard, 3-isobutyl-1-methylxanthine. After 5 min of preincubation at 30 °C, the reaction was initiated by the addition of prodrugs (3a–c, 4, 5, 9a,b) to a final concentration of 400 μM. The reactions were incubated at 30 °C, and 40 μL samples were taken out at various time points. The time courses of prodrug hydrolysis were linear for the time points chosen for each compound. Reaction samples were quenched with two volumes of ice-cold acetonitrile/water (1:1, v/v) containing 0.1% TFA. Agilent Zorbax SB-Aq (3.5 μL, 4.6 × 150 mm) column was used for analysis of 100 μL of the filtered reaction sample, and prodrugs were detected at 254 nm.
Prodrug Stability in Rat and Human Plasma
After collecting rat blood in tubes containing heparin, plasma was obtained by centrifuging the blood at 5000 rpm for 10 min. The stability of GCV prodrugs 3a–c, 4, 5, 9a, and 9b in rat and human plasma were determined using total plasma. The plasma was incubated at 37 °C for 5 min prior to the addition of prodrugs; GCV prodrugs (300 μM final concentration) in DMSO (less than 1% DMSO in the reaction) were added to initiate the reaction. For rat plasma, 50 μL samples were taken at 0, 5, 15, 30, 45, 60, 90, 120, 180, and 240 min. For human plasma, 50 μL samples were taken at 0, 15, 30, 60, 120, and 240 min. Samples were added to 100 μL of ice-cold acetonitrile/water (1:1, v/v) containing 0.1% TFA to stop the reaction and centrifuged at 10000g for 10 min. The supernatants were filtered using Microplate PVDF filters, and the sample filtrates were analyzed by HPLC.
Prodrug Assay and Data Analysis
The concentrations of prodrugs and parent drugs were determined using an Agilent HPLC system (Foster City, CA) and Waters HPLC system (Milford, MA). The Waters HPLC system was composed of a reversed-phase column (Xterra, C-18, 5 μm, 4.6 × 250 mm, Waters) and Agilent Zorbax Sb-Aq (3.5 μm, 4.6 μm x 150 mm), a Waters 515 pump, a 996 photodiode array UV detector, and a WiSP model 712 autosampler (Waters). The system was controlled by Waters Millennium 32 software (version 3.0.1). For all drugs and prodrugs, mobile phase A was water (0.1% TFA) and mobile phase B was acetonitrile (0.1% TFA). The compounds were eluted using a gradient method at 1 mL/min flow rate. The prodrugs and parent drug were detected at 254 nm. The apparent first-order degradation rate constants of the GCV prodrugs at 37 °C in human and rat plasma were determined with GraphPad Prism 5.0 (GraphPad Software, San Diego, CA).
Prism 5.0 was used for data analysis. Data was analyzed using one-way ANOVA with Dunnett’s post hoc test. In Figures 2, 3, and 4, statistical significance was denoted by * p < 0.05, ** p < 0.01. In all Figures, bar represents the standard error of the mean (SE) for n ≥ 3.
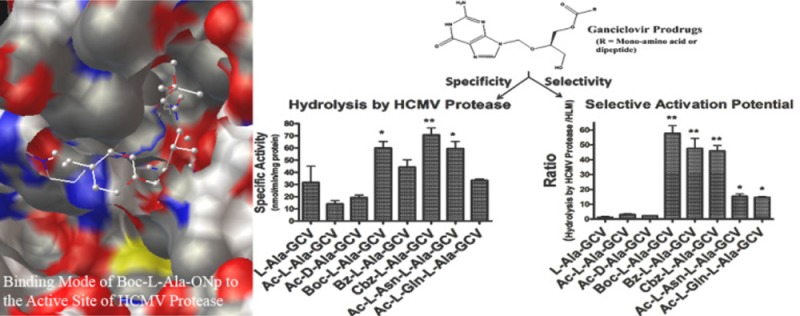
Figure 2.

Ganciclovir prodrug hydrolysis by HCMV A143T/A144T protease mutant. Ganciclovir prodrugs were subjected to hydrolysis by HCMV protease A143T/A144T mutant at 30 °C. The resulting samples were analyzed by HPLC, and the concentrations of prodrugs were determined. Data are expressed as mean specific activity ± SE (n = 6).
Figure 3.

The selectivity ratio of GCV prodrug hydrolysis: comparing hydrolysis by HCMV protease hydrolysis and Caco-2 cell homogenates. Ganciclovir prodrugs were subjected to hydrolysis by Caco-2 cell homogenate (1 mg/mL) at 37 °C. Samples were taken at different time points and analyzed by HPLC. The selectivity ratio was calculated by determining the ratio of GCV prodrug hydrolyzed by the HCMV A143T/A144T protease (from results shown in Figure 2) versus the extent of prodrug hydrolyzed by Caco-2 cell homogenates. Data are expressed as mean ± SE (n = 6).
Figure 4.

The selectivity ratio of GCV prodrug hydrolysis: comparing hydrolysis by HCMV protease hydrolysis and human liver microsomes. Ganciclovir prodrugs were subjected to hydrolysis by human liver microsomes (400 μg/mL) at 37 °C. Samples were taken at different time points and analyzed by HPLC. The selectivity ratio was calculated by determining the ratio of GCV-prodrug hydrolyzed by the HCMV A143T/A144T protease (from results shown in Figure 2) versus the extent of prodrug hydrolyzed by the human liver microsomes. Data are expressed as mean ± SE (n = 6).
Results
Cloning, Expression, and Purification of HCMV Protease
The wild type human cytomegalovirus protease with a C-terminal (His)6-tag was expressed in E. coli. Western blotting using an anti-His-tag monoclonal antibody confirmed the expression of the protein; however, the appearance of an intense band at ∼15 kDa indicated the presence of autoproteolysis products (data not shown). In order to reduce the self-degradation of HCMV protease, a more stable mutant, A143T/A144T,21 was successfully generated through site-directed mutagenesis. When this A143T/A144T mutant was overexpressed and purified by single-step nickel affinity to homogeneity, no autoproteolysis product(s) were observed (data not shown). SDS–PAGE revealed an apparent molecular weight of approximately 30 kDa for the recombinant mutant HCMV protease.
Esterase Activity toward p-Nitrophenol Substrates
HCMV protease is an endopeptidase for which alanine is the preferred amino acid at the P1 position of its endogenous substrates. To explore the esterase activity of the mutant HCMV protease, a commercially available model ester, Boc-l-Ala-ONp, was subjected to hydrolysis by HCMV A143T/A144T mutant. The hydrolysis was concentration-dependent; however, Boc-l-Ala-ONp has low solubility. Therefore, kinetics parameters of Boc-l-Ala-ONp were obtained at 10 μg/mL protease concentration in 50 mM Tris buffer (pH 7.4). HCMV protease exhibited saturable kinetics against Boc-l-Ala-ONp, with Km and kcat being 163 μM and 0.397 s–1, respectively. HCMV protease showed moderate affinity for Boc-l-Ala-ONp, which was about 10-fold less than that of the fluorogenic tetrapeptide substrate (Table 1), N-Ac-Tbg-Tbg-Asn(NMe2)-Ala-AMC, derived from HCMV protease endogenous substrates. However, the turnover rate (kcat) of Boc-l-Ala-ONp was 10-fold faster than that of the peptide compound, while the kcat/Km values of these two compounds were similar. To evaluate the ester substrate specificity, several other commercially available ONp esters, Boc-l-Xaa-ONp (Ala, Gly, Leu, Ile, Gln, Phe, or Val at the Xaa position) and Boc-l-Gly-l-Gly-ONp, were evaluated for hydrolysis by the A143T/A144T mutant (Figure 1). The specific activity for Boc-l-Ala-ONp was 190 nmol/min/mg protein, almost 5-fold higher than the specific activity for Boc-l-Gly-ONp and Boc-l-Phe-ONp. Boc-l-Ile-ONp, Boc-l-Leu-ONp, Boc-l-Val-ONp, and Boc-l-Gln-ONp were not hydrolyzed over the time course of the reaction by HCMV protease. When the dipeptide ester, Boc-l-Gly-l-Gly-ONp, was subjected to hydrolysis by HCMV protease, the rate of hydrolysis was slower than that of Boc-l-Gly-ONp, possibly indicating that a glycine at the P2 position of the substrate is not favored.
Table 1. Kinetics Parameters of Substrate Hydrolysis by HCMV A143T/A144T Protease Mutanta.
| Km (μM) | kcat (s–1) | kcat/Km (s–1 M–1) | |
|---|---|---|---|
| Boc-l-Ala-ONp | 163 | 0.397 | 2435 |
| N-Ac-Tbg-Tbg-Asn(NMe2)-Ala-AMC | 13.2b | 0.035b | 2650b |
Kinetic parameters of two substrates are listed in this table. Boc-l-Ala-ONp hydrolysis results were from our study, and N-Ac-Tbg-Tbg-Asn (NMe2)-Ala-AMC hydrolysis results were from the literature.
Data adopted from ref (20).
Figure 1.

Boc-Xaa-ONp hydrolysis by HCMV A143T/A144T protease mutant. 50 μM Boc-Xaa-ONp (Xaa represents amino acids Phe, Leu, Ile, Ala, Val, Gln, Gly) and Boc-l-Gly-l-Gly-ONp were incubated with 10 μg/mL HCMV A143T/A144T protease mutant for up to 10 min at 30 °C. p-Nitrophenol generated by the reaction was detected at 405 nm (n = 3).
Ganciclovir Prodrug Hydrolysis by HCMV Protease and Caco-2 Homogenates
The hydrolytic activity toward Ac-, Boc-, Bz-, Cbz-protected and unprotected alanine esters of ganciclovir by the HCMV A143T/A144T protease mutant was compared (Figure 2). HCMV protease demonstrated much better specific activity against Boc-, Bz-, and Cbz-protected prodrugs than the Ac-l-Ala-GCV prodrug, indicating that the active site of the enzyme prefers a larger hydrophobic substitution at the α-amino group. Given that Asn and Gln are preferred amino acids at the P2 position of its endogenous substrate,19 dipeptide prodrugs of GCV were also synthesized and evaluated. The specific activities of HCMV protease against both Ac-l-Asn-l-Ala-GCV and Ac-l-Gln-l-Ala-GCV are comparable to the Boc-, Bz-, and Cbz-protected prodrugs. It should be noted that Ac-l-Ala-GCV and Ac-d-Ala-GCV showed similar hydrolysis by HCMV protease, indicating that, at least with regard to the esterase activity of the protease, stereochemistry was not important.
To investigate the activation of the prodrugs to “endogenous cleavage”, we also examined their hydrolysis in the S10 fraction of Caco-2 homogenates. It was found that the Boc-, Bz-, and Cbz-protected mono amino acid prodrugs and the two dipeptide prodrugs were much more stable than the Ac-protected prodrug (Figure 3). The specificity ratio for the hydrolysis by HCMV protease to that by Caco-2 cell homogenates indicates that the Boc-, Bz-, and Cbz-protected prodrug and the dipeptide prodrugs, particularly Ac-l-Gln-l-Ala-GCV, have improved selectivity (Figure 3).
Prodrug Hydrolysis by Human Liver Microsomes
Examination of the hydrolysis of GCV prodrugs in human liver microsomes (HLMs) revealed that the unprotected l-Ala-GCV was the most unstable compound in HLMs (Figure 4), while Boc-, Bz-, Cbz-substituted alanine GCV prodrugs showed improved stability in HLMs. The stability ratios (i.e., the hydrolysis by HCMV protease divided by the hydrolysis by HLMs) indicate that the Boc-, Bz-, and Cbz-substituted prodrugs had more than a 20-fold higher relative activity than l-Ala-GCV, Ac-l-Ala-GCV, and Ac-d-Ala-GCV, and a 2- to 3-fold greater relative activity than dipeptide prodrugs that we tested (Figure 4).
Prodrug Stability in Rat and Human Plasma
Stability studies of GCV prodrugs in rat and human plasma showed that Cbz-l-Ala-GCV exhibited the poorest stability (half-lives of 2.9 and 54.7 min in rat and human plasma, respectively) among the prodrugs evaluated. The dipeptide prodrugs, especially Ac-l-Gln-l-Ala-GCV, showed better plasma stability, with half-lives of 338.4 and 356.0 min in rat and human plasma, respectively (Table 2).
Table 2. Half-Lives for the α-Amino Protected Mono Amino Acid and Dipeptide Prodrugs of Ganciclovir in Rat and Human Plasma at 37 °C (n = 6).
| half-lives, t1/2 (min) |
||||||
|---|---|---|---|---|---|---|
| plasma | Ac-l-Ala-GCV | Boc-l-Ala-GCV | Bz-l-Ala-GCV | Cbz-l-Ala-GCV | Ac-l-Asn-l-Ala-GCV | Ac-l-Gln-l-Ala-GCV |
| human | 253.3 | 177.6 | 129.5 | 54.7 | 79.5 | 356.0 |
| rat | 119.5 | 24.8 | 87.2 | 2.9 | 180.5 | 338.4 |
Discussion
This study was initiated with the HCMV protease wild type enzyme. However, as reported, the active 30 kDa enzyme undergoes autoproteolysis at an internal (or I-) recognition site to generate 13 kDa and 15 kDa fragments in solution.21 Several mutants have been reported to be more stable than wild type HCMV protease,21,22 and therefore site-directed mutagenesis was employed to generate the HCMV protease A143T/A144T mutant that was used throughout this study of small molecule substrates. Based on the crystal structure published, residues Ala143 and Ala144 are away from the catalytic pocket, suggesting that the A143T/A144T mutant is a viable platform in place of wild type enzyme for prodrug study.
Hydrolysis of Boc-Xaa-ONp esters was initially evaluated for the HCMV A143T/A144T protease mutant, and several small ester compounds were found to be good substrates (Figure 1). In particular, the kinetic parameter (Km/kcat) of Boc-l-Ala-ONp was comparable to that of a tetrapepetide substrate, N-Ac-Tbg-Tbg-Asn(NMe2)-Ala-AMC (Table 1). Both the Km and kcat values for the ester substrate were 10-fold higher than those for the amide substrate. Even though the ester substrate has much faster turnover rate than the amide substrate, the lower binding affinity of the mono amino acid ester toward HCMV protease suggests that dipeptide or tripeptide substrates may be better options for prodrug development. The ultimate goal of this study is to develop ganciclovir prodrugs targeting HCMV protease to achieve site-specific prodrug activation. We have synthesized several alanine ester prodrugs of ganciclovir with α-amino group of alanine blocked by different protection groups. Ganciclovir contains two hydroxyl groups. The synthesis yields both mono- and diester of ganciclovir. In this study, we have only focused on the monoester of ganciclovir. When the acetyl protection group on Ac-L-Ala-GCV was replaced with Boc-, Bz-, or Cbz-, the hydrolytic activity of the A143T/A144T mutant increased, suggesting that HCMV protease can accommodate larger hydrophobic functional groups at the amino-terminal of amino acid prodrug of ganciclovir. Furthermore, the Boc-, Bz-, and Cbz-substituted prodrugs were more stable in Caco-2 cell homogenates and human liver microsomes.
Endogenous substrates of HCMV protease contain Asn, Gln, or Lys at the P2 position of the scissile bond. Thus the dipeptide prodrugs, Ac-l-Asn/Gln-l-Ala-GCV, were evaluated for hydrolytic activity by HCMV protease. The hydrolysis rates of Ac-l-Asn-l-Ala-GCV and Ac-l-Gln-l-Ala-GCV were about 4-fold and 2.5-fold higher than that of Ac-l-Ala-GCV, respectively. Perhaps more noteworthy, the dipeptide prodrugs exhibited better stability in both Caco-2 cell homogenates and rat/human plasma (Table 2).
The results of these studies clearly demonstrated that α-amino substituted alanine esters of GCV have potential for CMV prodrug targeting. Alanine is preferred at the P1 position of the ester prodrugs, and the α-amino protected alanine ester prodrugs of GCV not only are hydrolyzed by HCMV protease but also exhibited improved stability in Caco-2 cell homogenates, HLMs, and human/rat plasma. The dipeptide prodrug of GCV, Ac-l-Gln-l-Ala-GCV, emerged as a promising candidate scaffold for further evaluation for selective prodrug targeting.
Acknowledgments
We thank Dr. Yasuhiro Tsume and Dr. Hollis H. D. Showalter for their excellent advice during the course of this work. This research was supported by NIH Grant 037188.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Ettmayer P.; Amidon G. L.; Clement B.; Testa B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47102393–404. [DOI] [PubMed] [Google Scholar]
- Dahan A.; Khamis M.; Agbaria R.; Karaman R. Targeted prodrugs in oral drug delivery: the modern molecular biopharmaceutical approach. Expert Opin. Drug Delivery 2102, 981001–13. [DOI] [PubMed] [Google Scholar]
- Han H. K.; Amidon G. L. Targeted prodrug design to optimize drug delivery. AAPS PharmSci 2000, 21E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller T. H. The cytomegaloviruses: ubiquitous agents with protean clinical manifestations. I. N. Engl. J. Med. 1971, 2854203–14. [DOI] [PubMed] [Google Scholar]
- Fields B. N., Knipe D. M., Howley P. M., Eds. Fields Virology 3rd ed.; Lippincott-Raven Publishers: 1996 [Google Scholar]
- Montgomery J. A. Purine nucleoside phosphorylase: a target for drug design. Med. Res. Rev. 1993, 133209–28. [DOI] [PubMed] [Google Scholar]
- Martin J. C.; Dvorak C. A.; Smee D. F.; Matthews T. R.; Verheyden J. P. 9-[(1,3-Dihydroxy-2-propoxy)methyl]guanine: a new potent and selective antiherpes agent. J. Med. Chem. 1983, 265759–61. [DOI] [PubMed] [Google Scholar]
- Cvetkovic R. S.; Wellington K. Valganciclovir: a review of its use in the management of CMV infection and disease in immunocompromised patients. Drugs 2005, 656859–78. [DOI] [PubMed] [Google Scholar]
- Sinclair J.; Sissons P. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87Part 71763–79. [DOI] [PubMed] [Google Scholar]
- Sia I. G.; Wilson J. A.; Groettum C. M.; Espy M. J.; Smith T. F.; Paya C. V. Cytomegalovirus (CMV) DNA load predicts relapsing CMV infection after solid organ transplantation. J. Infect. Dis. 2000, 1812717–20. [DOI] [PubMed] [Google Scholar]
- Lalezari J.; Lindley J.; Walmsley S.; Kuppermann B.; Fisher M.; Friedberg D.; Lalonde R.; Matheron S.; Nieto L.; Torriani F. J.; Van Syoc R.; Sutton M. A.; Buhles W.; Stempien M. J. A safety study of oral valganciclovir maintenance treatment of cytomegalovirus retinitis. J. Acquired Immune Defic. Syndr. 2002, 304392–400. [DOI] [PubMed] [Google Scholar]
- Matusick-Kumar L.; Newcomb W. W.; Brown J. C.; McCann P. J. 3rd; Hurlburt W.; Weinheimer S. P.; Gao M. The C-terminal 25 amino acids of the protease and its substrate ICP35 of herpes simplex virus type 1 are involved in the formation of sealed capsids. J. Virol. 1995, 6974347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch A. R.; Woods A. S.; McNally L. M.; Cotter R. J.; Gibson W. A herpesvirus maturational proteinase, assemblin: identification of its gene, putative active site domain, and cleavage site. Proc. Natl. Acad. Sci. U.S.A. 1991, 882310792–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum E. Z.; Bebernitz G. A.; Hulmes J. D.; Muzithras V. P.; Jones T. R.; Gluzman Y. Expression and analysis of the human cytomegalovirus UL80-encoded protease: identification of autoproteolytic sites. J. Virol. 1993, 671497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darke P. L.; Cole J. L.; Waxman L.; Hall D. L.; Sardana M. K.; Kuo L. C. Active human cytomegalovirus protease is a dimer. J. Biol. Chem. 1996, 271137445–9. [DOI] [PubMed] [Google Scholar]
- Qiu X.; Culp J. S.; DiLella A. G.; Hellmig B.; Hoog S. S.; Janson C. A.; Smith W. W.; Abdel-Meguid S. S. Unique fold and active site in cytomegalovirus protease. Nature 1996, 3836597275–9. [DOI] [PubMed] [Google Scholar]
- Chen P.; Tsuge H.; Almassy R. J.; Gribskov C. L.; Katoh S.; Vanderpool D. L.; Margosiak S. A.; Pinko C.; Matthews D. A.; Kan C. C. Structure of the human cytomegalovirus protease catalytic domain reveals a novel serine protease fold and catalytic triad. Cell 1996, 865835–43. [DOI] [PubMed] [Google Scholar]
- Liu F. Y.; Kildsig D. O.; Mitra A. K. Cyclodextrin/weak-electrolyte complexation: interpretation and estimation of association constants from phase solubility diagrams. Pharm. Res. 1992, 9121671–2. [DOI] [PubMed] [Google Scholar]
- LaPlante S. R.; Aubry N.; Bonneau P. R.; Cameron D. R.; Lagace L.; Massariol M. J.; Montpetit H.; Plouffe C.; Kawai S. H.; Fulton B. D.; Chen Z.; Ni F. Human cytomegalovirus protease complexes its substrate recognition sequences in an extended peptide conformation. Biochemistry 1998, 37279793–801. [DOI] [PubMed] [Google Scholar]
- Bonneau P. R.; Plouffe C.; Pelletier A.; Wernic D.; Poupart M. A. Design of fluorogenic peptide substrates for human cytomegalovirus protease based on structure-activity relationship studies. Anal. Biochem. 1998, 255159–65. [DOI] [PubMed] [Google Scholar]
- O’Boyle D. R. 2nd; Wager-Smith K.; Stevens J. T. 3rd; Weinheimer S. P. The effect of internal autocleavage on kinetic properties of the human cytomegalovirus protease catalytic domain. J. Biol. Chem. 1995, 27094753–8. [DOI] [PubMed] [Google Scholar]
- LaFemina R. L.; Bakshi K.; Long W. J.; Pramanik B.; Veloski C. A.; Wolanski B. S.; Marcy A. I.; Hazuda D. J. Characterization of a soluble stable human cytomegalovirus protease and inhibition by M-site peptide mimics. J. Virol. 1996, 7074819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]