

Abstract
Natural killer (NK) cells represent a key component of the innate immune system against cancer. Nevertheless, malignant diseases arise in immunocompetent individuals despite tumor immunosurveillance. Hodgkin lymphoma (HL) is characterized by CD30+ tumor cells and a massive infiltration of immune effector cells in affected lymph nodes. The latter obviously fail to eliminate the malignant cell population. Here, we tested for functional NK cell defects in HL and suggest an improvement of NK function by therapeutic means. We demonstrate that peripheral NK cells (pNK) from patients with HL fail to eliminate HL cell lines in ex vivo killing assays. Impaired NK cell function correlated with elevated serum levels of soluble ligands for NK cell receptors NKp30 (BAG6/BAT3) and NKG2D (MICA), factors known to constrict NK cell function. In vitro, NK cell cytotoxicity could be restored by an NKG2D/NKp30-independent bispecific antibody construct (CD30xCD16A). It artificially links the tumor receptor CD30 with the cytotoxicity NK cell receptor CD16A. Moreover, we observed that NK cells from patients treated with this construct were generally activated and displayed a restored cytotoxicity against HL target cells. These data suggest that reversible suppression of NK cell activity contributes to immune evasion in HL and can be antagonized therapeutically.
Introduction
There is a recent clinical and experimental evidence to show that natural killer (NK) cells are critically involved in the recognition and elimination of tumor cells. The NKG2D receptor and the group of natural cytotoxicity receptors (NCRs) (NKp46, NKp44, and NKp30) are regarded as the major NK cell receptors in tumor defence.1,2 NKG2D is a member of the C-type lectin superfamily which triggers NK cell activation. Ligands of the human NKG2D receptor are the major histocompatibility complex class I (MHC I)-related molecules MICA/MICB, and the UL16-binding proteins ULBP, which are rarely expressed in healthy tissues but induced on cells upon transformation. This induction alerts the immune system to the dangerous cell.3 The ligands for NKp46 and NKp44 are still enigmatic although heparan sulfate proteoglycans, vimentin, and proliferating nuclear antigen have been described as ligand structures.4,5,6 A member of the B7 family, B7-H6, and the nuclear factor BAG6 (also referred to as BAT3), which is released from tumor cells under stress conditions via the exosomal pathway, were recently identified as tumor cell-associated ligands for NKp30.7,8 Immune surveillance via NKG2D and the corresponding ligands appears to be particularly effective in the early stages of tumor growth. However, tumor cells develop escape mechanisms to evade NK cell surveillance, and it is obvious that NKG2D engagement by its ligands may result in either immune activation (tumor clearance) or immune silencing (tumor evasion).1,2 Silencing of NKG2D during tumor progression results from the persistent exposure of ligands expressed on the surface of target cells.9,10 Moreover, tumor cells release ligands into the environment by shedding. The soluble molecules not only block NKG2D but also induce receptor internalization and degradation.11,12,13 Soluble MICA molecules in addition engage NKG2D on CD4+ T cells to promote proliferation and differentiation of regulatory T cells that additionally inhibit the immune response.11
Plasma levels of soluble ligands correlate with disease progression in many hematological and solid tumors.14,15,16,17 Here, we demonstrate that peripheral NK cells from patients with Hodgkin lymphoma (HL) are functionally inactive, which is in line with former studies on NK cell function in HL.18,19 The observed NK cell dysfunction correlates to elevated serum levels for ligands engaging NKG2D (MICA) and NKp30 (BAG6/BAT3). We provide an evidence that immunotherapeutic strategies targeting NK cells are promising, because NK cell cytotoxicity could be restored in vitro and in patients by treatment with a novel human antibody construct that is designed for the treatment of HL and other CD30-expressing malignancies. The tetravalent, bispecific antibody used in this study targets CD30 on Hodgkin Reed-Sternberg (HRS) cells with two of its binding sites, whereas the activating receptor CD16A on NK cells (CD30xCD16A, AFM13) is targeted by the other two binding sites, thereby selectively cross-linking tumor and NK cells. CD16A (FCGR3A) is the human low-affinity IgG Fc receptor that is expressed on the surface of NK cells, macrophages, a subset of monocytes, and T cells. The engagement of CD16 triggers its interaction with both FcRI-γ and CD3-ζ immunoreceptor tyrosine-based activation motif complexes.20 This induces the recruitment and activation of phosphotyrosine kinases including Syk and ZAP70, finally resulting in the activation of NK cell-effector functions.20
CD30, a member of the tumor necrosis factor receptor family, is highly expressed on Hes1 cells, but rarely and faintly expressed in normal tissue and thus represents an excellent target structure for immunotherapy.21 Although more than 80% of patients with HL are cured by combined radio- and chemotherapy, there is still a high and unmet need for both treatment options for patients who relapse or fail to respond to front-line treatment and for therapies that have limited side effects.22 Our findings suggest that immunotherapeutic approaches are an effective and promising alternative to standard therapies.
Results
Function and phenotype of peripheral NK cells is altered in patients with HL
It is a hallmark of HL that the malignant cells in affected lymph nodes are surrounded by immune effector cells including lymphocytes, that are unable to recognize and kill the tumor cells.23 Here, we demonstrate that the recognition and killing of the HL-derived target cell line L428 was impaired in peripheral NK (pNK) cells isolated from patients with HL (Figure 1a), although this cell line was efficiently lyzed by NK cells from healthy donors. The difference between NK cell cytotoxicity from patients (samples were taken before therapy) and healthy donors was highly significant (P = 0.0001). Fluorescence activated cell sorter analysis confirmed published data24 that reported absence or very low expression of MHC I on L428 cells (Figure 1b, first panel) excluding an MHC I-mediated suppression of HL-NK cells in these assays. CD95 (APO-1/Fas) and CD262 (DR5), death receptors involved in NK cell-mediated killing and several other costimulatory adhesion molecules including ICAM-1 and ICAM-2 were expressed on L428 target cells (Figure 1b). The expression of ligands for the NCRs NKp30 and NKp46 was not detectable upon staining with recombinant receptors, whereas various ligands for NKG2D (MICA/B, members of the ULBP family) were assured using both, specific antibodies and NKG2D-Fc protein (Figure 1b). Lysis of L428 target cells by healthy NK cells was mainly dependent on NKG2D, as an NKG2D-blocking antibody was able to suppress NK cell-dependent killing (Figure 1c).
Figure 1.

The function and phenotype of peripheral NK cells (pNK) is altered in patients with Hodgkin lymphoma. (a) pNK from patients with HL (before therapy) or healthy donors were coincubated with the HL cell line L428 at different effector:target ratios for a cytotoxicity assay. Patient pNK cells were nearly unable to recognize and kill target cells that were lyzed by NK cells from healthy donors. One representative (left panel) and cumulative results of eight patients and six healthy donors of the effector:target ratio of 5:1 (right panel) are shown (mean ± SEM). Significant difference are indicated (Student's t-test, unpaired; P ≤ 0.0001). (b) Expression of various surface molecules on L428 cells was estimated by flow cytometry. Cells were incubated for 1 hour with 10 µg/ml isotype, primary antibody or the indicated Fc-fusion constructs followed by 30 minutes incubation with 10 µg/ml secondary antibodies (isotype control: filled histograms, specific staining: open histograms). (c) Cytoxicity assays with NK cells from healthy donors and L428 target cells were performed in the presence of an isotype control antibody (iso) or a NKG2D-blocking antibody (αNKG2D) (effector to target ratio was 5:1). Blocking of NKG2D resulted in a significant (P = 0.005, Student's t-test, unpaired) inhibition of killing. Depicted is the mean of four independent experiments; ***P ≤ 0.0001. **P ≤ 0.005. HL, Hodgkin lymphoma; NK, natural killer.
NKG2D surface expression on HL-derived pNK is reduced
We then analyzed the expression pattern of a panel of NK cell markers and receptors such as CD16, the NCRs including NKp30, NKp46, NKp44, and NKG2D. The samples were obtained from patients (mean age: 38) before (n = 40, BT), during (n = 39, DT), and on completion/after radio/chemotherapy (n = 17, AT). Surprisingly, no significant difference between the NK cells derived from healthy donors (n = 23) and those derived from untreated patients with HL was observed for CD16, NCRs, and for the activation markers CD25, CD69, and CD71, although NKp30 and NKp46 were significantly downregulated during therapy (Figure 2a and data not shown). The only distinctive feature of NK cells from patients with HL was a significantly decreased NKG2D surface expression (P = 0.0001 for healthy versus untreated patients with HL (Figure 2a, left panel)). It is well established that tumor cells release soluble ligands for NKG2D to inhibit antitumor immune responses via downregulation of the receptor.1,2 In fact, analysis of about 300 HL serum samples (Figure 2b) indicated that the NKD2D ligand MICA was significantly elevated in patients with HL before start of therapy. Regarding the NKD2D ligands ULBP2 and MICB, there was no significant difference between healthy donors and patients (data not shown). Of note, there was also a trend for a higher level of BAG6/BAT3 in patients (7988 ± 551 (SEM) pg/ml) versus healthy donors (5666 ± 822 (SEM) pg/ml). BAG6 is a novel ligand for NKp30 which is known to suppress NK cell function in soluble form.7 ELISA assays to measure soluble NKp30-ligand B7-H6 are not yet available. Elevated serum levels were also observed for macrophage migration inhibitory factor (MIF), a leaderless cytokine that contributes to NKG2D downregulation in ovarian cancer or malignant gliomas.25,26 After treatment, serum levels for all of these factors decreased, although some patients with later relapse were characterized by sustained and significantly higher MIF and BAG6 expression. The HL cell line L428 did not express surface ligands for NKp30, as binding of NKp30-Fc and anti-BAG6 antibody staining was not detectable by flow cytometry (Figure 1b), although soluble NKp30-ligand BAG6/BAT3 was traceable in L428 cell supernatant (data not shown). To address the in vivo expression profile, we analyzed primary HL tissue samples for BAG6 expression (Figure 2c). Staining of HL tissue (n = 21) revealed a strong BAG6 expression in HRS cells (21/21), and weaker expression of some bystander cells. The BAG6 staining was predominantly detectable either within the cytoplasm (HL 1) or in the cell nuclei (HL 2 and 3) probably reflecting its different intracellular functions that range from metabolic and chaperone activity to p53-dependent gene regulation.27,28,29
Figure 2.

The surface expression of NKG2D on HL-derived pNK cells is reduced. (a) Four-color flow cytometry was performed to determine the expression of the activating NK cell receptors NKG2D, NKp30, and NKp46 on gated NK cells (CD3−/CD56+/CD16+). Samples were analyzed before (BT), during (DT), and after (AT) therapy. Significant differences (Mann–Whitney U test) are indicated. (b) Serum levels of MICA, BAG6, and MIF are elevated in HL serum samples in comparison with samples of healthy controls (age-matched). HL samples were collected before therapy (BT), after therapy (AT), and from patients after therapy with later relapse (ATre). Significant differences are indicated (Mann–Whitney U test). (c) Histology: Tissue samples from patients with HL were stained with the BAG6-specific monoclonal antibody 3E4. Three samples from different patients are shown. ***P ≤ 0.0001; *P ≤ 0.05. HL, Hodgkin lymphoma; MIF, macrophage migration inhibitory factor; NK, natural killer.
HL serum derogates NK cell activity
Consequently, to gain insight into the impact of the aberrantly expressed serum factors, we analyzed the effect of patient serum on NK cell function. Interestingly, stimulation with interleukin 2 (IL-2) resulted in the activation of pNK cells isolated from patients (HL-NK) and led to a robust target cell killing. This indicates that NK cell “anergy” is reversible (Figure 3a, left panel). However, the IL-2–mediated activation was not observed in the presence of patient serum (HL-NK/HLs), suggesting that HL serum factors suppress IL-2 function or IL-2 signaling pathways of NK cells in this disease. Supporting this conclusion, we demonstrate that HL serum was sufficient to suppress the cytotoxicity of normal (healthy donor) NK cells against target cells, whereas serum from healthy donors (NK/healthy serum) had no effect on NK cell activity (Figure 3a, right panel). In line, we observed that overnight incubation of freshly isolated NK cells with patient serum with HL (n = 5) resulted in a significant downregulation of NKG2D, NKp30, and the activation marker CD69 (Figure 3b) in comparison with control sera of healthy donors (n = 5). A significant reduction was also observed for CD56, whereas the expression of CD16 and NKp46 remained stable rendering them as promising therapeutic targets. These data correlated with the elevated serum level for BAG6 targeting NKp30 and for MICA targeting NKG2D (Figure 2b) and may explain the reduced cytotoxicity of patient peripheral NK cells and healthy NK cells upon incubation with HL sera (Figures 1a and 3a). This ex vivo cocultivation experiment allowed a direct estimation of the HL sera-mediated effects on a given polyclonal NK cell population. However, the results were not exactly reflected by the comparison of the NK cell phenotype of healthy donors versus patients with HL (Figure 2a), which is probably due to the pattern of heterogeneous receptor expression among the donors.
Figure 3.

The impaired cytotoxicity of HL-derived NK cells is reversible. (a) Left panel: Peripheral patient NK cells can be activated with IL-2. The relative cytotoxicity of patient pNK cells was assessed using europium release assays with L428 target cells. NK cells were either left untreated (HL-NK) or stimulated over night with 50 U/ml IL-2 with (HL-NK/HLs + IL-2) or without (HL-NK + IL-2) the presence of autologous patient serum with HL. Right panel: NK cells from healthy donors were incubated with HL serum (NK/HL serum) and serum from healthy donors (NK/healthy serum) over night. Preincubated NK cells were used in a europium release assay with L428 target cells. All HL sera samples were taken before therapy. (b) Sera from patients with HL shape the phenotype of normal NK cells. NK cells were incubated with 25% sera from healthy donors (ctr) or patients with HL (five samples each, HL samples before therapy) for 19 hours, and the expression of the receptors was analyzed by flow cytometry. Results for each sample are represented by mean fluorescence intensity (MFI). *** P ≤ 0.0001; **P ≤ 0.005; *P ≤ 0.05. Significances are indicated. HL, Hodgkin lymphoma; IL-2, interleukin 2; NK, natural killer.
HL-NK cells can be activated in vitro and in vivo with an CD30xCD16A
Provided that the inhibition of HL-derived pNK cells is not intrinsic but depends on their environment—soluble serum factors—NK cells might be excellent targets for immunotherapy. Therefore, we tested the impact of a tetravalent, bispecific antibody construct CD30xCD16A (TandAb AFM13; Affimed Therapeutics, Heidelberg, Germany) on HL-derived pNK cells in vitro and in vivo. AFM13 is designed for the treatment of Hodgkin lymphoma and is currently tested in a phase I clinical study (ClinicalTrials.gov identifier NCT01221571) to assess the safety in patients with HL. The construct targets CD16A on NK cells and binds simultaneously to the surface receptor CD30, a member of the tumor necrosis factor family. CD30 is overexpressed on malignant HRS cells and contributes to the proinflammatory tumor microenvironment in this malignancy.30 Previous studies showed promising clinical results for a bivalent, bispecific monoclonal antibody targeting CD16 and CD30;31 however, the potency to trigger patient NK cell function and target cell killing has not been analyzed so far.
Cytotoxicity assays revealed that the bispecific protein, but not a αCD30 single chain control protein, significantly enhanced lymphocyte and HL-NK cell-dependent killing (Figure 4a) of L428 target cells. The lysis of CD30− 293T cells remained unaffected. Control experiments proved that NK cell activation was dependent on the presence of both target cells and CD30xCD16A, because neither L428 cells nor CD30xCD16A alone were sufficient to induce the expression of the activation marker CD69 on NK cells (Figure 4b). CD69 belongs to the family of C-type lectin receptors and triggers cytotoxicity and cytokine production of NK cells via pathways activating PLC and vav1.32 CD69 is induced after activation with IL-2, IL-12, or tumor necrosis factorα and also in response to anti-CD16 monoclonal antibodies; however, the CD30xCD16A failed to upregulate CD69 in the absence of target cells, arguing against nonspecific activity of this antibody construct (Figure 4b).
Figure 4.

CD30xCD16A activates effector cells from patients with HL in vitro and in vivo. (a) The cytotoxicity of HL-PBMCs (left panel) or purified patient NK cells (right panel) was analyzed by a standard 3 hours europium release assay with L428 target cells in the presence of CD30xCD16A (AFM13) or a control α-CD30 single chain (αCD30sv control). NK cell cytotoxicity against CD30− target cells (293T) in the presence of AFM13 is indicated (AFM/CD30-target). (b) NK cells from healthy donors were incubated with either AFM13 (10 µg/ml), L428 target cells (ratio 5:1), or AFM in combination with L428 for 16 hours, and the expression of CD69 on NK cells was estimated by flow cytometry. (c) CD69 is upregulated on NK cells of patients with HL in response to CD30xCD16A therapy. Flow cytometry: The percentage of CD69+ NK cells in the peripheral blood of CD30xCD16A-treated patients (two different doses) was estimated at the following time points: before therapy (pre), 24 hours after first (d1), second (d8), third (d15), and last (d22) CD30xCD16A infusion. Each curve represents one patient. (d) Ex vivo cytotoxicity assay: NK cells were isolated from the peripheral blood from patients before start of the therapy and 24 hours after the end of first AFM13 infusion to assess the cytotoxicity against the Hodgkin lymphoma cell line L428 (left panel). Left panel: one representative of four patients; right panel: mean of six patients tested. NK cells from two out of six patients tested remained inactive upon treatment. (e) The same killing assay was performed in the presence of a MHC I-blocking antibody (a-HLA I), an isotype control (iso), or without antibodies (ctr) (right panel). (f) The binding of AFM13 to peripheral NK cells was estimated before and after (24 hours and 48 hours) the first AFM13 infusion by flow cytometry. The percentage of NK cells loaded with AFM13 is indicated. d, day; HL, Hodgkin lymphoma; MFI, mean fluorescence intensity; MHC I, major histocompatibility complex class I; NK, natural killer; pre, pretreatment.
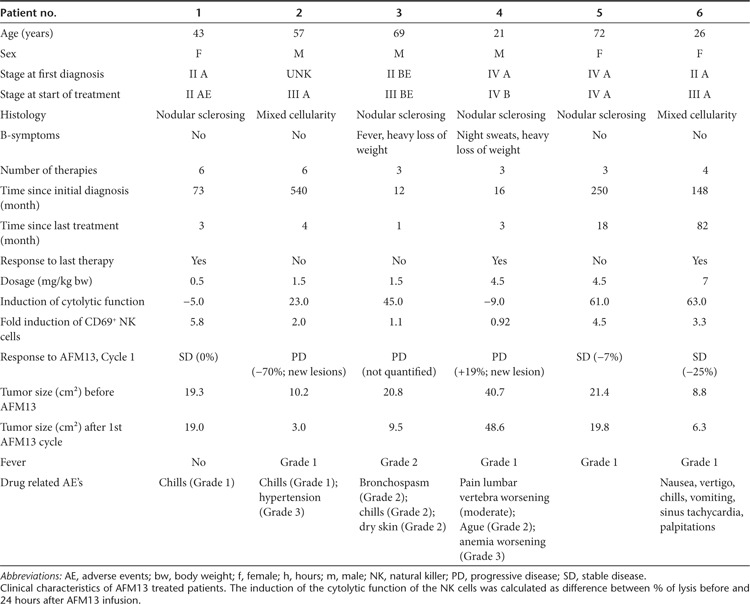
In the next step, we asked whether an activation of NK cells monitored by CD69 upregulation was detectable in study patients treated with CD30xCD16A (Figure 4c). Patient NK cells were isolated and analyzed before the start of therapy (pre) and 24 hours after each of 4 weekly infusions. After the first treatment, CD69 expression was induced in all patients analyzed. The initial induction showed a decline in response to further treatments; however, the percentage of CD69+ NK cells was still higher in five out of seven patients compared with the pretreatment level. Upregulation of CD69 suggests an enhanced cytotoxic potential of the affected NK cells. In fact, ex vivo killing assays demonstrated these functional consequences: patient NK cells isolated before therapy were inactive, whereas NK cells isolated 24 hours after a single AFM13 infusion were nearly as cytotoxic as NK cells from healthy donors in four out of six patients tested (Figure 4d). To further prove that the killing of the target cells was independent of MHC I-missing self-activation, any remaining inhibitory self-antigens were blocked through the addition of an MHC I-specific antibody (Figure 4e). This blocking of MHC I antigens had no impact on target cell killing; therefore, it can also be expected in the autologous patient setting. The binding of AFM13 to the NK cells was detectable on the isolated patient cells by fluorescence activated cell sorter analysis using an antibody with specificity for the construct (Figure 4f). AFM13 was thus present in the cytotoxicity assays with patient NK cells as effector cells. Thus, the high cytotoxic activity of these cells might be due to an antibody-dependent cellular cytotoxicity-like–mediated mechanism. The enhanced killing, mediated through CD30xCD16A, is most likely restricted to CD30+ target cells as shown for NK cells from healthy donors, although this could not be directly tested due to the limitation of patient cells. Taken together, the treatment with an antibody construct targeting CD16A on NK cells can overcome NK cell inhibition in vivo, despite the presence of inhibitory factors in the patient blood. The overall objective of the AFM13 study (ClinicalTrials.gov identifier NCT01221571 ongoing study, 28 patients treated so far), which is a single arm phase I dose escalation trial for patients with relapsed or refractory HL, is to evaluate safety, tolerability, pharmacokinetics, immunogenicity, antitumor activity, the maximum tolerated dose, and the optimal biological dose of AFM13. The study drug was well tolerated and adverse events were generally mild (Table 1). Three out of six patients that were heavily pretreated achieved stable disease (SD), reflecting antitumor activity of AFM13. These patients showed clear upregulation of CD69 on NK cells, and two of them showed efficient ex vivo killing of HL cells after treatment. One patient with SD but no enhanced NK cell-mediated tumor cell killing ex vivo was treated with the least dose of 0.5 mg/kg body weight of all patients tested in this setting. This dose was obviously sufficient to trigger upregulation of CD69 but might not be high enough to preactivate the NK cells above the necessary stimulus threshold with one single infusion. Thus, repeated NK cell stimulation for 4 weeks by AFM13 treatment on a weekly basis could be sufficient to reach this threshold leading to the observed efficacy of the drug in vivo. Moreover, the cytotoxicity of patient NK cells was also enhanced in two patients with SD probably indicating that NK cell activation by AFM13 is a prerequisite for the biological activity of AFM13 but alone not sufficient for the therapy response. So far, we do not know much about homing of the activated NK cells to the tumor tissue and the influence of the HL tumor microenvironment on NK cell activity in these patients, which should be addressed in further studies. Recent data showed that the HL microenvironment might compromise immune effector functions.33 Taken together, AFM13 has demonstrated encouraging biologic activity and seems to be a new feasible targeted therapy for heavily pretreated patients with HL. These findings strongly support the high demand for therapeutic strategies aimed at restoring NK cell function to avoid tumor evasion from the patient immune system.
Table 1. Clinical characteristics and response.
Discussion
NK cells represent the main component within the innate immune system involved in recognition and elimination of cancer cells. Here, we report that NK cells from patients with HL have defects in target cell killing and reveal an “anergic” phenotype. The impaired cytotoxicity was attributed to an impaired NKG2D-mediated recognition. The reduced lysis of the target cells was reflected by a downregulation of the NKG2D receptor on peripheral NK cells that correlated to elevated levels of sMICA in patient serum samples. There is an experimental and clinical evidence showing that the NKG2D/NKG2D-L system represents a major part of tumor cell rejection and recent studies demonstrated that there is an impaired immunosurveillance in NKG2D-deficient mice.34 For diseases such as chronic lymphocytic leukemia, a positive correlation of soluble NKG2D ligands to disease stage and bad prognosis was reported.35 Shedding of NKG2D-L from the tumor cell surface is one of the best studied mechanisms that circumvent detection by the innate immune system through reducing activating signals from the cell surface.1,2 Moreover, the released soluble ligands for NKG2D are known to counteract immunosurveillance in two ways: they inhibit NKG2D-cytotoxicity by binding/blocking the receptor and induce its downregulation on the cell surface of T cells and NK cells.1,2 Recently, it was shown that patient leukemia sera with high sNKG2D-L levels reduce NKG2D on NK cells promoting immune evasion,17 whereas other serum factors and their effects on NK cell receptor expression were not addressed in this study. The prominent role for soluble NKG2D ligands in the evasion of NK cell recognition is widely accepted, although not reported for HL so far. Aside from that, our data suggest that not only MICA, the ligand for NKG2D, contributes to NK cell inhibition, but also the factors MIF and BAG6 reveal elevated serum levels in patients versus healthy donors. MIF, known as the MIF, reduces NK cell function also by NKG2D downregulation.25,26 High level of BAG6, the soluble ligand for NKp30, might further constrict NK cell function as incubation of healthy NK cells with HL serum resulted not only in NKG2D downregulation but also in a decreased NKp30-surface detection.
A reduced expression of NCRs on peripheral or tumor-associated NK cells has already been described for tumors such as leukemia, multiple myeloma, and breast cancer.36,37 A critical role for NKp30 for immune surveillance was already proposed but only recently demonstrated in gastrointestinal tumors38 shedding light on its potential role in antitumor immunity. Analysis of larger patient cohorts is needed to uncover potential diagnostic or prognostic value of MIF and BAG6 as novel markers for the treatment outcome in HL. However, a direct correlation of ligand serum level and receptor expression level could not be established in this study, because NK cell phenotype and ligand serum level data were only available from different patients.
The impaired NKG2D-dependent killing of target cells by cytotoxic T cells that were obtained from HL tissue was recently attributed to the release of MICA and ULBP3 in combination with TGFβ from bystander and HRS cells, which caused downregulation of NKG2D on effector lymphocytes in the microenvironment.33 The data presented here indicate that tissue-derived factors of tumors shape the phenotype and function of peripheral immune cells, which may further inhibit antitumor immune responses.
However, the inhibition of NK cell activity of patient NK cells could be restored with IL-2, a cytokine that is well known to trigger NK cell activity via Ras/MAPK, JAK/Stat, and PI 3-kinase/Akt signaling pathways. Interestingly, IL-2 failed to activate NK cells in the presence of patient serum, suggesting that serum factors act against NK cell stimulation. In line with our data, a reversible suppression of peripheral NK cells, that promote self tolerance, was recently reported for breast cancer.37,39
Killing dysfunction of NK cells from patients with HL could be restored with AFM13, a bispecific antibody construct that targets CD16A on effector cells and binds simultaneously to the surface receptor CD30. AFM13 belongs to a new group of antibody formats that has two binding sites for each antigen as it is a homodimer consisting of two polypeptides pairing head-to-tail with each other. This allows tetravalent binding and leads to a comparable avidity as for IgG. In contrast to the previously used CD30-CD16 targeting antibody HRS-3/A9,40 AFM13 consist solely of variable domains and therefore avoids Fc-mediated side effects. A previous study showed promising results against xenotransplanted solid human HL for a bivalent, bispecific monoclonal antibody targeting CD16 and CD30,31 indicating that these targets are well suited for an NK cell activating approach.
CD30, a member of the tumor necrosis factor family is overexpressed on malignant HL cells and associated with constitutive CD30 signaling, known to contribute to the proinflammatory tumor microenvironment.30,41
Enhancement of patients NK cell cytotoxicity against L428 target cells using AFM13 worked in both settings, after direct addition of the construct to the cytotoxicity assay and after treatment of patients. CD16, the low-affinity IgG receptor, is responsible for antibody-dependent cellular cytotoxicity and the engagement of the receptor causes degranulation of NK cells thus triggering target cell lysis.20,42 Both in vitro and in vivo studies indicate that the CD16-mediated antibody-dependent cellular cytotoxicity is the predominant mode by which an antitumor response is achieved. In vivo studies using mice with defects in Fc receptor expression demonstrated that the activity of therapeutic antibodies was dependent on the expression of Fc receptors on immune cells.43,44
The general activation of NK cells by the bispecific construct was exclusively observed in the presence of CD30+ target cells and was reflected by the induced expression of the NK cell activation marker CD69. CD69 cross-linking induces the cytotoxic activity and costimulates cytokine production via phosphotyrosine kinases of the Src family. Engagement of CD69 through either antibodies or—under physiological conditions—with undefined ligands on target cells initiates the activation of PLC and Vav1, both involved in the development of cytotoxicity.32 The treatment with the bispecific tetravalent antibody CD30xCD16A that induced CD69 expression on the effector cells may thus be regarded a first step in NK cell activation that results in the target cell killing upon direct interaction of NK cells with target cells via the CD30 antibody fragment. Evidence for NK cell activation as a two-stage process by means of first an unspecific activation and second target cell specific activation step was described previously.45,46 Other groups also reported that antibody-based approaches with antibody-drug conjugates like Brentuximab vedotin47 or bispecific antibodies31,48 with promising results against B-cell lymphomas, confirming the principle efficiency of novel immunological therapeutics.
Taken together, the impaired function of patient NK cells that are characterized by a decreased NKG2D expression does not reflect a general cytolytic dysfunction. We were able to show that the impaired NK cell function in patients with HL can be restored: NK cells from patients with HL were activated upon treatment with a construct targeting CD16 and CD30 (ex vivo and in vivo), resulting in an improved recognition of an HL-derived target cell line. This data warrant further studies on the feasibility, toxicity, and the clinical response of patients with HL. Furthermore, the NK cell population—in contrast to the T cells—is reconstituted quickly after the end of standard therapies in HL (data not shown). Innovative immune therapies for patients following chemo/radiotherapy that target NK cells49 rather than T cells are therefore extremely promising.
Materials and Methods
Patients. The research was approved by the Ethics Committee of the University Clinic of Cologne and all human participants gave written informed consent. NK cells were isolated with the NK Cell Isolation Kit and AutoMACS (Miltenyi, Bergisch-Gladbach, Germany) as described previously50 from healthy humans (blood or buffy coats), from patients before/during/after standard chemo/radiotherapy (patients participating in HD14–18 trials of the German Hodgkin Study Group), or from patients participating in the clinical study to assess AFM13 safety in patients with HL (Figure 4c–e; Study Identifier NCT01221571 (ClinicalTrials.gov)). The purity of NK cells used (CD3−, CD56+) was evaluated by flow cytometry (≥94%).
Cells. The HL cell line L428 (B cell, nodular sclerosis, defective IκBα, and IκBε, MHC I low or absent, obtained from DSMZ ACC-197) was cultured in RPMI 1640 with 10% fetal calf serum (Invitrogen, Carlsbad, CA), 50 µg/ml penicillin, 50 µg/ml streptomycin at 37 °C in a 5% (v/v) CO2 atmosphere.
Flow cytometry. EDTA whole blood was stained for four-color flow cytometry and subsequent lysis of erythrocytes using IOTest 3 Lysing Solution (BeckmanCoulter, Brea, CA). T and NK cells were discriminated by CD3-PerCp-Cy5.5 and CD16-AlexaFluor647 + CD56-AlexaFluor647 (BioLegend, San Diego, CA) staining. NKG2D (Abcam, Cambridge, UK), NKp30 (BioLegend), NKp44 (BeckmanCoulter), NKp46 (BioLegend), and the activation marker CD25, CD69, and CD71 (all BD Biosciences, San Jose, CA) were detected with FITC- or PE-labeled antigen-specific antibodies. AFM13 binding on patient NK cells was detected with AlexaFluor488-labeled rat-monoclonal IgG anti-AFM13 from Affimed Therapuetics AG. For the analysis of surface expression pattern of L428, cells staining was performed with PE-labeled antibodies against CD262, ICAMs, and HLA-A,B,C (BioLegend) and PE anti-CD95 (BD Biosciences). Antibodies against MICA and ULBPs were from Bamomab, whereas anti-MICB was from R&D Systems (Minneapolis, MN). PE goat antimouse from BioLegend was used as secondary antibody. Respective isotype controls were from BioLegend. Binding to recombinant NKG2D-Fc, NKp30-Fc, and NKp46-Fc proteins (R&D Systems) was detected by Cy3 antihuman Fc from Dianova.
Serum incubation. NK cells were incubated with 25% (v/v) serum in Iscove's modified Dulbecco's medium overnight prior analysis to determine the expression level of NKG2D, NKp30, CD16, CD56, NKp46, and CD69.
Cytotoxicity assays. NK cell-mediated cytotoxicity was analyzed by a standard 3 hours europium release assay in a 96-well microtiter plate as previously described.50 Briefly, NK effector cells were mixed with europium chloride labeled (Fluka, Buchs, Switzerland) 5 × 103 target cells at different ratios. NK cells were preincubated over night with IL-2 as indicated.
Blocking of NKG2D was performed by preincubation of NK cells (30 minutes, 4 °C) with 10 µg/ml of the blocking antibody clone 1D11 (BioLegend) or equivalent amount of an isotype control (ms IgG1; BioLegend).
The ex vivo activation with a tetravalent, bispecific CD30xCD16A antibody (TandAb AFM13; Affimed Therapeutics) was performed with purified NK cells without any costimuli. AFM13 or a control monospecific bivalent anti-CD30 diabody comprising the same anti-CD30 variable domains as AFM13 were added at 10 µg/ml final concentration to the NK cells before adding target cells. The CD30− cell line 293T was used as a control. Supernatant was assayed for europium release after 3 hours in a Wallac Victor 1420 multilabel counter (Wallac Oy, Turku, Finland). The percentage of specific lysis was calculated as 100 × ((experimental release − spontaneous release)/(maximal release − spontaneous release)).
ELISA. Sandwich ELISAs were performed using the commercially available R&D Duo Sets for ULBP2, MICB, MIF, and MICA, except that AM01 (5 µg/ml; Bamomab, Gräfelfing, Germany) was used as a capture antibody for MICA. The mouse monoclonal 3E4 (raised against the BAG6 N-terminus) and the BAG6-specific rabbit serum 41607 were used to detect BAG6.
Immunohistology. Immunohistological staining was performed by the alkaline phosphatase anti-alkaline phosphatase method.
Statistics. Analysis (nonparametric Mann–Whitney U test or unpaired Student's t-test) was performed using the GraphPad prism5 software (GraphPad Software, San Diego, CA). (***P < 0.0001; **P < 0.005, and *P < 0.05).
Acknowledgments
We thank particularly the blood donors for their generous contribution and Birgit Gathof, Director of the Institut für Transfusionsmedizin, University Clinic Cologne for support. U.R. and C.H. are employees of Affimed Therapeutics AG. This study was supported by a grant from the Deutsche Forschungsgemeinschaft (SFB832, TP19) to E.P.v.S. The other authors declared no conflicts of interest.
References
- Waldhauer I., and, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27:5932–5943. doi: 10.1038/onc.2008.267. [DOI] [PubMed] [Google Scholar]
- Nausch N., and, Cerwenka A. NKG2D ligands in tumor immunity. Oncogene. 2008;27:5944–5958. doi: 10.1038/onc.2008.272. [DOI] [PubMed] [Google Scholar]
- Raulet DH., and, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol. 2006;6:520–531. doi: 10.1038/nri1863. [DOI] [PubMed] [Google Scholar]
- Hershkovitz O, Jarahian M, Zilka A, Bar-Ilan A, Landau G, Jivov S.et al. (2008Altered glycosylation of recombinant NKp30 hampers binding to heparan sulfate: a lesson for the use of recombinant immunoreceptors as an immunological tool Glycobiology 1828–41. [DOI] [PubMed] [Google Scholar]
- Rosental B, Brusilovsky M, Hadad U, Oz D, Appel MY, Afergan F.et al. (2011Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44 J Immunol 1875693–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershkovitz O, Jivov S, Bloushtain N, Zilka A, Landau G, Bar-Ilan A.et al. (2007Characterization of the recognition of tumor cells by the natural cytotoxicity receptor, NKp44 Biochemistry 467426–7436. [DOI] [PubMed] [Google Scholar]
- Pogge von Strandmann E, Simhadri VR, von Tresckow B, Sasse S, Reiners KS, Hansen HP.et al. (2007Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells Immunity 27965–974. [DOI] [PubMed] [Google Scholar]
- Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B.et al. (2009The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans J Exp Med 2061495–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim DE, Roberts SJ, Clarke SL, Filler R, Lewis JM, Tigelaar RE.et al. (2005Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance Nat Immunol 6928–937. [DOI] [PubMed] [Google Scholar]
- Coudert JD, Zimmer J, Tomasello E, Cebecauer M, Colonna M, Vivier E.et al. (2005Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells Blood 1061711–1717. [DOI] [PubMed] [Google Scholar]
- Groh V, Wu J, Yee C., and, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419:734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH.et al. (2007Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands Nature 447482–486. [DOI] [PubMed] [Google Scholar]
- Salih HR, Antropius H, Gieseke F, Lutz SZ, Kanz L, Rammensee HG.et al. (2003Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia Blood 1021389–1396. [DOI] [PubMed] [Google Scholar]
- Salih HR, Holdenrieder S., and, Steinle A. Soluble NKG2D ligands: prevalence, release, and functional impact. Front Biosci. 2008;13:3448–3456. doi: 10.2741/2939. [DOI] [PubMed] [Google Scholar]
- Paschen A, Sucker A, Hill B, Moll I, Zapatka M, Nguyen XD.et al. (2009Differential clinical significance of individual NKG2D ligands in melanoma: soluble ULBP2 as an indicator of poor prognosis superior to S100B Clin Cancer Res 155208–5215. [DOI] [PubMed] [Google Scholar]
- Li K, Mandai M, Hamanishi J, Matsumura N, Suzuki A, Yagi H.et al. (2009Clinical significance of the NKG2D ligands, MICA/B and ULBP2 in ovarian cancer: high expression of ULBP2 is an indicator of poor prognosis Cancer Immunol Immunother 58641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilpert J, Grosse-Hovest L, Grünebach F, Buechele C, Nuebling T, Raum T.et al. (2012Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses J Immunol 1891360–1371. [DOI] [PubMed] [Google Scholar]
- Tursz T, Dokhelar MC, Lipinski M., and, Amiel JL. Low natural killer cell activity in patients with malignant lymphoma. Cancer. 1982;50:2333–2335. doi: 10.1002/1097-0142(19821201)50:11<2333::aid-cncr2820501119>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Konjevic G, Jurisic V, Banicevic B., and, Spuzic I. The difference in NK-cell activity between patients with non-Hodgkin's lymphomas and Hodgkin's disease. Br J Haematol. 1999;104:144–151. doi: 10.1046/j.1365-2141.1999.01129.x. [DOI] [PubMed] [Google Scholar]
- Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foyil KV., and, Bartlett NL. Anti-CD30 Antibodies for Hodgkin lymphoma. Curr Hematol Malig Rep. 2010;5:140–147. doi: 10.1007/s11899-010-0053-y. [DOI] [PubMed] [Google Scholar]
- Eichenauer DA, Fuchs M, Borchmann P., and, Engert A, German Hodgkin Study Group Hodgkin's lymphoma: current treatment strategies and novel approaches. Expert Rev Hematol. 2008;1:63–73. doi: 10.1586/17474086.1.1.63. [DOI] [PubMed] [Google Scholar]
- Steidl C, Connors JM., and, Gascoyne RD. Molecular pathogenesis of Hodgkin's lymphoma: increasing evidence of the importance of the microenvironment. J Clin Oncol. 2011;29:1812–1826. doi: 10.1200/JCO.2010.32.8401. [DOI] [PubMed] [Google Scholar]
- Van den Berg A, Visser L, Eberwine J, Dadvand L., and, Poppema S. Frequent lack of translation of antigen presentation-associated molecules MHC class I, CD1a and Beta(2)-microglobulin in Reed-Sternberg cells. Int J Cancer. 2000;86:548–552. doi: 10.1002/(sici)1097-0215(20000515)86:4<548::aid-ijc17>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Krockenberger M, Dombrowski Y, Weidler C, Ossadnik M, Hönig A, Häusler S.et al. (2008Macrophage migration inhibitory factor contributes to the immune escape of ovarian cancer by down-regulating NKG2D J Immunol 1807338–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelbronn M, Platten M, Zeiner P, Dombrowski Y, Frank B, Zachskorn C.et al. (2011Macrophage migration inhibitory factor (MIF) expression in human malignant gliomas contributes to immune escape and tumour progression Acta Neuropathol 122353–365. [DOI] [PubMed] [Google Scholar]
- Hessa T, Sharma A, Mariappan M, Eshleman HD, Gutierrez E., and, Hegde RS. Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature. 2011;475:394–397. doi: 10.1038/nature10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Wang S, Xiao M, Lin Y, Zhou L, Lei Q.et al. (2011Acetylation regulates gluconeogenesis by promoting PEPCK1 degradation via recruiting the UBR5 ubiquitin ligase Mol Cell 4333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Gan EC, Wakeham A, Kornbluth S, Mak TW., and, Okada H. HLA-B-associated transcript 3 (Bat3)/Scythe is essential for p300-mediated acetylation of p53. Genes Dev. 2007;21:848–861. doi: 10.1101/gad.1534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenko M., and, Sidorenko SP. Hodgkin's lymphoma: the role of cell surface receptors in regulation of tumor cell fate. Exp Oncol. 2010;32:214–223. [PubMed] [Google Scholar]
- Arndt MA, Krauss J, Kipriyanov SM, Pfreundschuh M., and, Little M. A bispecific diabody that mediates natural killer cell cytotoxicity against xenotransplantated human Hodgkin's tumors. Blood. 1999;94:2562–2568. [PubMed] [Google Scholar]
- Pisegna S, Zingoni A, Pirozzi G, Cinque B, Cifone MG, Morrone S.et al. (2002Src-dependent Syk activation controls CD69-mediated signaling and function on human NK cells J Immunol 16968–74. [DOI] [PubMed] [Google Scholar]
- Zocchi MR, Catellani S, Canevali P, Tavella S, Garuti A, Villaggio B.et al. (2012High ERp5/ADAM10 expression in lymph node microenvironment and impaired NKG2D ligands recognition in Hodgkin lymphomas Blood 1191479–1489. [DOI] [PubMed] [Google Scholar]
- Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N.et al. (2008NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy Immunity 28571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nückel H, Switala M, Sellmann L, Horn PA, Dürig J, Dührsen U.et al. (2010The prognostic significance of soluble NKG2D ligands in B-cell chronic lymphocytic leukemia Leukemia 241152–1159. [DOI] [PubMed] [Google Scholar]
- Fauriat C, Just-Landi S, Mallet F, Arnoulet C, Sainty D, Olive D.et al. (2007Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction Blood 109323–330. [DOI] [PubMed] [Google Scholar]
- Mamessier E, Sylvain A, Bertucci F, Castellano R, Finetti P, Houvenaeghel G.et al. (2011Human breast tumor cells induce self-tolerance mechanisms to avoid NKG2D-mediated and DNAM-mediated NK cell recognition Cancer Res 716621–6632. [DOI] [PubMed] [Google Scholar]
- Delahaye NF, Rusakiewicz S, Martins I, Ménard C, Roux S, Lyonnet L.et al. (2011Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors Nat Med 17700–707. [DOI] [PubMed] [Google Scholar]
- Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R.et al. (2011Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity J Clin Invest 1213609–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann F, Renner C, Jung W, Deisting C, Juwana M, Eichentopf B.et al. (1997Treatment of refractory Hodgkin's disease with an anti-CD16/CD30 bispecific antibody Blood 892042–2047. [PubMed] [Google Scholar]
- Kennedy MK, Willis CR., and, Armitage RJ. Deciphering CD30 ligand biology and its role in humoral immunity. Immunology. 2006;118:143–152. doi: 10.1111/j.1365-2567.2006.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL. Natural killer cell receptor signaling. Curr Opin Immunol. 2003;15:308–314. doi: 10.1016/s0952-7915(03)00039-6. [DOI] [PubMed] [Google Scholar]
- Clynes RA, Towers TL, Presta LG., and, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- Clynes R, Takechi Y, Moroi Y, Houghton A., and, Ravetch JV. Fc receptors are required in passive and active immunity to melanoma. Proc Natl Acad Sci USA. 1998;95:652–656. doi: 10.1073/pnas.95.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North J, Bakhsh I, Marden C, Pittman H, Addison E, Navarrete C.et al. (2007Tumor-primed human natural killer cells lyse NK-resistant tumor targets: evidence of a two-stage process in resting NK cell activation J Immunol 17885–94. [DOI] [PubMed] [Google Scholar]
- Brenner CD, King S, Przewoznik M, Wolters I, Adam C, Bornkamm GW.et al. (2010Requirements for control of B-cell lymphoma by NK cells Eur J Immunol 40494–504. [DOI] [PubMed] [Google Scholar]
- Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ.et al. (2012Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma J Clin Oncol 302183–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pörtner LM, Schönberg K, Hejazi M, Brünnert D, Neumann F, Galonska L.et al. (2012T and NK cells of B cell NHL patients exert cytotoxicity against lymphoma cells following binding of bispecific tetravalent antibody CD19 × CD3 or CD19 × CD16 Cancer Immunol Immunother 611869–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terme M, Ullrich E, Delahaye NF, Chaput N., and, Zitvogel L. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol. 2008;9:486–494. doi: 10.1038/ni1580. [DOI] [PubMed] [Google Scholar]
- von Strandmann EP, Hansen HP, Reiners KS, Schnell R, Borchmann P, Merkert S.et al. (2006A novel bispecific protein (ULBP2-BB4) targeting the NKG2D receptor on natural killer (NK) cells and CD138 activates NK cells and has potent antitumor activity against human multiple myeloma in vitro and in vivo Blood 1071955–1962. [DOI] [PubMed] [Google Scholar]