

Abstract
Oncolytic virotherapy is an emerging treatment modality that uses replication-competent viruses to destroy cancers. Many naturally occurring viruses have a preferential, although nonexclusive, tropism for tumors and tumor cells. In addition, specific targeting of cancer cells can be achieved at the virus entry level. We optimized retargeting of cell entry by elongating the measles virus attachment protein with designed ankyrin repeat proteins (DARPins), while simultaneously ablating entry through the natural receptors. DARPin-targeted viruses were strongly attenuated in off-target tissue, thereby enhancing safety, but completely eliminated tumor xenografts. Taking advantage of the unique properties of DARPins of being fused without generating folding problems, we generated a virus simultaneous targeting two different tumor markers. The bispecific virus retained the original oncolytic efficacy, while providing proof of concept for a strategy to counteract issues of resistance development. Thus, DARPin-targeting opens new prospects for the development of personalized, targeted therapeutics.
Introduction
Despite all available current treatment regimens, cancer is still the second leading cause of death in industrialized countries.1 Even with approved targeted and efficacious stratified therapies as e.g., chemotherapy in conjunction with trastuzumab for HER2/neu-positive breast or ovarian carcinomas, not all patients benefit.2 Viruses that infect and destroy tumor cells, so called oncolytic viruses (OVs), are developed as future complementary strategy to fight cancer.3 This killing strategy is not redundant to existing regimens and rarely cross-resistance is to be expected. One candidate virus in the clinic is recombinant measles virus (MV),3,4 because remission of tumors had already been reported about 40 years ago in patients with hematologic cancers becoming MV infected.5 MV is a nonintegrating negative-strand RNA virus from the family Paramyxoviridae, which destroys infected cells by induction of intercellular fusion and formation of multinucleated giant cells, so called syncytia, causing apoptosis.6 Three cellular surface proteins have been identified as entry receptors for MV: the signaling lymphocyte activation molecule (SLAM) on activated lymphocytes and myeloid cells,7 nectin-4 on epithelial cells,8,9 and—just for vaccine strains and derivatives thereof—the complement attenuation receptor CD46, expressed on all human nucleated cells.10,11 Interestingly, a broad variety of tumors overexpress CD46, which protects tumor cells from complement-mediated lysis,12 but renders them especially susceptible for infection and killing by attenuated MV.6
To enhance tumor-targeting, oncolytic MV is the most advanced replication-competent system that can be fully genetically retargeted to specified surface receptors. For this purpose, protein domains that specifically bind to a variety of targets have been fused to the mutated MV attachment protein hemagglutinin (H) that cannot bind to its natural receptors anymore.13 Usually, single-chain variable fragments (scFvs) of antibodies have been used for this purpose.14,15,16 However, certain scFvs might be suboptimal targeting domains, because of the intrinsic aggregation tendencies of many scFvs.17 In addition, therapies relying on just one targeted marker carry the risk of resistance development.18 Targeting more than one epitope using scFvs is challenging, because the linking of several scFvs in series or as diabody constructs19 will potentially impair their correct folding, and even more so as H fusions. Therefore, designed ankyrin repeat proteins (DARPins) with at least similar versatility and affinity could be an alternative to scFvs as targeting domain for MV circumventing these drawbacks.20 DARPins are ankyrins in which residues potentially interacting with targets have been randomized and the others are derived from a consensus design, yielding elongated, rigid protein domains. High-affinity binders have been selected from these libraries.20,21 Importantly, DARPins can be linked to each other without any negative effect on folding or expression.22,23,24,25
Here, we aimed to establish retargeting of replicating MVNSe with the help of DARPins. In addition to efficacious monospecific targeting against one out of three relevant tumor markers (EGFR, HER2/neu, or EpCAM), we demonstrated for the first time rational efficient simultaneous bispecific targeting of an OV using coupled DARPins specific for either one of two receptors. Thus, this study reveals that DARPins are very effective targeting domains for oncolytic MV, demonstrating potent tumor-lytic efficacy, enhanced safety, and excellent specificity of fully retargeted monospecific and even of bispecific oncolytic DARPin-MV for next generation targeted OV.
Results
Generation and characterization of DARPin-MVs
To start generation of replication-competent DARPin-targeted MV, we substituted the αCD20-scFv coding sequence of the expression plasmid pCG-Hmut-αCD20-6His by one out of four HER2/neu-specific, three EGFR-specific, or two EpCAM-specific DARPins (Table 1). All chimeric H-DARPin proteins were stably expressed and transported to the cell surface in 293T cells (Supplementary Figure S1). The Hmut-DARPin fragments were cloned into the genome of attenuated Edmonston MV strain NSe encoding eGFP as reporter protein (Figure 1a), and the corresponding recombinant MVs were rescued, propagated, and titrated. All viruses could be rescued and gave titers of up to 1 × 107 TCID50/ml. While demonstrating the efficient expression of the respective H proteins in virus-infected cells we noticed that all H-DARPin proteins showed a higher molecular weight than those of the parental MVNSe virus due to the size of the individual DARPins (14–20 kDa) (Table 1, Figure 1b). Furthermore, the ratios of N- and H- protein expression did not differ significantly between the different viruses (Figure 1b), indicating viral expression of the different DARPin-H proteins without altering the typical MV protein expression profile.
Table 1. Properties of applied DARPins.
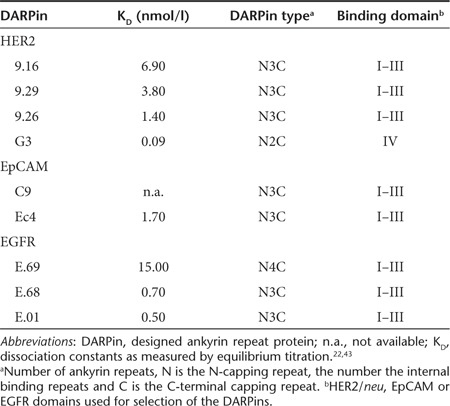
Figure 1.

Generation and specificity of DARPin-MV. (a) Schematic depiction of the MV genome and the cloning strategy for insertion of DARPin sequences into mutated H ORF with a carboxy-terminal His6 tag (H6) and into the MV genome. Point mutations in H ablating the natural receptor tropism are indicated. Viral transcription units are displayed as boxes. Specific restriction sites used for cloning are depicted in italics. (b) Analysis of rescued MV by immunoblot analysis of infected Vero-αHis cell lysates. Proteins separated on 10% SDS-gels (EpCAM-MV: 7.5%) were detected by antibodies directed against the indicated viral or cellular proteins. Uninfected cell lysate: mock. (c) Specificity of retargeted MV. Transgenic CHO cells stably expressing SLAM, CD46, nectin-4, HER2/neu, EGFR, or EpCAM (as indicated) were infected with DARPin-retargeted viruses (DARPins with highest affinities for HER2/neu, EGFR, or EpCAM, as indicated; MOI = 0.3) and analyzed by fluorescence microscopy 72 hours after infection. Parental CHO-K1 cells naturally lacking any of the designated receptors served as controls. Scale bar: 400 µm. DARPin, designed ankyrin repeat protein; MOI, multiplicity of infection; MV, measles virus; SLAM, signaling lymphocyte activation molecule.
To confirm the specific receptor usage of the retargeted DARPin-MV, we used a transgenic CHO cell panel expressing either the natural MV receptors, or the targeted receptors. Each of these cell lines were infected with one DARPin-MV specific for each receptor (i.e., DARPins with the highest respective affinity), an unspecific DARPin that is binding the maltose-binding protein, or with the nontargeted, parental MVNSe. The infected monolayers were analyzed 72 hours after infection (Figure 1c). All retargeted DARPin-MV had lost their tropism for CD46 and SLAM as well as for nectin-4 expressing cells. In contrast, transgenic CHO cells expressing the targeted receptors were not infected by the nontargeted parental virus, but selectively and efficiently by the respective DARPin-targeted MV. These data confirm target specificity of DARPin-retargeted MVs.
Furthermore, using panels of stably transfected CHO clones expressing different amounts of HER2/neu, EGFR, or EpCAM (Supplementary Table S1), and the whole panel of targeted MV displaying DARPins differing in their respective receptor affinities (Table 1), we analyzed cell–cell fusion after infection as read-out for viral cytotoxicity. We observed that the size of the syncytia formed correlated directly to the receptor density on target cells as well as to the affinity of the DARPins for the target receptors (Figure 2). Thus, the DARPin affinities can be used to regulate the cytotoxicity of DARPin-MV. These data confirm target specificity and potential for tailoring the cytotoxicity of DARPin-retargeted MVs.
Figure 2.

Cell fusion activity depends on receptor affinity of DARPins and receptor density. (a–c) Viral spread and virus-induced cell–cell fusion activity in transgenic CHO cells expressing different amounts of (a) HER2/neu, (b) EGFR, or (c) EpCAM infected with matching retargeted DARPin-MV displaying DARPins with different receptor affinities as targeting domains. Subconfluent cells were infected (MOI = 0.3) and analyzed by fluorescence microscopy 48 hours after infection. Viruses were arranged from left to right with increasing receptor affinity, cell lines from top to bottom with increasing receptor density. Scale bar: 400 µm. DARPin, designed ankyrin repeat protein; MOI, multiplicity of infection; MV, measles virus.
Infection of human cancer cell lines by DARPin-MV and selective spread
We next addressed whether the DARPin-MV effectively infect human carcinoma cell lines. Many carcinoma cell lines reveal upregulation of EGFR, HER2/neu, or EpCAM. We determined the receptors' densities, including CD46 (used by nontargeted MV) on a set of different human cancer cell lines by quantitative flow cytometry analysis (Table 2) and used these cell lines subsequently. Infection of these cancer cells with the matching retargeted DARPin-MV caused intercellular fusion and revealed again the correlation between the surface densities of the receptors and the extent of virus-caused intercellular fusion, depending as well on the binding affinity of the respective DARPins (Figure 3a–c). For example, MV-G3 infection in “low” HER2/neu expressing MCF-7 cells (1.2 × 104/cell) showed only weak virus infection, whereas high HER2/neu expressing SK-OV-3 cells (5.7 × 105/cell) formed large syncytia. Most interestingly, the HER2/neu-targeted MV-G3 virus seemed to infect certain cell lines (BT-474, SK-BR-3) better and to induce a higher cytopathic effect than the nontargeted MVNSe, which represented the standard for our experiments, because a closely related MV is currently tested in clinical trials.3 Replication and spread of targeted MV with high-affinity DARPins were not affected in matching target cells in comparison with nontargeted MVNSe (data not shown).
Table 2. Quantification of cell surface molecules.
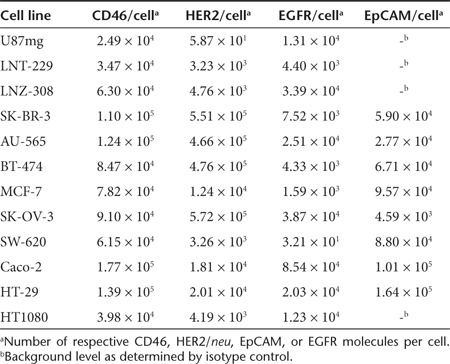
Figure 3.
Infection and spread in receptor-positive human cancer cell lines. (a–c) Different human cancer cell lines expressing target receptors with different densities on their surface were infected with matching retargeted MV or nontargeted, parental MVNSe (MOI = 0.1) and analyzed 48 hours after infection by fluorescence microscopy. Retargeted viruses were arranged from left to right with increasing receptor affinity. Original magnification: 50×. (a) HER2/neu-positive breast (MCF-7, SK-BR-3, BT-474, AU-565) or ovarian carcinoma (SK-OV-3) cells were infected with HER2/neu-targeted DARPin-MV, or MVNSe. (b) EGFR-positive ovarian carcinoma (SK-OV-3), glioblastoma (LNT-229, LNZ-308), or fibrosacroma (HT1080) cells were infected with EGFR-targeted DARPin-MV or MVNSe. (c) EpCAM-positive ovarian (SK-OV-3) or colon (SW-620, HT-29, Caco-2) carcinoma cells were infected with EpCAM-targeted DARPin-MV or MVNSe. (d) Infection and spread of HER2/neu-targeted MV-G3 (top panel) and nontargeted MVNSe (bottom panel) in near confluent 1:1 mixed cultures of U87mg-Katushka (red fluorescent, HER2/neu-negative) and SK-OV-3 (nonfluorescent, HER2/neu-positive) cells. Cells were infected (MOI = 0.1) and cultured for 15 days. Cultures were analyzed by fluorescence microscopy at indicated time points after infection. Yellow fluorescence indicates infection of red-fluorescent U87mg-Katushka cells with GFP-encoding MV. Scale bars: 400 µm. DARPin, designed ankyrin repeat protein; MOI, multiplicity of infection; MV, measles virus.
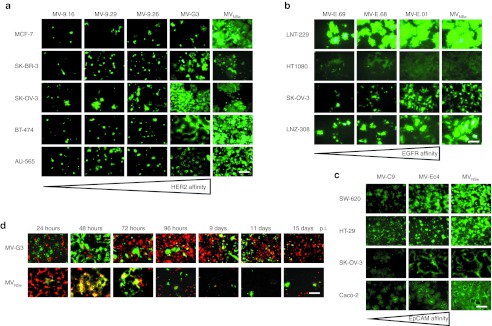
To further determine the stability and the selectivity of the targeted MV-G3 virus, we infected a coculture of HER2/neu-negative (an U87mg clone stably marked by the Katushka protein (data not shown)) and -positive (SK-OV-3) cancer cell lines. In fact, nontargeted MVNSe showed an indiscriminate infection evidenced by yellow fluorescence due to the red fluorescence protein in U87mg-Katushka cells and the eGFP encoded by the virus (Figure 3d, lower panel). MV-G3 almost exclusively spread in the nonfluorescent HER2/neu-positive target cells as revealed by exclusively green fluorescent syncytia in this mixed culture of red U87mg-Katushka and nonfluorescent SK-OV-3 cells (Figure 3d, upper panel). Furthermore, MVNSe wiped out nearly all cells in the mixed culture, whereas MV-G3 killed just the nonfluorescing SK-OV-3 target cells and spared the red-fluorescing, nontarget U87mg-Katushka cells, as expected. Most interestingly, even over a cultivation period of more than 2 weeks with ongoing viral replication in isolated SK-OV-3 nests, the target specificity of MV-G3 remained, indicating the stunning stability of the DARPin-targeting strategy for oncolytic MV (Figure 3d, upper panel). Only single yellow cells became apparent during the whole time of cultivation (starting 72 hours after infection) that suggest rare, unspecific off-target background infection events by an otherwise specific virus, which is not able to further spread among nontarget cells. In summary, the retargeted DARPin-MV revealed the same patterns of selectivity and spreading on human carcinoma cell lines as in receptor-transgenic cell panels, accompanied by extraordinary stability of the targeted virus' specificity.
DARPin-MV reveal potent cytolytic efficacy
Next, we aimed to determine the cytolytic potential of the retargeted MV. For the HER2/neu-targeted viruses, we compared the DARPin-MV side-by-side with previously published scFv-targeted MV directed against the same target.14 SK-OV-3 (Figure 4a) or SK-BR-3 (Supplementary Figure S3) cells were infected with each virus and the cytotoxic effect was assessed after 72 hours by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay. Our results indicate an enhanced oncolytic efficacy of the DARPin-MV compared with the scFv-MV. Direct comparison of MV-G3 and MV-MH3B1, with a comparable receptor affinity of approximately 0.09 and 0.12 nmol/l, respectively, revealed a significantly higher cytolytic activity for the DARPin-MV (42 versus 75% residual viability after infection with MV-G3 or MV-MH3B1, respectively) (Figure 4a). To identify discrepancies between scFv- and DARPin-targeted viruses potentially causative for the superior cytotoxicity of DARPin-MV, we determined the incorporation of DARPin-H and scFv-H into purified viral particles that affects the avidity of the viruses (Figure 4f). The immunoblot data reveal better incorporation of DARPin-H into viral particles and indicate a higher stability of DARPin-H than scFv-H proteins. The latter seem to be cleaved in a considerable fraction by an unknown mechanism when presented on viral particles, thereby presumably losing their C-terminal targeting domain. Both mechanisms would result in higher avidity of DARPin-MV in comparison with the scFv-targeted MV and thus could explain enhanced efficacy of DARPin-MV. Interestingly, MV-G3 was about as cytotoxic as nontargeted MVNSe control virus (42 versus 50%) (Figure 4a).
Figure 4.
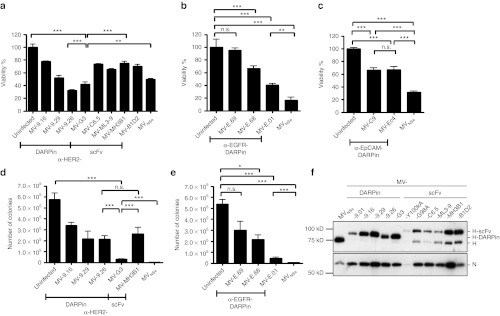
Oncolytic potency of DARPin-MV in vitro. (a–e) Oncolytic potency of DARPin-targeted MV was assessed in comparison with nontargeted MVNSe and scFv-targeted MV via measuring cytotoxicity in SK-OV-3 cells by (a–c) determining metabolic activity (MTT assay) or (d,e) colony forming capacity of infected cultures. (a–c) SK-OV-3 cells were infected (MOI = 1) with nontargeted MVNSe, or viruses targeted against (a) HER2/neu, (b) EGFR, or (c) EpCAM, and the cytolytic effect was assessed 72 hours after infection. Metabolic activity of uninfected cultures was set to 100%. (d,e) SK-OV-3 cells were infected (MOI = 0.1) with nontargeted MVNSe or viruses targeted against (d) HER2/neu, or (e) EGFR, and colony forming capacity of infected cultures was assessed 72 hours after infection. Colony numbers were counted after further 11 days. Mean ± SD. 1 representative out of 3 experiments. Bonferroni test: *P < 0.05, **P < 0.01 and ***P < 0.001, n.s. = P > 0.05. (f) Analysis of purified virus particles by immunoblot analysis of supernatant of infected Vero-αHis cells. Proteins separated on 10% SDS-gel were detected by antibodies directed against the indicated viral proteins. DARPin, designed ankyrin repeat protein; MOI, multiplicity of infection; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide; MV, measles virus; n.s., not significant; scFv, single-chain variable fragment.
A similar performance was also observed for the MV variant retargeted against EGFR using the DARPin with highest EGFR affinity (MV-E.01) (Figure 4b), but the oncolytic efficacy of the MV-E.01 seemed somewhat impaired as compared with the nontargeted MVNSe on EGFR-expressing SK-OV-3 cells (Table 2) (40 versus 17% residual viability after infection with MV-E.01 or MVNSe, respectively) (Figure 4b). For EpCAM-targeted viruses, both MV-C9 and MV-Ec4 revealed significant oncolytic activity, but diminished oncolysis on SK-OV-3 cells (67 versus 31% viability with MV-C9/-Ec4 or MVNSe, respectively) (Figure 4c). To confirm these results, we further analyzed viral cytotoxicity by colony formation assay. Altogether, colony formation assay results were consistent with the MTT assays. Again, the DARPin-MV with the highest affinity for the respective receptor (MV-G3, MV-E.01) showed high cytotoxicity which is comparable with the nontargeted, parental MVNSe (Figure 4d,e). Just 3.1 × 104 versus 1.2 × 103 colonies could be formed after infection with either MV-G3 or MVNSe, and 4.3 × 104 versus 4.8 × 103 after infection with MV-E.01 or MVNSe, respectively, whereas the mock controls were at 5.8 × 105 or 5.4 × 105 colonies. This corresponds to a killing efficacy in excess of 90% for all three viruses. Both assays underline the excellent cytotoxicity for tumor cells and thereby suggest a high oncolytic potential of DARPin-targeted MVs, especially HER2/neu-targeted MV-G3, which we subsequently chose for detailed efficacy studies in vivo.
Potent oncolytic efficacy of DARPin-MV and robust attenuation in vivo
To evaluate the oncolytic potency of the DARPin-targeted MV in vivo, we analyzed the two most cytotoxic HER2/neu-targeted MV (MV-9.26, MV-G3) in direct comparison with the scFv-targeted MV-MH3B1 and the nontargeted MVNSe control virus. SCID mice harboring HER2/neu-positive SK-OV-3 xenograft tumors subcutaneously in their left flanks were treated by intratumoral injections of the respective viruses or controls, when the tumors had reached a defined volume (~35 mm3) (Figure 5a). The tumor volumes of the control groups with or without UV-inactivated MV-9.26 revealed exponential tumor growth, and all mice had to be killed within the first 50 days after tumor implantation (Figure 5b, I and II). Nearly all tumors of mice which were treated with either DARPin-MV or nontargeted MV underwent complete remission (Figure 5b, IV–VI), in contrast to the animals treated with scFv-targeted MV-MH3B1, which experienced only partial remission of the injected tumors (Figure 5b, III).
Figure 5.

Efficacy and safety of HER2/neu-targeted MV in vivo. (a–c) Analysis of oncolytic efficacy in a HER2/neu / EGFR / EpCAM triple-positive, subcutaneous (s.c.) tumor model in immunodeficient mice (a) Schematic depiction of treatment schedule in a human xenograft tumor model in SCID mice implanted subcutaneously with SK-OV-3 cells. Ten days thereafter, mice were injected i.t. on 5 consecutive days with viruses (1 × 106 TCID50/injection) or controls. n = 9–12. Tumor volume was monitored and animals were killed when reaching predefined end points. (b) Growth of tumors. Each line represents tumor burden of one animal. Individual animals with no tumor relapse are indicated by red arrows. (c) Tumor progression as defined by tumor growth of >5 mm3 after reaching minimal volumes upon treatment. Kaplan-Meyer plots for time to progression (TTP). (d) Survival of tumor-bearing animals. Kaplan-Meyer survival plots of treated animals. (e,f) Analysis of virus safety in neurotoxicity assay (e) Kaplan-Meyer survival plot of IFNARtm-CD46Ge mice inoculated i.c. with retargeted MV-G3 (n = 9) or MV-MH3B1 (n = 9), nontargeted MVNSe (n = 10), or medium (mock) (n = 7), monitored for appearance of neurologic symptoms and killed reaching predefined endpoints or 30 days after infection for histologic analysis. Logrank test, sequentially tested: *P < 0.05, **P < 0.01 and ***P < 0.001. (f) Paraffin-sections of representative brains from mice inoculated i.c. were HE-stained for standard histology (I, II, VII, VIII). Consecutive tissue sections were stained for MV-N (III, IV, IX, X), or counterstained with hematoxylin (blue) revealing infiltration of inflammatory cells in MV-infected areas (V, VI, XI, XII). Scale bars: 100 µm (top panel) or 20 µm (bottom panel). Dpi, days post infection; i.c., intracranial; i.t., intratumoral; MV, measles virus; n.s., not significant.
The lack of full remission in MV-MH3B1 treated animals was reflected by a median time to progression (as defined of tumor growth exceeding 5 mm3 after reaching minimal volume upon treatment) of 38 days (Figure 5c) and a reduced prolongation of median survival time to 71 days after tumor implantation, which indicates a significantly weaker efficacy as compared with DARPin-MV (Figure 5c,d and Supplementary Table S2). Mice treated by MV-G3 or nontargeted MVNSe experienced a doubling of median survival time in comparison with the control mice (Supplementary Table S2). Remarkably, oncolytic efficacy and survival in DARPin-MV treated animals were comparable with mice treated with the nontargeted virus MVNSe. In contrast to the controls and the scFv-targeted MV-MH3B1 treated group, we observed 1–2 long-term (>160 days) survivors per group indicating potential cure of these animals (Figure 5d, Supplementary Table S2). Comparing the in vivo efficacy of MV-G3 and MVNSe starting with a mean tumor volume of approximately 85 mm3 demonstrated median survival times of 60 and 65 days, respectively (data not shown). These data underline that MV-G3 is comparably effective as the nontargeted MVNSe. In summary, these results indicate that the DARPin-MV possesses higher anti-tumoral efficacy in vivo than the appropriate scFv-targeted control MV and are comparably potent as the nontargeted control MVNSe in moderate (~35 mm3) and large (85 mm3) xenograft tumors in this experimental setting.
We next aimed to analyze whether the retargeting of oncolytic MV using DARPins resulted in further attenuation of the oncolytic agent's off-target toxicity, as intended. Pathogenic MV has neurotoxic potential manifesting in acute or delayed MV-induced encephalitis.26 This neurotoxicity is usually not found in vaccine strains and oncolytic strains derived thereof,27 but toxicity can re-emerge in heavily immunosuppressed patients.28 However, in Ifnartm-CD46Ge mice, which express the human MV receptor CD46 and miss the type I interferon receptor, lethal encephalitis is caused by intracranial injection of attenuated MV in adult mice,29 thus creating a very sensitive in vivo model for the unspecific toxicity and virulence of MV. We generated targeted and parental MV without the reporter gene GFP, because its additional expression at the start of the viral genome attenuates MV virulence.30
These viruses were used to inject Ifnartm-CD46Ge mice intracranially, and mice were subsequently monitored for neurologic symptoms and death. All but one mouse injected with nontargeted MVNSe had to be killed or died 3–10 days after infection, as expected (Figure 5e). These animals showed clinical signs of neurological disease (e.g., tremor, convulsion, paresis, paralysis). Neither the mock-injected mice nor the mice injected with the DARPin- or scFv-targeted MV had to be killed or showed any signs of neurotoxicity (Figure 5e). To evaluate the neurotoxicity by histopathology, we collected brain tissues of injected mice at time of death or 30 days after infection. Fixed mouse brains were stained for histological features and MV infection. Brain tissue of mice injected with MVNSe showed spatially restricted inflammation and infiltration of inflammatory cells (like lymphocytes and granulocytes) within the white matter of the cerebrum (Figure 5f, I and II). In addition, we documented MV infection by viral nucleoprotein staining in consecutive sections (Figure 5f, III and IV) colocalizing with cellular infiltration (Figure 5f, V and VI). In contrast, the brain tissue of mice injected with DARPin- or scFv-targeted MV showed no pathologic alterations and absence of MV-specific staining (Figure 5f, VII–XII) similar to the mock-injected control group (data not shown). This very sensitive in vivo model indicates that the retargeted MVs are strongly attenuated in nontarget tissue and analysis of neurotoxicity of retargeted MV in vivo confirmed their enhanced attenuation despite conserved oncolytic potency.
Bispecific DARPin-MV for counteracting resistance development
To assess the risk of resistance development of tumor cells treated with monotargeted MV, we infected SK-OV-3 cells with monospecific MV-G3, and cultivated the infected cells in parallel to untreated cells. Both cultures were split when reaching confluence and assessed every 4 weeks for expression of the targeted receptor HER2/neu and CD46 as nontargeted control. Among SK-OV-3 cells inoculated with HER2/neu-targeted MV-G3, we observed a mixed culture phenotype with infected (GFP-positive) and noninfected (GFP-negative) cells (data not shown). CD46 and HER2/neu expression were found to remain stable in uninfected cultures, and CD46 expression in infected SK-OV-3 cells revealed only minimal and variable downregulation (Figure 6a). In contrast, HER2/neu expression continuously declined over time in SK-OV-3 cells incubated with MV-G3, with an approximately 8-fold reduced HER2/neu density after 20 weeks of infection (Figure 6a). These data clearly indicate the risk of resistance development for monotargeted OVs as also observed for other biomedical therapies such as adoptive T cell transfer.31
Figure 6.

MV simultaneously retargeted against two tumor-antigens to avoid resistance development. (a) Selection of low-level target receptor expressing cells after infection with monospecific MV. Kinetic flow cytometry analysis of SK-OV-3 cells infected with MV-G3 (MOI = 1, bottom panel) or left uninfected (top panel). Diagrams depict HER2/neu- (right column) or CD46-expression (left column) over several weeks of culture, as color coded. Black, unstained cells; gray, isotype controls. (b) Schematic depiction of the bispecific H. Point mutations in H ablating the natural receptor tropism are indicated, as well as DARPins, linker ([(G4S)2]), and His-tag (H6). Specific restriction sites are depicted in italics. (c) Immunoblot analysis of the rescued MV protein expression, including bispecific MV-Ec4-G3. Detected viral or cellular proteins are indicated. Uninfected lysate: mock. (d) Specificity of recombinant MV receptor usage. Transgenic CHO cells stably expressing SLAM, nectin-4, CD46, HER2/neu, or EpCAM (as indicated) were infected with DARPin-retargeted viruses (MOI = 0.1) and analyzed by fluorescence microscopy 72 hours after infection. Parental CHO-K1 cells served as controls. Scale bar: 400 µm. (e) Oncolytic potency of bispecific MV-Ec4-G3 was assessed in SK-OV-3 cells by MTT assay. Cells were infected (MOI = 1), and metabolic activity of surviving cells was assessed 72 hours after infection. Metabolic activity of uninfected cultures: 100% Logrank test, sequentially tested: ***P < 0.001, n.s. = P > 0.05. DARPin, designed ankyrin repeat protein; MOI, multiplicity of infection; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide; MV, measles virus; n.s., not significant; p.i., post infection; SLAM, signaling lymphocyte activation molecule.
To avoid this potential tumor escape, we attempted to target MV simultaneously at two different essential tumor epitopes. In line with our approach, recently published data indeed suggest that targeting of two different tumor markers can limit antigen loss even in recurrent tumors.32 We chose HER2/neu and EpCAM as designated receptors, two markers of aggressive breast cancers. Postulating that the DARPins' versatility, small size and lateral location of the binding surface might allow generation of bispecific MVs, we used the two DARPins of highest affinity (G3 and Ec4), fused them genetically to each other by a glycin-serin-linker [(G4S)2] encoding sequence, and subsequently to receptor-blind MV-H (Figure 6b). We could show receptor-specific fusion helper function of the DARPin-linker-DARPin-H attachment protein in cells expressing either target receptor (Supplementary Figure S4) and inserted the ORF encoding these bispecific attachment proteins into the MV genome which was used to rescue recombinant MV, resulting in the putative bispecific recombinant virus MV-Ec4-G3 (Figure 6c–e). This virus containing the 2-DARPin Ec4-G3-targeting domain was genetically stable over at least 3 passages, and replicated efficiently on Vero-αHis cells. Expression of the Ec4-G3-H attachment protein by MV-Ec4-G3 was demonstrated by immunoblot analysis of virus-infected cells, revealing expression of a recombinant H with an increased apparent molecular weight of approximately 100 kDa, as expected (Figure 6c).
To assess receptor usage of the bispecific MV-Ec4-G3, we took advantage of the transgenic receptor CHO cell panel. Each cell line was incubated with the HER2/neu- and EpCAM-directed bispecific MV-Ec4-G3, monospecific MV-G3 and MV-Ec4, or with the nontargeted MVNSe (Figure 6d). MV-Ec4-G3 had lost its tropism for SLAM, nectin-4, and CD46, but infected the cells via both targeted receptors. In contrast, the nontargeted, parental virus could not infect the transgenic CHO cells expressing any of the targeted receptors. The cytolytic potential of this bispecific MV was assessed in SK-OV-3 cells expressing both designated receptors cells (Table 2) by MTT assay. The cytolytic activity of the bispecific virus was at least as high as of both monotargeted virus, and the nontargeted MV strain (Figure 6e) (27 versus 25 versus 24% residual viability after infection with MV-Ec4-G3, MV-G3, or MVNSe, respectively). In conclusion, our data demonstrate for the first time specificity and efficacy of a fully retargeted, bispecific OV.
Discussion
Our analysis reveals that targeting of oncolytic measles virus can be critically enhanced by the use of designed ankyrin repeat proteins as targeting domains. Most important, DARPins allow for the first time selective bispecific targeting of a replicating OV to critical tumor markers, thereby offering the potential to suppress resistance of tumor cells against targeted virotherapy during cell entry. In addition, DARPin-targeting of MV is versatile and effective, revealing conservation of curative potential in vivo accompanied by enhanced safety.
Targeting strategies give the chance to personalize or stratify cancer treatment. Among OV developed and tested clinically for treating cancer,3 MV offers a special platform. MV is up to now the viral system, which can be most easily genetically retargeted to cells expressing surface receptors rationally selected as targets to further enhance tumor-specificity.33
Here, we present retargeted MV using DARPins as targeting domain of a replicating MV in analogy to their use for nonreplicating vectors.24,34 We demonstrate that DARPin-targeted MVs are very selective in receptor recognition in vitro and more cytolytic than previously published scFv-MV in vitro and in vivo.14 Furthermore, DARPin-MVs are comparably efficacious as the nontargeted virus, which is closely related to a clinically evaluated MV (phase I),35 but retargeted MVs are significantly more attenuated, thus revealing a favorable safety profile. Although safety was demonstrated in current clinical trials,3,4,35 based on a vaccine strain-lineage MV further attenuated by defective interactions with innate immunity proteins,36 future clinical trials may be based on less attenuated MV armed with effector proteins. Enhanced targeting specificity may be necessary to guarantee safety of these viruses while sustaining their improved anti-tumor efficacy. Although there is no reason to believe that the virus loose target selectivity in vivo, analysis of in vivo targeting and biodistribution of DARPin-targeted viruses will be in the focus of future experiments. Our data thus support DARPins to be at least as suitable targeting domains as scFv for MV. This could be due to several parameters: DARPins are very stable, their folding is robust even in fusion proteins, and they reveal a very low tendency to aggregate. As they consist of only one domain, domain-swapping as in scFv19 will not occur even in high local densities as on the virus envelope. Due to their smaller size than scFv their access to membrane-proximal or otherwise poorly reachable epitopes may be feasible. Altogether, DARPins are robust and versatile targeting domains which perfectly match replicating MV, especially because DARPins allow generation of bispecific and probably multispecific DARPin-MV, as multispecific DARPins have been demonstrated in a variety of different systems and formats.22,23,25 Indeed, we found evidence for more efficient incorporation and less cleavage of DARPin-H in MV particles compared with scFv-H, resulting in higher avidity of DARPin-MV. Thus, higher oncolytic efficacy can be correlated to structural advantages of DARPins when displayed on MV particles as H fusions.
We chose HER2/neu, EGFR, or EpCAM as receptors, three markers of aggressive tumors. The HER2/neu and EGFR signaling pathways are dominant drivers in many cancers,37 whereas the third target receptor, EpCAM, is widely expressed on carcinomas,38 but also discussed as frequent marker of cancer stem cells.39 Thus, by aiming at the tumor's Achilles heels, be it critical driver mutations or stem cells, targeted agents will represent the future of personalized cancer therapy.
Nevertheless, this class of monotargeted therapies highlights also current limitations, notably tumor resistance a priori or during treatment, e.g., trastuzumab resistance2 or tumor escape under adoptive T cell therapy.31 As a proof of concept, we aimed for active agents that simultaneously target two different critical tumor markers. Thereby, fast resistance due to mutation, ablation, or inconsistent expression of a single targeted surface antigen will be impaired as just demonstrated for adoptive T cell transfer.32 Thus, MV targeting HER2/neu and EpCAM simultaneously seems to be very attractive: First, it would enhance tumor remission because bispecific MV could still infect cancer cells with ablation or downregulation of just one of the targeted receptors. Second, EpCAM as a potential marker of cancer stem cells could enhance the oncolytic efficacy by even killing the tumor initiating cells. Third, targeting one MV simultaneously at two markers may be advantageous over administering two distinct MV, because issues of e.g., bioavailability and -distribution, toxicity, processing, and licensing have to be cleared for just one active substance. Finally, the DARPin technology may even allow easily extending specificity to tri- or tetravalence by linking additional DARPins in the same way.
For further studies it will be important to consider, which tumor markers should be combined for targeting also other OV, e.g., vesicular stomatitis virus.40 Coupling of the DARPins and their spatial arrangements are parameters to be optimized. Moreover, it is difficult to judge the impact of pre-existing or induced anti-MV immunity on therapeutic efficacy using current models.
In summary, this study provides proof of concept that retargeting of OV is feasible with DARPins, that DARPin-targeting works efficiently for different receptors, and that it is even possible to target at least two different receptors with one virus. Our achievements underline the clinical potency of the DARPin-targeting concept as a strategy to generate safer OVs for the treatment of e.g., breast and ovarian cancers and the opportunity to overcome resistance development against targeted therapies.
Materials and Methods
Cells. AU-565 (CRL-2351), BT-474 (HTB-20), Caco-2 (HTB-37), CHO-K1 (CCL-61), 293T (CRL-11268), HT1080 (CLL-121), HT-29 (HTB-38), MCF-7 (HTB-22), SK-BR-3 (HTB-30), SK-OV-3 (HTB-77), SW-620 (CCL-227), and U87mg (HTB-14) cells were purchased from ATCC (Manassas, VA) and grown in recommended media at 37 °C in a humidified atmosphere containing 6% CO2 for not longer than 6 months of culture after thawing of the original stock. CHO-SLAM, CHO-CD46, Vero-αHis, CHO-HER2, CHO-nectin-4, LNT-229, and LNZ-308 cells and their maintenance have been described.7,13,34,41,42 CHO-EpCAM and CHO-EGFR cell clones were established by transfection of pcDNA3.1-EpCAM or pcDNA3.1-EGFR, respectively, into CHO-K1 and selected as described.34 U87mg-Katushka cells were stably transduced by lentiviral vectors pseudotyped with VSV-G encoding the Katushka protein (pS-TFP635-W) and single cell clones were selected.
Plasmids. The cDNAs encoding DARPins binding to HER2/neu (9_16, 9_26, 9_29, G3),43 EGFR (E_01, E_68, E_69),43 or EpCAM (Ec4, C9)22,44 were amplified by PCR with flanking SfiI/NotI sites. Fully sequenced DARPin genes were inserted into pCG-Hmut-αCD20-6His45 via SfiI/NotI to yield the expression plasmids for retargeted H (pCG-Hmut-9.16, pCG-Hmut-9.29, pCG-Hmut-9.26, pCG-Hmut-G3, pCG-Hmut-E.69, pCG-Hmut-E.68, pCG-Hmut-E.01, pCG-Hmut-C9, and pCG-Hmut-Ec4). All H-DARPin-encoding genes were transferred via PacI/SpeI into plasmid p(+)PolII-MVNSe-GFP(N), to yield p(+)PolII-MVNSe-GFP(N)-9.16, p(+)PolII-MVNSe-GFP(N)-9.29, p(+)PolII-MVNSe-GFP(N)-9.26, p(+)PolII-MVNSe-GFP(N)-G3, p(+)PolII-MVNSe-GFP(N)-E.69, p(+)PolII-MVNSe-GFP(N)-E.68, p(+)PolII-MVNSe-GFP(N)-E.01, p(+)PolII-MVNSe-GFP(N)-C9, and p(+)PolII-MVNSe-GFP(N)-Ec4.
To generate the bispecific DARPin-targeting domains, the sequences encoding DARPins Ec4 or G3 were amplified by PCR with flanking SfiI/BstBI or BstBI/NotI sites, respectively. The reverse primer for DARPin Ec4 additionally encoded a (G4S)2 linker. PCR fragments were ligated into pCR2.1-TOPO (Invitrogen, Carlsbad, CA) and sequenced. The DARPin sequences were combined using the BstBI restriction site to yield pCR2.1-Ec4-[(G4S)2]-G3. The combined DARPin-linker-DARPin fragment was then inserted into pCG-Hmut-αCD20-6His and further handled as described above. Detailed description of primers, templates, and PCR are available upon request.
Viruses. For rescue of recombinant MV the PolII rescue system46 was used with modifications. In short, the plasmids encoding recombinant MVs' genomes were cotransfected with expression plasmids pCA-MV-N, pCA-MV-P, and pCA-MV-L into 293T cells using Lipofectamine 2000 (Invitrogen). Two days after transfection, transfected 293T cells were overlaid onto 50% confluent Vero-αHis cells13 allowing rescue of retargeted viruses. The viruses were propagated in Vero-αHis cells and titers were determined by 50% tissue culture infective dose (TCID50) titration on Vero-αHis cells as described.47
Immunoblotting. Vero-αHis cells were infected with recombinant MV at an multiplicity of infection (MOI) of 0.1 and cells were lysed 48 hours after infection using RIPA lysis-buffer and processed as described before to detect MV-H, MV-N, or β-actin as standard.48
Infection assays. Cells were seeded in six-well tissue culture plates (Nunc, Wiesbaden, Germany), infected with recombinant MV at an MOI of 0.1, 0.3, or 1.0, and subsequently cultured at 37 °C. Syncytia formation was analyzed 48–72 hours after infection by fluorescence microscopy (Axiovert 25 200M; Zeiss, Göttingen, Germany).
Virus particle purification. Vero-αHis cells were seeded in 10 cm dishes and infected with respective MV (MOI = 0.03). The culture supernatants were collected 2 days after infection, clarified and pelleted in an SW28 rotor (25,000 rpm, 2.5 hours) through 20% sucrose onto a 60% sucrose cushion in STE buffer (10 mmol/l Tris (pH 8.0), 100 mmol/l NaCl, and 1 mmol/l EDTA (pH 8.0)). Purified viral particles were pelleted in an SW41 rotor (35,000 rpm, 1.5 hours) through 20% sucrose, resuspended in RIPA lysis buffer, and subjected to immunoblot analysis.
Colony forming assay. SK-OV-3 cells were infected in triplicates with recombinant MV (MOI = 0.1) or left untreated. 72 hours after infection, the surviving cells were trypsinized, serially diluted, replated in six-well plates, and incubated for 11 days to allow colony formation. Cells were then fixed with 10% PFA (w/v) for 4 hours and subsequently stained with crystal violet solution (PBS, 18% ethanol (v/v), 0.1% crystal violet (w/v)). Only colonies that were well separated from each other and contained >50 cells were counted.
MTT assay. SK-OV-3 or SK-BR-3 cells were seeded into 96-well plates (Nunc) and infected with recombinant MV (MOI = 1), or left uninfected (mock). Viability of the cells after infection was determined using MTT (Cell Proliferation Kit I; Roche Diagnostics, Mannheim, Germany). Cells were incubated with the MTT solution for 4 hours and then solubilization solution was added 72 hours after infection. Following overnight incubation, a formazan dye was formed, which was quantified in quadruplicates using an ELISA reader (Multiskan RC; Thermo Labsystems, Dreieich, Germany).
Animal experiments. Experimental mouse work was carried out in compliance with the regulations of the German animal protection law. To evaluate the oncolytic efficacy in vivo, 5 × 106 SK-OV-3 cells were subcutaneously injected into the left flanks of 6- to 8-week-old SCID mice (Charles River, Köln, Germany). Ten days after tumor inoculation (tumor size: 30–50 mm3), mice were randomized into groups. They received intratumoral injections with a dose of 1 × 106 TCID50/injection MV in 100 µl OptiMEM (Invitrogen) on 5 consecutive days. Control animals were injected with 100 µl OptiMEM (mock), or with 100 µl UV-inactivated MV-9.26 (120,000 µJ/cm2 UV light (254 nm), 90 min). Tumor volumes were determined twice a week. Animals were euthanized, when the tumor volume reached 500 mm3, mice lost more than 20% of body weight, or tumor ulceration occurred. To determine off-target toxicity, Ifnartm-CD46Ge mice were analyzed for neurotoxicity after intracranial injection, as described.29 In brief, 5- to 8-week-old animals were injected intracranially with 2 × 104 TCID50 MV in 10 µl. Mice were euthanized when grading of neurological symptoms (e.g., tremor, convulsion, paresis, paralysis) exceeded a predefined threshold.
Histology. Mouse brains were isolated directly after euthanazation, fixed in 4% paraformaldehyde for 48 hours, and embedded in paraffin. The samples were cut into consecutive 2 µm sections using a rotation microtome RM 2255 (Leica, Wetzlar, Germany). Sections were stained with Mayer's hematoxylin and eosin, or by immune staining using a monoclonal antibody against MV nucleoprotein (NB100-1856; Novus Biologicals, Littleton, CO). For that purpose samples were blocked with Avidin Biotin blocking Kit (X0590; Dako, Hamburg, Germany), then incubated with α-MV-N mAb o/n, subsequently incubated with Vectastain ABC Kit (Vector Laboratories, Burlingame, CA) and counterstained with hematoxylin.
Flow cytometry analysis. Flow cytometry was performed on an LSRII-SORP FACS (BD, Heidelberg, Germany) and data were analyzed using the FACSDiva version 6.1.3 or FCS Express version 3. Cells were stained and analyzed as described before49 using the following antibodies: mu α-hu HER2/neu-APC or -PE (Neu 24.7; BD); mu α-hu CD46-AlexaFluor700 (MEM-258; Exbio, Vestec, Czech Republic); mu α-hu CD46-PE (MEM-258; AbDSerotec, Düsseldorf, Germany), mu α-huEpCAM-PE (EBA-1; BD); mu α-hu EGFR-PE (582; Santa Cruz, Heidelberg, Germany). The number of the respective receptors per cell was calculated as described.50 To detect H-DARPin proteins, transfected 293T were analyzed as described.34
Statistical analysis. Data presented in Figures 4 and 6 are presented as mean ± SD and analyzed by t-test; P values were adjusted for multiple comparisons. Pairwise statistical comparison of survival between treatment groups by Logrank test (sequentially tested). For statistical significant results, the following convention was used: n.s. = P > 0.05, *P < 0.05, **P < 0.01, and ***P < 0.001. The statistical analysis was performed with SAS/STAT software, version 9.3, SAS System for Windows (SAS Institute, Cary, NC).
SUPPLEMENTARY MATERIAL Figure S1. Determination of H-DARPin protein surface expression. Chimeric H-DARPin proteins were stably expressed and transported to cell surface. Figure S2. Quantification of virus-induced cell-cell fusion by retargeted DARPin-MV. Figure S3. Cytolytic effect of DARPin-MV in SK-BR-3 cells. Figure S4. Quantification of cell-cell fusion induced by bispecific DARPin-H proteins. Table S1. Surface densities of transgenic CHO cell lines. Table S2. Median survival of animals.
Acknowledgments
We thank U Schneider for providing the PolII Rescue System for measles viruses, Y Yanagi for providing CHO-hSLAM and T Abel for plasmid pS-TFP635-W. We are indebted to A Geyer, M Selbert, and D Kreuz for excellent technical assistance and to R Marschalek for helpful comments. This work has been supported by a grant from the German Cancer Aid (109614) to M.D.M and the LOEWE Center for Cell and Gene Therapy Frankfurt funded by Hessisches Ministerium für Wissenschaft und Kunst (III L 4- 518/17.004 (2010)) to C.J.B. This work was performed in Langen, Germany. A.P. is a shareholder of Molecular Partners A6, who are commercializing the DARPin technology.
Supplementary Material
References
- Heron M, Hoyert DL, Murphy SL, Xu J, Kochanek KD., and, Tejada-Vera B. Deaths: final data for 2006. Natl Vital Stat Rep. 2009;57:1–134. [PubMed] [Google Scholar]
- Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F., and, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- Russell SJ, Peng KW., and, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA.et al. (2010Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer Cancer Res 70875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluming AZ., and, Ziegler JL. Regression of Burkitt's lymphoma in association with measles infection. Lancet. 1971;2:105–106. doi: 10.1016/s0140-6736(71)92086-1. [DOI] [PubMed] [Google Scholar]
- Anderson BD, Nakamura T, Russell SJ., and, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64:4919–4926. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- Tatsuo H, Ono N, Tanaka K., and, Yanagi Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 2000;406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- Mühlebach MD, Mateo M, Sinn PL, Prüfer S, Uhlig KM, Leonard VH.et al. (2011Adherens junction protein nectin-4 is the epithelial receptor for measles virus Nature 480530–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS.et al. (2011Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus PLoS Pathog 7e1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörig RE, Marcil A, Chopra A., and, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C.et al. (1993Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus J Virol 676025–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan K. Oncolytic viruses move forward in clinical trials. J Natl Cancer Inst. 2010;102:590–595. doi: 10.1093/jnci/djq165. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD.et al. (2005Rescue and propagation of fully retargeted oncolytic measles viruses Nat Biotechnol 23209–214. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Hu C, Nakamura T, Marks JD, Russell SJ., and, Peng KW. Affinity thresholds for membrane fusion triggering by viral glycoproteins. J Virol. 2007;81:13149–13157. doi: 10.1128/JVI.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond AL, Plemper RK, Zhang J, Schneider U, Russell SJ., and, Cattaneo R. Single-chain antibody displayed on a recombinant measles virus confers entry through the tumor-associated carcinoembryonic antigen. J Virol. 2001;75:2087–2096. doi: 10.1128/JVI.75.5.2087-2096.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng KW, Donovan KA, Schneider U, Cattaneo R, Lust JA., and, Russell SJ. Oncolytic measles viruses displaying a single-chain antibody against CD38, a myeloma cell marker. Blood. 2003;101:2557–2562. doi: 10.1182/blood-2002-07-2195. [DOI] [PubMed] [Google Scholar]
- Wörn A., and, Plückthun A. Stability engineering of antibody single-chain Fv fragments. J Mol Biol. 2001;305:989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M.et al. (2012Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer Nat Med 18221–223. [DOI] [PubMed] [Google Scholar]
- Todorovska A, Roovers RC, Dolezal O, Kortt AA, Hoogenboom HR., and, Hudson PJ. Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J Immunol Methods. 2001;248:47–66. doi: 10.1016/s0022-1759(00)00342-2. [DOI] [PubMed] [Google Scholar]
- Boersma YL., and, Plückthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol. 2011;22:849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P.et al. (2004High-affinity binders selected from designed ankyrin repeat protein libraries Nat Biotechnol 22575–582. [DOI] [PubMed] [Google Scholar]
- Stefan N, Martin-Killias P, Wyss-Stoeckle S, Honegger A, Zangemeister-Wittke U., and, Plückthun A. DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J Mol Biol. 2011;413:826–843. doi: 10.1016/j.jmb.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Eggel A, Baumann MJ, Amstutz P, Stadler BM., and, Vogel M. DARPins as bispecific receptor antagonists analyzed for immunoglobulin E receptor blockage. J Mol Biol. 2009;393:598–607. doi: 10.1016/j.jmb.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Dreier B, Mikheeva G, Belousova N, Parizek P, Boczek E, Jelesarov I.et al. (2011Her2-specific multivalent adapters confer designed tropism to adenovirus for gene targeting J Mol Biol 405410–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahnd C, Kawe M, Stumpp MT, de Pasquale C, Tamaskovic R, Nagy-Davidescu G.et al. (2010Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size Cancer Res 701595–1605. [DOI] [PubMed] [Google Scholar]
- Bellini WJ, Rota JS, Lowe LE, Katz RS, Dyken PR, Zaki SR.et al. (2005Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized J Infect Dis 1921686–1693. [DOI] [PubMed] [Google Scholar]
- Myers R, Harvey M, Kaufmann TJ, Greiner SM, Krempski JW, Raffel C.et al. (2008Toxicology study of repeat intracerebral administration of a measles virus derivative producing carcinoembryonic antigen in rhesus macaques in support of a phase I/II clinical trial for patients with recurrent gliomas Hum Gene Ther 19690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention Measles pneumonitis following measles-mumps-rubella vaccination of a patient with HIV infection, 1993. MMWR Morb Mortal Wkly Rep. 1996;45:603–606. [PubMed] [Google Scholar]
- Mrkic B, Pavlovic J, Rülicke T, Volpe P, Buchholz CJ, Hourcade D.et al. (1998Measles virus spread and pathogenesis in genetically modified mice J Virol 727420–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Messling V, Milosevic D., and, Cattaneo R. Tropism illuminated: lymphocyte-based pathways blazed by lethal morbillivirus through the host immune system. Proc Natl Acad Sci USA. 2004;101:14216–14221. doi: 10.1073/pnas.0403597101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluza KM, Thompson JM, Kottke TJ, Flynn Gilmer HC, Knutson DL., and, Vile RG. Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer. 2012;131:844–854. doi: 10.1002/ijc.26447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluza KM, Kottke T, Diaz RM, Rommelfanger D, Thompson J., and, Vile R. Adoptive transfer of cytotoxic T lymphocytes targeting two different antigens limits antigen loss and tumor escape. Hum Gene Ther. 2012;23:1054–1064. doi: 10.1089/hum.2012.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo R, Miest T, Shashkova EV., and, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch RC, Mühlebach MD, Schaser T, Kneissl S, Jost C, Plückthun A.et al. (2011DARPins: an efficient targeting domain for lentiviral vectors Mol Ther 19686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msaouel P, Iankov ID, Dispenzieri A., and, Galanis E. Attenuated oncolytic measles virus strains as cancer therapeutics. Curr Pharm Biotechnol. 2012;13:1732–1741. doi: 10.2174/138920112800958896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux P, Hudacek AW, Hodge G, Reyes-Del Valle J, McChesney MB., and, Cattaneo R. A recombinant measles virus unable to antagonize STAT1 function cannot control inflammation and is attenuated in rhesus monkeys. J Virol. 2011;85:348–356. doi: 10.1128/JVI.00802-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y., and, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12:553–563. doi: 10.1038/nrc3309. [DOI] [PubMed] [Google Scholar]
- Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G.et al. (2004Frequent EpCam protein expression in human carcinomas Hum Pathol 35122–128. [DOI] [PubMed] [Google Scholar]
- Gires O, Klein CA., and, Baeuerle PA. On the abundance of EpCAM on cancer stem cells. Nat Rev Cancer. 2009;9:143; author reply 143. doi: 10.1038/nrc2499-c1. [DOI] [PubMed] [Google Scholar]
- Ayala-Breton C, Barber GN, Russell SJ., and, Peng KW. Retargeting vesicular stomatitis virus using measles virus envelope glycoproteins. Hum Gene Ther. 2012;23:484–491. doi: 10.1089/hum.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre-Lafay S, Garrido-Urbani S, Reymond N, Gonçalves A, Dubreuil P., and, Lopez M. Nectin-4, a new serological breast cancer marker, is a substrate for tumor necrosis factor-alpha-converting enzyme (TACE)/ADAM-17. J Biol Chem. 2005;280:19543–19550. doi: 10.1074/jbc.M410943200. [DOI] [PubMed] [Google Scholar]
- Albertoni M, Shaw PH, Nozaki M, Godard S, Tenan M, Hamou MF.et al. (2002Anoxia induces macrophage inhibitory cytokine-1 (MIC-1) in glioblastoma cells independently of p53 and HIF-1 Oncogene 214212–4219. [DOI] [PubMed] [Google Scholar]
- Steiner D, Forrer P., and, Plückthun A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J Mol Biol. 2008;382:1211–1227. doi: 10.1016/j.jmb.2008.07.085. [DOI] [PubMed] [Google Scholar]
- Winkler J, Martin-Killias P, Plückthun A., and, Zangemeister-Wittke U. EpCAM-targeted delivery of nanocomplexed siRNA to tumor cells with designed ankyrin repeat proteins. Mol Cancer Ther. 2009;8:2674–2683. doi: 10.1158/1535-7163.MCT-09-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerechts G, Springfeld C, Frenzke ME, Lampe J, Johnston PB, Parker WB.et al. (2007Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine Cancer Res 6710939–10947. [DOI] [PubMed] [Google Scholar]
- Martin A, Staeheli P., and, Schneider U. RNA polymerase II-controlled expression of antigenomic RNA enhances the rescue efficacies of two different members of the Mononegavirales independently of the site of viral genome replication. J Virol. 2006;80:5708–5715. doi: 10.1128/JVI.02389-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kärber G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch Exp Pathol Pharmakol. 1931;162:480–483. [Google Scholar]
- Mühlebach MD, Schaser T, Zimmermann M, Armeanu S, Hanschmann KM, Cattaneo R.et al. (2010Liver cancer protease activity profiles support therapeutic options with matrix metalloproteinase-activatable oncolytic measles virus Cancer Res 707620–7629. [DOI] [PubMed] [Google Scholar]
- Funke S, Maisner A, Mühlebach MD, Koehl U, Grez M, Cattaneo R.et al. (2008Targeted cell entry of lentiviral vectors Mol Ther 161427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong HT, Timm MM, Greipp PR, Witzig TE, Dispenzieri A, Russell SJ.et al. (2006Oncolytic measles virus targets high CD46 expression on multiple myeloma cells Exp Hematol 34713–720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.