

Abstract
Vesicular stomatitis virus (VSV) is an oncolytic virus which selectively infects and kills cancer cells. The goal of the present study was to determine whether VSV is capable of targeting metastatic lesions that arise in situ in the transgenic adenocarcinoma of the mouse prostate (TRAMP) model. The interferon (IFN)-responsive luciferase containing VSV(AV3) strain was injected intraprostatically into both control and TRAMP mice. Distribution, infectivity, apoptosis, and status of the IFN response were evaluated at the site of viral injection (prostate), as well as in metastatic lesions (lymph nodes), through plaque, polymerase chain reaction (PCR), and immunohistochemical analysis. Bioluminescence analyses demonstrated that VSV(AV3) persisted at high levels in the prostate region of TRAMP mice for up to 96 hours, but at relatively low levels and for only 48 hours in control mice. Live virus was discovered in the lymph nodes of TRAMP mice, but not in control mice. TUNEL staining revealed increased cell death in VSV(AV3) infected metastatic cells present in the lymph nodes of TRAMP mice. There was an evidence of IFN activation in lymph nodes containing metastatic cells. Our results indicate that intraprostatic injections of VSV(AV3) can be used as a means to infect and kill metastatic lesions associated with advanced prostate cancer.
Introduction
Metastatic prostate cancer is the second leading cause of cancer-related death in North American men.1 Currently, the therapeutic options available for treatment of advanced, metastatic prostate cancer are limited to androgen-ablation therapy followed by chemotherapy, both of which are designed to extend the life expectancy of patients. However, neither treatment is curative; hence, there is a clear need for development of new and more effective therapies for controlling advanced prostate cancer.2,3,4,5
In the last decade, oncolytic virotherapy, using either naturally occurring or genetically engineered viruses, has been explored in the context of an alternative treatment for several cancers.6,7,8 Oncolytic viruses have several features that make them ideal anti-cancer therapeutics. They are self-replicating, generally very efficient at entering cells, and capable of indiscriminately killing cancer cells in a tumor mass through cell lysis as well as immune-mediated mechanisms. The self-replication aspect allows amplification in the host cell and hence raises the effective “dose ” level achievable. Also, there is virtually no cross-resistance with hormone or drug resistance mechanisms. Oncolytic viruses are generally able to target and kill malignant cells through specific intrinsic differences between malignant and normal cells.9,10,11 Due to its anatomical location, the prostate gland is a particularly good candidate for oncolytic virotherapy because it allows for direct intratumoral injection (via trans-rectal or trans-urethral routes).
Vesicular stomatitis virus (VSV) is a negative strand RNA oncolytic virus that preferentially replicates in malignant cells which have an incapacitated or dysfunctional interferon (IFN) response.3 IFN induced genes are involved in the pathogenesis of prostate cancer with downregulation of IFN response genes seen in approximately 30% of clinical samples.12,13 VSV's matrix M-protein is involved in the cytopathic effects of the virus, which lead to rapid apoptosis in an infected cell. Deletion of the methionine at position 51 of the viral M-protein makes the virus (referred to as VSV(AV3)) more susceptible to a host cell's antiviral response, allowing the virus to preferentially infect and kill malignant cells which frequently have a defective IFN response.14,15
Previously, we reported that intraprostatic injection of VSV(AV3) into prostate-specific PTEN−/− transgenic mice led to selective infection and oncolysis of malignant prostate cells while sparing normal prostate tissue.16 Although the prostate-specific PTEN−/− transgenic mouse model produces invasive carcinoma, typically the incidence of metastatic lesions is low. Hence, to study the effects of VSV(AV3) in advanced metastatic prostate cancer, the immunocompetent transgenic adenocarcinoma of the mouse prostate (TRAMP) model was used. The main advantages of using the TRAMP model is that it follows a pattern of prostate cancer progression similar to that which is seen in the clinical disease and that it develops reproducible metastatic lesions.17 Consequently, by 24 weeks of age, virtually 100% of mice exhibit poorly differentiated and invasive tumors, with metastasis observed predominantly in the lumbar lymph nodes.18,19
The aim of the present study was to determine the early dynamics of infection and viral distribution during the initial days following intraprostatic injections of VSV(AV3) into TRAMP mice. We found that VSV(AV3) rapidly accumulates in the lymph nodes and lungs of TRAMP mice, but not in comparable tissues of control mice. Furthermore, after 72 hours, there is another round of viral replication and amplification in the prostate as well as increased apoptotic activity in the lymph nodes of TRAMP mice. Our results indicate that VSV is able to selectively target and kill metastatic prostate cancer cells in an immunocompetent host and may provide an alternative non-cross resistant treatment for dealing with castration and drug resistant disease.
Results
Cytopathic effect of VSV(AV3) on TRAMP-C2 cells
To determine whether VSV(AV3) is capable of infecting TRAMP tumor cells, the TRAMP-C2 cell line was infected with increasing titers of virus over a 48-hour period. The TRAMP-C2 cell line is derived from a heterogeneous prostate of a 32-week-old TRAMP mouse.20 Cell viability, measured via MTS assay, indicated that there was a direct relationship between cell death and increasing viral titer, with a 55% decrease in cell viability seen as the viral multiplicity of infection increased from 0 to 0.1 plaque-forming unit (pfu)/cell. These results demonstrate that TRAMP tumor cells are susceptible to VSV(AV3) infection and oncolysis (Figure 1a).
Figure 1.

Cytotoxicity of VSV(AV3) and the IFN response in TRAMP-C2 cells. (a) TRAMP-C2 cells were infected with VSV(AV3) at viral titers ranging from 0–100 pfu/cell. MTS assays were performed at 48 hours after viral infection. (b) TRAMP-C2 cells were preincubated with increasing concentrations of IFN (0–100,000 IU/ml) for 16 hours and then challenged with VSV(AV3) at an MOI of 0.01 for 48 hours. Control represents untreated cells. Each point is the mean ± SD (n = 4) of three independent experiments with a minimum of four replicates each. IFN, interferon; MOI, multiplicity of infection; pfu, plaque-forming unit; TRAMP, transgenic adenocarcinoma of the mouse prostate; VSV, vesicular stomatitis virus.
The AV3 strain of VSV is particularly sensitive to IFN.14,15 To determine the status of the IFN response, TRAMP-C2 cells were preincubated with increasing doses of type I universal IFN for 16 hours and were then challenged with 0.1 pfu/cell of VSV(AV3). The results indicated that the IFN response pathway is not completely disrupted in TRAMP-C2 cells, because the mean cell viability increased by 26% in cells that were pretreated with 10,000 IU/ml of universal type I IFN (Figure 1b).
Viral distribution after intraprostatic injection of VSV(AV3) into TRAMP mice
In vivo studies were conducted to compare VSV(AV3) distribution in tumor-bearing TRAMP mice and in control nontransgenic mice. The prostates of both TRAMP and control mice were injected with 100 µl of 5 × 108 pfu/ml VSV(AV3). Viral distribution in both cohorts of mice was monitored over a 96-hour period by i.p. injection of luciferin and visualization using the IVIS100 system (Figure 2a).
Figure 2.

In vivo bioluminescence after intraprostatic injection of VSV(AV3). TRAMP and control mice were intraprostatically injected with 100 µl of 5 × 108 pfu/ml of VSV(AV3). (a) Visual representation of viral distribution, tracked by i.p. injections of luciferin over a 3–96 hours time period. UVI (UV-inactivated virus) was used as control. (b) Bioluminescence in the prostate regions of control and TRAMP mice were averaged and are presented as mean ± SEM (n = 3). TRAMP, transgenic adenocarcinoma of the mouse prostate; VSV, vesicular stomatitis virus.
Bioluminescence intensity was shown to be much higher in the prostate region of the TRAMP tumor-bearing mice as compared with controls (Figure 2b). The strongest signal was seen at 24 hours after viral inoculation with the bioluminescence level detected in the prostate region >75-fold higher in TRAMP mice than in control mice. In addition, the bioluminescence was maintained at higher levels in TRAMP prostates over a much longer period than in controls. For example, in control mice, the detected signal in the prostate region was diminished by 90% after 48 hours and only traces amounts of signal was seen at 72 hours after viral injection. By comparison, in TRAMP mice there was only a 35% reduction between 48 hours and 72 hours after viral inoculation. At 72 hours, bioluminescence was >275-fold higher in TRAMP prostates compared with controls, indicating that VSV(AV3) was retained and amplified to higher concentrations in TRAMP prostates for at least 72 hours after viral infection. By 96 hours, there was no detectable bioluminescence observed in the prostates of control mice, whereas there was a 266% increase in bioluminescence seen in TRAMP mice prostates compared with the 72 hours time point, indicating a subsequent increase in the amount of virus.
Presence of live virus in organs of TRAMP mice
To evaluate whether the bioluminescent data correlated with the presence of live virus, both TRAMP and control mice were killed at 24, 48, 72, and 96 hours following treatment with VSV(AV3) and various organs (kidney, liver, lung, lymph, prostate, and spleen) were harvested and snap-frozen. Equal amounts of tissue were homogenized and plaque assays were performed to quantify viral delivery and replication (Figure 3).
Figure 3.

Tissue distribution of replication competent virus. (a) TRAMP and (b) control mice were injected with 100 µl of 5 × 108 pfu/ml of VSV(AV3). Mice were killed at 24, 48, 72, and 96 hours after viral injection. Prostate, lymph, lung, spleen, kidney, and liver tissues were harvested at each time point. Organs were snap frozen and then plaque assays were performed. Results are presented as mean pfu/g ± SEM (n = 3). pfu, plaque-forming unit; TRAMP, transgenic adenocarcinoma of the mouse prostate; UV-inactivated virus; VSV, vesicular stomatitis virus.
In TRAMP mice (Figure 3a), a substantial amount of live virus (160.0 ± 5.8 pfu/g) was observed in the prostate tumors at 24 hours after viral injection. The amount of live virus decreased over time, such that by 48 and 72 hours, there were approximately 138.0 ± 1.5 pfu/g and 48.7 ± 17.9 pfu/g respectively. However, at 96 hours, there was an increase in viral load to 520.0 ± 60.8 pfu/g, indicating a tenfold amplification in viral replication.
A similar trend was observed with the amount of live virus found in lymph nodes and lung tissue from TRAMP mice. In lymph nodes, there was approximately 90.0 ± 0.15 pfu/g seen at 24 hours after intraprostatic injection of VSV(AV3), with the amount of live virus increasing to 170.0 ± 0.06 pfu/g by 48 hours. However, the amount of virus significantly dropped (p ≤ 0.05) by 72 hours after infection to 10.0 ± 1.4 pfu/g, but increased at 96 hours to 80.0 ± 12.1 pfu/g, suggesting another round of viral replication and amplification had occurred in the lymph nodes of these mice.
Lung tissue from TRAMP mice showed the same trend in viral titers as seen with the lymph nodes. At 24 hours after intraprostatic injection of VSV(AV3), 73.3 ± 15.3 pfu/g was seen, with the number increasing to 123.3 ± 25.2 pfu/g by 48 hours, but decreasing to 12.5 ± 3.5 pfu/g by 72 hours and once again increasing to 48.0 ± 83.7 pfu/g at 96 hours. The amount of live viral particles in the lungs was not statistically different (p ≥ 0.05) from that measured in the lymph nodes at all corresponding time points. In the liver, kidney, and spleen, there was no significant amount of virus detected at any time (p ≥ 0.05), suggesting that these organs were not readily infected or able to support viral replication.
In control mice (Figure 3b), VSV(AV3) was mainly present at the prostate injection site (131.0 ± 37.3 pfu/g), with a small amount detected in the spleen (5.3 ± 0.58 pfu/g) at 24 hours after viral injection. This initial concentration of live virus was reduced in these prostates by almost 90% (15.3 ± 4.8 pfu/g) by 48 hours and undetectable by 72 hours after infection. In the spleen there was no virus detected after 24 hours. Also, there was no virus detected in the lymph, lung, liver, and kidney tissues of control mice.
Presence and distribution of VSV(AV3) in prostate and lymph nodes
The presence and distribution of VSV(AV3) in both prostate and lymph node tissues of TRAMP mice were analyzed by immunohistochemistry for viral antigens and scored by a pathologist (L.F.). Although anti-VSV staining in the prostates of control mice was positive at 24 hours after VSV(AV3) injection, the intensity of staining lessened over time to nearly undetectable levels by 72 hours (Figure 4a). By comparison, anti-VSV staining intensity in the prostates of TRAMP mice gradually increased in a time-dependent manner, with strong staining intensity by 24 hours, which then decreased by 48 hours. However, by the 96 hours time point, anti-VSV staining was at its highest level, a pattern analogous to that seen in the plaque assays.
Figure 4.

Immunohistochemical analysis for the presence of VSV(AV3). The presence of VSV was established by immunohistochemical staining of (a) prostate and (b) lymph nodes from TRAMP and control mice using an anti-VSV antibody. Representative slides were prepared and visualized at 40× magnification. Results were scored by a pathologist (n = 3). TRAMP, transgenic adenocarcinoma of the mouse prostate; VSV, vesicular stomatitis virus.
Immunohistochemical analysis of lymph nodes, the primary metastatic site in TRAMP mice, revealed a different staining pattern for VSV (Figure 4b). In control mice, anti-VSV staining was not observed at any time point, whereas strong staining was observed in the enlarged lymph nodes from tumor-bearing TRAMP mice. The intensity of VSV staining increased over time with the maximum staining observed at the 48 hours time point, indicating the presence of VSV(AV3) in metastatic cells present in the TRAMP lymph nodes.
VSV(AV3) treatment leads to apoptotic cell death in TRAMP lymph nodes
Previously, we demonstrated that intraprostatic infection of VSV(AV3) leads to cancer-specific cell death in prostates of tumor-bearing PTEN−/− mice.16 To determine whether VSV(AV3) is capable of destroying metastatic malignant cells after it finds and infects them, TUNEL analysis was performed on the enlarged lymph nodes of TRAMP mice and compared with controls (Figure 5a,b). Our results indicate that after intraprostatic injection of VSV(AV3), there was a substantial increase in cell death observed in the pelvic lymph nodes of TRAMP mice compared with lymph nodes from control mice. In the TRAMP lymph nodes, there was a significant increase (p ≤ 0.05) in cell death observed at 24 hours after viral injection (75 ± 10 mean apoptotic cells) compared with that seen in lymph nodes from TRAMP mice treated with UV-inactivated virus (22 ± 7 mean apoptotic cells). However, there was no significant (p >0.05) change in apoptotic cell death observed at 48 hours (91 ± 7 mean apoptotic cells) compared with either 24 or 72 hours (93 ± 10 mean apoptotic cells). In lymph nodes from control mice, there was no significant change in apoptosis observed in UV-inactivated (6 ± 7 mean apoptotic cells) treated mice compared with those treated with live VSV(AV3) for 24 hours (14 ± 12 mean apoptotic cells), indicating that virus-driven cell death only occurred in tumor-bearing animals.
Figure 5.

TUNEL analysis of apoptotic cell death in the lymph nodes of tumor-bearing TRAMP and control mice. (a) Paraffin-embedded lymph node tissues from TRAMP and control mice were stained with TUNEL, and visualized at 40× magnification. (b) Apoptotic cells were counted in 10 fields of view and presented as mean apoptotic cells ± SD. TRAMP, transgenic adenocarcinoma of the mouse prostate.
Status of IFN pathway after intraprostatic injection of VSV(AV3)
As VSV(AV3) infection leads to activation of a type I IFN response,15,21 the status of IFN activity in the prostates and metastatic lumbar lymph nodes of TRAMP mice was assessed. Prostates and lymph nodes were extracted from control and TRAMP mice treated with VSV(AV3) over a 72 hours time period. Tissues were stained with IFN response factor-7 (IRF-7), because activation of IRF-7 has been shown to be an indicator synonymous with IFN type I activity.22,23,24,25 Immunohistochemical staining of control prostate tissues showed an extensive increase in the intensity of IRF-7 staining after VSV(AV3) injection, implying activation of a type I IFN response (Figure 6a). However, in the prostates of TRAMP mice treated with VSV(AV3), no IRF-7 staining was seen, indicating that the type I IFN pathway was not activated.
Figure 6.
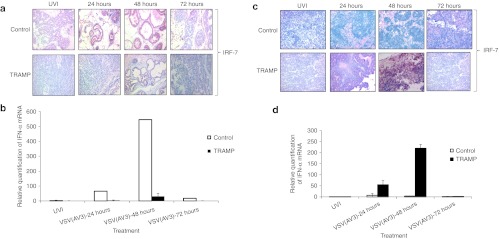
Analysis of IRF-7 and IFN-α expression in prostates and lymph nodesof control and TRAMP mice. (a) Paraffin-embedded prostate tissues were stained with anti-IRF-7 antibody. Representative slides were prepared and visualized at 40× magnification. Results were scored by a pathologist (n = 3). (b) IFN-α mRNA levels in the prostates of control and TRAMP mice were compared after VSV(AV3) injection. UVI is UV-inactivated control virus. IFN-α mRNA levels were first normalized to rRNA level (δCT = CT IFN-α − CT UVI). The results were expressed as mean relative quantification = 2−δδCT, ± D (n = 3). (c) Paraffin-embedded lymph nodes were stained with IRF-7. Representative slides were prepared and visualized at 40× magnification. Results were scored by a pathologist (n = 3). (d) IFN-α mRNA levels in lymph nodes of control and TRAMP mice were compared after VSV(AV3) injection. UVI is UV-inactivated control virus. IFN-α mRNA levels were first normalized to rRNA level (δCT = CT IFN-α − CT UVI). The results were expressed as mean relative quantification = 2−δδCT, ±SD (n = 3). IFN, interferon; IRF-7, interferon response factor-7; TRAMP, transgenic adenocarcinoma of the mouse prostate; VSV, vesicular stomatitis virus.
Message RNA transcript levels for IFN-α in the prostates from VSV(AV3)-infected TRAMP and control mice were determined and compared by quantitative real-time polymerase chain reaction (PCR) analysis. Our results (Figure 6b) demonstrated that there was a >500-fold increase in IFN-α mRNA levels 48 hours after intraprostatic injection of VSV(AV3) into the control prostates. By comparison, there was only about a 30-fold increase in IFN-α mRNA levels in TRAMP prostates at that time, indicating a much reduced and almost negligible IFN response.
Immunohistochemical staining of lymph nodes derived from control mice demonstrated a slight increase in IRF-7 staining intensity by 24 hours after viral injection (Figure 6c). These data were supported by a >20-fold elevation of IFN-α mRNA levels seen in control lymph nodes 24 hours after intraprostate injection with VSV(AV3) (Figure 6d). However, there was a substantially greater increase in IRF-7 staining intensity in the enlarged lymph nodes derived from TRAMP mice seen by 48 hours following viral administration. Similarly, there was over a 200-fold increase in IFN-α mRNA transcript levels by 48 hours in TRAMP derived lymph nodes. These data demonstrate that intraprostatic administration of VSV(AV3) can trigger an IFN response in lymph node tissue from TRAMP mice.
Discussion
Taxane chemotherapeutic agents, such as docetaxel, are the main forms of treatment for metastatic, castration-resistant prostate cancers.26,27,28 However, due to the emergence of drug resistant cells, this line of treatment is largely palliative and not curative.29,30 Drug and hormone resistance can occur in a variety of ways including epigenetic or mutational alterations.31 To successfully overcome these numerous molecular-resistance mechanisms, an entirely new class of non cross-resistant treatments is required. Recently, oncolytic viruses have been tested in phase I clinical trials as a possible treatment modality for prostate cancer.32,33,34 Oncolytic viruses can intrinsically kill malignant cells while sparing normal healthy cells. These viruses influence the host cell through activation of gene products that enable manipulation of apoptotic pathways and ultimately activation of the host's immune response.15 Coincidentally, viruses use many of the same biological pathways that are deregulated during the process of tumor progression.35 For example, several studies have shown that most cancer cells, including prostate cancer cells, have a defective IFN response.13,14,36 VSV is an oncolytic virus that exploits defective IFN pathways for selective infection and destruction of tumor cells.
Previously, we demonstrated that VSV(AV3) is capable of selectively targeting and killing IFN-defective prostate cancer cells in vivo.16 In the present study, we explored the possibility of using VSV(AV3) for targeted killing of metastatic prostate cancer using the well-established TRAMP mouse model. To demonstrate that VSV(AV3) is able to infect TRAMP mouse tumor cells, TRAMP-C2 cells were infected with increasing viral titers. It was evident from MTS cell viability analysis that there was a direct correlation between increasing viral titer and decreased cell survival (Figure 1a), indicating that the prostate tumor cells from TRAMP mice are susceptible to VSV(AV3) infection. As VSV(AV3) infection exploits a defective IFN antiviral pathway, TRAMP-C2 cells were pretreated with increasing doses of universal type I IFN before treatment with VSV(AV3) to determine the status of the IFN response. Interestingly, we found that increasing doses of IFN rescued the TRAMP-C2 cells from cell death induced by this virus. This finding suggests that the IFN response is not fully nonfunctional in TRAMP-C2 cells (Figure 1b). It has been previously shown that some prostate tumors, such as the PC-3 xenograft model, are more resistant to VSV infection.37 From this, it was concluded that the presence of IFN bioactivity reduced the oncolytic activity of VSV in these tumors. Furthermore, it was reported that advanced metastatic PC-3 cell lines required a higher dose of VSV compared with LNCaP prostate cancer cells for infection.37,38 In a similar fashion, it is likely that TRAMP-C2 cells also require higher doses of VSV for efficient infection. Also, the production and escape of VSV progeny from the surface of the infected cell may be compromised, as was seen in PC-3 cells.38 Alternatively, TRAMP-C2 cells may be unable to produce IFN due to faulty IFN production pathways. However, from our results it is evident that they are still able to use IFN to launch secondary IFN pathways to protect themselves from further viral infection.
Bioluminescence results obtained after intraprostatic administration of VSV(AV3) demonstrated much higher levels of virus in the prostate region of tumor-bearing TRAMP mice compared with controls (Figure 2). These data were corroborated by immunohistochemical staining for VSV, which clearly showed a higher intensity of VSV staining in the TRAMP tumor-bearing prostates relative to controls (Figure 4a). With respect to live viral progeny, plaque assays revealed a significant increase in viral titer 24 hours after virus administration to the prostates of TRAMP mice (Figure 3). These results agree with our previous work using PTEN−/− transgenic mice, where comparable higher bioluminescence and viral amplification were seen in prostate tumors compared with normal controls.16 Although there have been some reports mentioning possible central nervous system toxicity with wild-type VSV in immunocompromised mice,39,40 we did not observe any symptoms of neurotoxicity in immunocompetent animals treated with our highly IFN-sensitive VSV(AV3) strain of virus.
The amount of live virus seen in plaque assays of lymph node and lung tissues from TRAMP mice was similar in magnitude (Figure 3a). Both of these tissues have previously been implicated as key locations for growth of metastatic lesions in the TRAMP model.41 The intensity of VSV staining in the enlarged lymph nodes extracted from TRAMP mice was extremely high relative to lymph nodes obtained from virally infected control mice (Figure 4b). In addition, TUNEL analysis on lymph nodes taken from both control and TRAMP mice after VSV(AV3) infection demonstrated a substantially higher increase in apoptotic cell count in tumor-bearing mice (Figure 5). Most metastatic tumors in viral infected TRAMP mice were undergoing considerably higher cell death rates compared with those infected with UV-inactivated virus. These results indicate that VSV(AV3) can target and kill metastatic prostate cancer cells found in the lymph nodes of TRAMP mice.
As VSV infection is enabled in cells with a defective IFN response, the status of the IFN response pathway was investigated in both prostate and lymph nodes derived from TRAMP mice. IRF-7 plays a key role in overall transcription of IFN-α/β by alerting neighboring cells to activate their innate immune response and to elevate their antiviral status.22 Immunohistochemical analysis of prostates from TRAMP mice showed that IRF-7 was not produced after VSV infection (Figure 6a). However, in prostates from control mice, there was a prominent increase in IRF-7 staining intensity upon viral infection. Also, by 48 hours, there was a tenfold rise in IFN-α mRNA levels in control prostates treated with virus, but almost no change in the TRAMP prostates (Figure 6b). By comparison, an increase in IRF-7 levels was observed in lymph nodes derived from TRAMP mice (Figure 6c). These results were substantiated by an increase in IFN-α mRNA levels in the TRAMP derived metastatic lymph nodes (Figure 6d). As lymph nodes are largely comprised of lymphocytes,18 the presence of high levels of VSV in TRAMP lymph nodes could potentially lead to hyper-activation of an IFN response in the resident nonmalignant lymphocytes. In contrast, there was no remarkable change in either IRF-7 protein or IFN-α mRNA levels in lymph nodes from control mice. This was presumably due to the absence of a sustainable, systemic viral infection of control mice (Figures 3 and 4).
In conclusion, this study is the first to show the efficacy of intraprostatic injection of VSV(AV3) for targeting and killing metastatic lesions in an immunocompetent host. The proof of principle presented here sets the stage for further investigation of VSV(AV3) as an oncolytic agent for treatment of advanced, metastatic prostate cancer in patients, particularly for those with a malignant phenotype that is castration and drug resistant.
Materials and Methods
Tissue culture. TRAMP-C2 cells were purchased from the American Type Culture Collection and maintained in DMEM (Invitrogen, Burlington, Ontario, Canada). Medium was supplemented with 0.005 mg/ml bovine insulin (Sigma, Oakville, Ontario, Canada), 10 nmol/l dehydroisoandrosterone (Sigma), 5% Nu-Serum IV (BD-bioscience, Mississauga, Ontario, Canada) and 5% fetal bovine serum (Invitrogen).
Virus propagation. VSV(AV3), a recombinant IFN-inducing mutant of the Indiana serotype, was propagated in Vero cells. VSV(AV3) was constructed to express the GFP-firefly luciferase fusion gene as previously described.15 Virons were collected and purified as previously described;42 pfu were counted and used for calculating infectious titers.15,42,43
TRAMP model. TRAMP mice hemizygous for the ARR2BP (rat probasin derived gene promoter)-SV40Tag transgenes maintained in a pure C57BL/6 background were purchased from Jackson Laboratories (Bar Harbor, ME). C57BL/6 TRAMP mice were bred in house with an inbred strain of FVB mice (Jackson Laboratories). To confirm the presence of the transgene in male mice, genomic DNA was removed from tail clips of F1 male progeny from C57BL/6 TRAMP × FVB cross and tested using PCR (MβCx7F:5′-GAT-GTG-CTC-CAG-GCT-AAA-GTT-3′,MβCx7R:5′-AGA- AAC-GGA-ATG-TTG-TGG-AGT-3′, Pb-1F:5′-CCG-GTC-GAC-CGG- AAG-CTT-CCA-CAA-GTG-CAT-TTA-3′, and SV40TagR: 5′-CTC-CTT-TCA-AGA-CCT-AGA-AGG-TCC-A-3′) primers. For this study, all mice used were 25 weeks of age.
In vivo studies. A small incision was made in the abdomen of 25-week-old TRAMP male mice and 100 µl of VSV(AV3) at 5 × 108 pfu/ml was delivered by intraprostatic injection. For quantification of viral uptake and distribution, animals were injected through the intraperitoneal (i.p.) route with 150 mg/kg luciferin at 3, 6, 24, 48, 72, and 96 hours after viral inoculation and imaged with an IVIS100 Imaging System (Caliper Life Sciences, Xenogen, Hopkinton, MA). Data were analyzed using Living Image 2.50 (Caliper Life Sciences) software. At least four mice per group were used and then killed at the indicated time points. Organs (kidney, liver, lung, prostate, and spleen) were extracted and then either fixed in 10% buffered formalin and paraffin-embedded or snap-frozen in liquid nitrogen. Animal procedures were performed according to the Canadian Council on Animal Care guidelines.
Titration of VSV from infected tissue. Tissues were removed at the indicated time points and approximately equal amounts of tissues were weighed and homogenized in 1 ml phosphate-buffered saline using a Polytron homogenizer. Serial dilutions were prepared in serum free media and added to confluent Vero cells for 60 minutes. Cells were subsequently overlaid with 1% methyl cellulose (Sigma) and plaques were grown for 72 hours. Infected Vero cells were then fixed in 4% formaldehyde and stained with crystal violet. Plaques were counted by visual inspection and titers were calculated as pfu/ml.44,45
Immunohistochemical staining. Five micrometer sections were prepared from paraffin-embedded tissues and extracted from paraffin, as described previously.46 TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) assays were performed according to the manufacturer's instructions (ROCHE, Mississauga, Ontario, Canada). Tissues were stained with primary antibody to IRF-7 (Abcam, Cambridge, MA) and VSV (from Dr. John Bell) at 1:75 and 1:500 dilutions, respectively. All sections were reviewed by a pathologist (L. Fazli).
Quantitative real-time PCR. Total RNA from mouse tissue was isolated using the Trizol method (Invitrogen), and was reverse transcribed and amplified with IFN-α primers47 on an Applied Biosystems (Carlsbad, CA) 7900HT Fast Real-time PCR System following the SYBR Green PCR Master Mix protocol. Relative quantification of gene expression was performed using ribosomal RNA as control.20
In vitro cell proliferation assay. TRAMP-C2 cells (1 × 104) were plated into each well of a 96-well plate. Cells were grown for 48 hours, stimulated with universal type I IFN (PBL Biomedical Laboratories, Piscataway Township, NJ) for 16 hours and then challenged with VSV(AV3) at a multiplicity of infection of 1.0 pfu/cell. MTS[3-(4,5-dimethylthiaol-2-yl)5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-tetrazolium (250 µg/ml) inner salt] (Promega, Madison, WI) and 20 µg of PMS (Gibco, Burlington, Ontario, Canada) solutions were mixed together and added to each well for 2 hours. Colorimetric analysis was carried out using an ELISA plate reader at an absorbance of 490 nm.48
Statistical analysis. Student's t test was used to assess statistical significance between groups. To measure statistical differences between mouse stained tissues, the nonparametric Wilcoxon rank test was used, as there was no assumption of a normal distribution of scores.
Acknowledgments
This work was supported by a grant from the Terry Fox Foundation of Canada. Maryam Moussavi was supported by a scholarship from the Department of Defense USA, PC073406. We thank Latif Wafa and Eric Leblanc for critical reading of this manuscript. The authors declare no conflict of interest.
REFERENCES
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T.et al. (2008Cancer statistics, 2008 CA Cancer J Clin 5871–96. [DOI] [PubMed] [Google Scholar]
- Andrieu C, Taieb D, Baylot V, Ettinger S, Soubeyran P, De-Thonel A.et al. (2010Heat shock protein 27 confers resistance to androgen ablation and chemotherapy in prostate cancer cells through eIF4E Oncogene 291883–1896. [DOI] [PubMed] [Google Scholar]
- Chang G, Xu S, Watanabe M, Jayakar HR, Whitt MA., and, Gingrich JR. Enhanced oncolytic activity of vesicular stomatitis virus encoding SV5-F protein against prostate cancer. J Urol. 2010;183:1611–1618. doi: 10.1016/j.juro.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Fusi A, Procopio G, Della Torre S, Ricotta R, Bianchini G, Salvioni R.et al. (2004Treatment options in hormone-refractory metastatic prostate carcinoma Tumori 90535–546. [DOI] [PubMed] [Google Scholar]
- Petrylak DP. Future directions in the treatment of androgen-independent prostate cancer. Urology. 2005;65 suppl. 6:8–12. doi: 10.1016/j.urology.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Huang B, Sikorski R, Kirn DH., and, Thorne SH. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 2011;18:164–172. doi: 10.1038/gt.2010.121. [DOI] [PubMed] [Google Scholar]
- Lun X, Chan J, Zhou H, Sun B, Kelly JJ, Stechishin OO.et al. (2010Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma Mol Ther 181927–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchefeu Y, Harrington KJ, Galmiche JP., and, Vassaux G. Review article: gene therapy, recent developments and future prospects in gastrointestinal oncology. Aliment Pharmacol Ther. 2010;32:953–968. doi: 10.1111/j.1365-2036.2010.04424.x. [DOI] [PubMed] [Google Scholar]
- Alajez NM, Mocanu JD, Shi W, Chia MC, Breitbach CJ, Hui AB.et al. (2008Efficacy of systemically administered mutant vesicular stomatitis virus (VSVDelta51) combined with radiation for nasopharyngeal carcinoma Clin Cancer Res 144891–4897. [DOI] [PubMed] [Google Scholar]
- Ribacka C., and, Hemminki A. Virotherapy as an approach against cancer stem cells. Curr Gene Ther. 2008;8:88–96. doi: 10.2174/156652308784049372. [DOI] [PubMed] [Google Scholar]
- Zhang W, Cai R, Luo J, Wang Y, Cui Q, Wei X.et al. (2007The oncolytic adenovirus targeting to TERT and RB pathway induced specific and potent anti-tumor efficacy in vitro and in vivo for hepatocellular carcinoma Cancer Biol Ther 61726–1732. [DOI] [PubMed] [Google Scholar]
- Nagano K, Masters JR, Akpan A, Yang A, Corless S, Wood C.et al. (2004Differential protein synthesis and expression levels in normal and neoplastic human prostate cells and their regulation by type I and II interferons Oncogene 231693–1703. [DOI] [PubMed] [Google Scholar]
- Shou J, Soriano R, Hayward SW, Cunha GR, Williams PM., and, Gao WQ. Expression profiling of a human cell line model of prostatic cancer reveals a direct involvement of interferon signaling in prostate tumor progression. Proc Natl Acad Sci USA. 2002;99:2830–2835. doi: 10.1073/pnas.052705299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N.et al. (2000Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus Nat Med 6821–825. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty BD, tenOever BR, Paterson JM, Power AT, Knowles S.et al. (2003VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents Cancer Cell 4263–275. [DOI] [PubMed] [Google Scholar]
- Moussavi M, Fazli L, Tearle H, Guo Y, Cox M, Bell J.et al. (2010Oncolysis of prostate cancers induced by vesicular stomatitis virus in PTEN knockout mice Cancer Res 701367–1376. [DOI] [PubMed] [Google Scholar]
- Winter SF, Cooper AB., and, Greenberg NM. Models of metastatic prostate cancer: a transgenic perspective. Prostate Cancer Prostatic Dis. 2003;6:204–211. doi: 10.1038/sj.pcan.4500655. [DOI] [PubMed] [Google Scholar]
- Gingrich JR, Barrios RJ, Morton RA, Boyce BF, DeMayo FJ, Finegold MJ.et al. (1996Metastatic prostate cancer in a transgenic mouse Cancer Res 564096–4102. [PubMed] [Google Scholar]
- Greenberg NM, DeMayo FJ, Sheppard PC, Barrios R, Lebovitz R, Finegold M.et al. (1994The rat probasin gene promoter directs hormonally and developmentally regulated expression of a heterologous gene specifically to the prostate in transgenic mice Mol Endocrinol 8230–239. [DOI] [PubMed] [Google Scholar]
- Foster BA, Gingrich JR, Kwon ED, Madias C., and, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–3330. [PubMed] [Google Scholar]
- Faul EJ, Lyles DS., and, Schnell MJ. Interferon response and viral evasion by members of the family rhabdoviridae. Viruses. 2009;1:832–851. doi: 10.3390/v1030832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Weerd NA, Samarajiwa SA., and, Hertzog PJ. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282:20053–20057. doi: 10.1074/jbc.R700006200. [DOI] [PubMed] [Google Scholar]
- Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- Oldak M, Tolzmann L, Wnorowski A, Podgórska MJ, Silling S, Lin R.et al. (2011Differential regulation of human papillomavirus type 8 by interferon regulatory factors 3 and 7 J Virol 85178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor P, Tamura T., and, Ozato K. IRF family proteins and type I interferon induction in dendritic cells. Cell Res. 2006;16:134–140. doi: 10.1038/sj.cr.7310018. [DOI] [PubMed] [Google Scholar]
- Caffo O, Fratino L, Barbieri R, Perin A, Martini T, Sava T.et al. (2011Pemetrexed as second-line chemotherapy for castration-resistant prostate cancer after docetaxel failure: Results from a phase II study Urol Oncol (in the press) [DOI] [PubMed]
- Gleave M, Goldenberg SL, Bruchovsky N., and, Rennie P. Intermittent androgen suppression for prostate cancer: rationale and clinical experience. Prostate Cancer Prostatic Dis. 1998;1:289–296. doi: 10.1038/sj.pcan.4500260. [DOI] [PubMed] [Google Scholar]
- Yap TA, Zivi A, Omlin A., and, de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8:597–610. doi: 10.1038/nrclinonc.2011.117. [DOI] [PubMed] [Google Scholar]
- Chen FL, Armstrong AJ., and, George DJ. Cell signaling modifiers in prostate cancer. Cancer J. 2008;14:40–45. doi: 10.1097/PPO.0b013e318161d40f. [DOI] [PubMed] [Google Scholar]
- Damber JE., and, Aus G. Prostate cancer. Lancet. 2008;371:1710–1721. doi: 10.1016/S0140-6736(08)60729-1. [DOI] [PubMed] [Google Scholar]
- Shen MM., and, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freytag SO, Khil M, Stricker H, Peabody J, Menon M, DePeralta-Venturina M.et al. (2002Phase I study of replication-competent adenovirus-mediated double suicide gene therapy for the treatment of locally recurrent prostate cancer Cancer Res 624968–4976. [PubMed] [Google Scholar]
- Freytag SO, Movsas B, Aref I, Stricker H, Peabody J, Pegg J.et al. (2007Phase I trial of replication-competent adenovirus-mediated suicide gene therapy combined with IMRT for prostate cancer Mol Ther 151016–1023. [DOI] [PubMed] [Google Scholar]
- Freytag SO, Stricker H, Pegg J, Paielli D, Pradhan DG, Peabody J.et al. (2003Phase I study of replication-competent adenovirus-mediated double-suicide gene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment of newly diagnosed, intermediate- to high-risk prostate cancer Cancer Res 637497–7506. [PubMed] [Google Scholar]
- Ottolino-Perry K, Diallo JS, Lichty BD, Bell JC., and, McCart JA. Intelligent design: combination therapy with oncolytic viruses. Mol Ther. 2010;18:251–263. doi: 10.1038/mt.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyên TL, Abdelbary H, Arguello M, Breitbach C, Leveille S, Diallo JS.et al. (2008Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis Proc Natl Acad Sci USA 10514981–14986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M, Cramer SD., and, Lyles DS. Sensitivity of prostate tumors to wild type and M protein mutant vesicular stomatitis viruses. Virology. 2004;330:34–49. doi: 10.1016/j.virol.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Carey BL, Ahmed M, Puckett S., and, Lyles DS. Early steps of the virus replication cycle are inhibited in prostate cancer cells resistant to oncolytic vesicular stomatitis virus. J Virol. 2008;82:12104–12115. doi: 10.1128/JVI.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun X, Senger DL, Alain T, Oprea A, Parato K, Stojdl D.et al. (2006Effects of intravenously administered recombinant vesicular stomatitis virus (VSV(deltaM51)) on multifocal and invasive gliomas J Natl Cancer Inst 981546–1557. [DOI] [PubMed] [Google Scholar]
- Muik A, Dold C, Geiß Y, Volk A, Werbizki M, Dietrich U.et al. (2012Semireplication-competent vesicular stomatitis virus as a novel platform for oncolytic virotherapy J Mol Med 90959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan-Lefko PJ, Chen TM, Ittmann MM, Barrios RJ, Ayala GE, Huss WJ.et al. (2003Pathobiology of autochthonous prostate cancer in a pre-clinical transgenic mouse model Prostate 55219–237. [DOI] [PubMed] [Google Scholar]
- Hadaschik BA, Zhang K, So AI, Fazli L, Jia W, Bell JC.et al. (2008Oncolytic vesicular stomatitis viruses are potent agents for intravesical treatment of high-risk bladder cancer Cancer Res 684506–4510. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, Paterson JM, Lemay CG, Falls TJ, McGuire A, Parato KA.et al. (2007Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow Mol Ther 151686–1693. [DOI] [PubMed] [Google Scholar]
- Oliere S, Arguello M, Mesplede T, Tumilasci V, Nakhaei P, Stojdl D.et al. (2008Vesicular stomatitis virus oncolysis of T lymphocytes requires cell cycle entry and translation initiation J Virol 825735–5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumilasci VF, Olière S, Nguyên TL, Shamy A, Bell J., and, Hiscott J. Targeting the apoptotic pathway with BCL-2 inhibitors sensitizes primary chronic lymphocytic leukemia cells to vesicular stomatitis virus-induced oncolysis. J Virol. 2008;82:8487–8499. doi: 10.1128/JVI.00851-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margiotti K, Wafa LA, Cheng H, Novelli G, Nelson CC., and, Rennie PS. Androgen-regulated genes differentially modulated by the androgen receptor coactivator L-dopa decarboxylase in human prostate cancer cells. Mol Cancer. 2007;6:38. doi: 10.1186/1476-4598-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T.et al. (2005IRF-7 is the master regulator of type-I interferon-dependent immune responses Nature 434772–777. [DOI] [PubMed] [Google Scholar]
- Moussavi M, Assi K, Gómez-Muñoz A., and, Salh B. Curcumin mediates ceramide generation via the de novo pathway in colon cancer cells. Carcinogenesis. 2006;27:1636–1644. doi: 10.1093/carcin/bgi371. [DOI] [PubMed] [Google Scholar]