

Abstract
Restriction factors constitute a newly appreciated line of innate immune defense, blocking viral replication inside of infected cells. In contrast to these antiviral proteins, some cellular proteins, such as the CD4, CCR5, and CXCR4 cell surface receptors, facilitate HIV replication. We have used zinc finger nucleases (ZFNs) to insert a cocktail of anti-HIV restriction factors into the CCR5 locus in a T-cell reporter line, knocking out the CCR5 gene in the process. Mirroring the logic of highly active antiretroviral therapy, this strategy provides multiple parallel blocks to infection, dramatically limiting pathways for viral escape, without relying on random integration of transgenes into the genome. Because of the combination of blocks that this strategy creates, our modified T-cell lines are robustly resistant to both CCR5-tropic (R5-tropic) and CXCR4-tropic (X4-tropic) HIV-1. While zinc finger nuclease–mediated CCR5 disruption alone, which mimics the strategy being used in clinical trials, confers 16-fold protection against R5-tropic HIV, it has no effect against X4-tropic virus. Rhesus TRIM5α, chimeric human-rhesus TRIM5α, APOBEC3G D128K, or Rev M10 alone targeted to CCR5 confers significantly improved resistance to infection by both variants compared with CCR5 disruption alone. The combination of three factors targeted to CCR5 blocks infection at multiple stages, providing virtually complete protection against infection by R5-tropic and X4-tropic HIV.
Introduction
One of the major obstacles to treating HIV infection is the virus's ability to mutate and evade therapy.1 This has led to a broad interest in developing alternative treatment strategies to disrupt the host-virus interaction, including cell-based gene therapy approaches to restrict infection.2,3,4,5,6,7 Cellular entry of HIV is mediated through binding to the CD4 receptor and either the CCR5 (CCR5-tropic virus) or CXCR4 (CXCR4-tropic virus) coreceptor on the surface of CD4+ T-cells, the primary target cells in vivo. In patients, early infection is typically established by CCR5-tropic (R5-tropic) virus, while CXCR4-tropic (X4-tropic) or dual-tropic variants predominate in late stage disease.8 Interestingly, individuals who are homozygous for the truncated δ32 variant of the CCR5 gene are resistant to HIV infection and are otherwise healthy,9 making CCR5 an intriguing target for HIV therapy. This has been done both by the small molecule approaches to inhibit binding of HIV to the CCR5 receptor10 and by genetic manipulation to create HIV resistant cells that do not express CCR5 on the cell surface.11,12 Moreover, the demonstration of an apparent cure of a patient infected by HIV by allogeneic bone marrow transplantation from a matched CCR5 δ32 donor was recently reported.13,14 Although it is not known whether it was the donor cells alone or a combination of ablative therapy and transplantation with HIV resistant cells that led to the apparent cure, it strongly supports the idea that using genetically modified cells is a promising approach for altering the course of HIV infection.
Specific genome modification can be achieved with engineered proteins called zinc finger nucleases (ZFNs).15 ZFNs are composed of a zinc finger DNA binding domain fused to a FokI endonuclease domain. Each zinc finger recognizes and binds to a three-nucleotide sequence, such that a four-fingered protein recognizes 12 base pairs. Antiparallel binding of two ZFNs to contiguous sites separated by a short DNA spacer leads to dimerization of the endonuclease domain and creation of a site-specific DNA double-strand break which can be repaired either by potentially mutagenic nonhomologous end joining (NHEJ) or high-fidelity homologous recombination with a homologous DNA donor template. ZFNs have been developed that target the CCR5 gene, and upon induction of a site-specific double-strand break and mutagenic repair by NHEJ, populations of HIV resistant T-cells11 and hematopoietic stem cells (HSCs)12 have been created which phenotypically mimic CCR5 δ32 cells. The potential limitation of this approach is that, in patients infected with both X4- and R5-tropic virus, mutating CCR5 in a fraction of T-cells or HSCs may not be sufficient to alter the course of the disease. Instead, cells that are genetically resistant to both coreceptor tropisms of HIV need to be created.
One way to generate cells that are resistant to both R5-tropic and X4-tropic HIV is to simultaneously knock out expression of CCR5 and CXCR4. In fact, recent reports16,17 have described a ZFN-mediated CXCR4 disruption strategy effective in protecting human CD4+ T-cells against X4-tropic but not R5-tropic infection. To achieve dual-tropic resistance, Wilen et al. disrupted CXCR4 in T-cells from CCR5 δ32 patients, suggesting a potential double knockout strategy using two pairs of ZFNs against CXCR4 and CCR5.
Besides being used for targeted gene disruption, ZFNs can also be used to stimulate precise targeting of a gene therapy payload to a specific genomic locus by homologous recombination (Figure 1a). Gene targeting minimizes the risk of uncontrolled genomic insertion, which has caused serious adverse events including leukemia in several clinical gene therapy trials.18,19,20 Taking advantage of the benefits of gene targeting by homologous recombination, we have developed a novel method for creating multiple genetic resistances to HIV infection with a single gene-targeting event. Our approach mimics the current pharmacological treatment strategy, highly active antiretroviral therapy. A highly active antiretroviral therapy regimen typically consists of three or more drugs that inhibit HIV infection at multiple stages of the virus lifecycle. It has been shown that treatment with two antiviral drugs is better than one and that a three-drug regimen is superior.21 By analogy, we hypothesize that by creating multiple layers of genetic resistance to HIV (“stacking” genetic traits), we can generate broader and more robust inhibition of both R5- and X4-tropic HIV compared with CCR5 disruption alone. The ultimate goal would be to develop an approach that, when translated into humans, would create a reservoir of protected T-cells that would stave off immune collapse and the onset of AIDS, either alone or in combination with antiretroviral drugs.
Figure 1.

Targeting GFP to the endogenous CCR5 locus using ZFN-mediated homologous recombination in K562 cells. (a) Schematic of ZFN-mediated gene targeting by homologous recombination. (b) Examples of homologous targeting vectors used. For complete list see Supplementary Figure S4. (c) K562 cells were transfected with the CCR5-GFP targeting vector with or without CCR5 ZFNs delivered as plasmid DNA or mRNA, and GFP expression was followed by FACS for 16 days. (d) FACS plots from day 16 after transfection of CCR5-GFP, and with or without CCR5 ZFNs. (e) Diagram of genomic PCR and Southern blot strategies used to show targeting at CCR5. Probe, DNA probe used for Southern blot; arrows, PCR primers. (f) K562 cells transfected with the CCR5-GFP targeting vector, and without (−) or with (+) CCR5 ZFNs were analyzed for evidence of targeting at CCR5 by genomic PCR using the primers in e. (g) Southern blot analysis of GFP positive clones following targeting. “-“ untargeted, B – biallelically targeted. See also Supplementary Figure S3. APO, APOBEC3G D128K; FACS, fluorescence-activated cell sorting; GFP, green fluorescent protein; hrhTRIM5α, human-rhesus hybrid TRIM5α HSV-TK, herpes simplex virus thymidine kinase; hTRIM5α, human TRIM5α IRES-puro, internal ribosome entry site–puromycin N-acetyltransferase; PCR, polymerase chain reaction; Rev, Rev M10; rhTRIM5α rhesus TRIM5α Ubc, ubiquitin C promoter; 2A, 2A translation skipping peptide; GFP, green fluorescent protein; WT, wild type; ZFN, zinc finger nuclease.
Genetic studies have revealed several cellular proteins that confer effective resistance to HIV. TRIM5α and APOBEC3G are restriction factors that constitute a newly appreciated arm of the mammalian innate immune system.22,23,24 Interestingly, rhesus macaque TRIM5α25,26,27 and an engineered human-rhesus hybrid TRIM5α6 inhibit effective HIV capsid disassembly in the cytoplasm of infected cells, while the human version of TRIM5α is significantly less effective. APOBEC3G is a cytidine deaminase that is packaged with newly formed viral particles and causes hypermutation of the viral genome.28,29,30 The APOBEC3G D128K mutant escapes depletion by the viral protein Vif7 and thus successfully interferes with HIV replication. Rev M10 is a dominant negative form of a viral protein that prevents the export of early viral RNAs from the nucleus5,31 (Supplementary Figure S1). Rev M10 differs from the other two proteins in that it is not a naturally occurring molecule in human cells, although it effectively behaves as a restriction factor when expressed in cells.
Recent reports showed lentiviral integration of a cassette of three anti-HIV genes, including human-rhesus hybrid TRIM5α, into HSCs to confer robust protection against infection.32,33 Here, the strengths of that approach are combined with the clinical success and specific genome modification of the CCR5 disruption strategy. We precisely targeted a combination of anti-HIV genes to the CCR5 locus, generating the first example of specifically modified T-cells that are broadly resistant to HIV infection. In this way, we created multilayered genetic resistance to HIV by disrupting CCR5 while simultaneously integrating a cassette of anti-HIV restriction factors. We demonstrated that this approach conveys complete suppression of viral replication as well as full-spectrum resistance to both R5-tropic and X4-tropic HIV forms.
Results
ZFN-mediated gene targeting at the CCR5 locus
To determine the efficiency of targeting to the CCR5 locus by homologous recombination, we constructed a green fluorescent protein (GFP) expression cassette with one kilobase arms of homology from the CCR5 gene centered at the CCR5 ZFN cut site,11 and included a negative selectable herpes simplex virus thymidine kinase (HSV-TK) domain outside the homology (“CCR5-GFP” targeting vector, Figure 1b). For initial proof-of-principle experiments, we chose human erythroleukemic K562 cells, which have been extensively used to validate ZFN activity.34,35,36 Delivery of the CCR5-GFP targeting vector along with CCR5 ZFN DNA or mRNA into K562 cells by nucleofection resulted in stable GFP expression in up to 29.6% of the transfected cells (20.2% overall) compared with 2.2% (1.4% overall) in the absence of ZFNs (Figure 1c,d). As 2.2% of the cells in the samples without ZFNs remained GFP positive once expression from the episomal plasmid was lost by dilution through serial passaging, we used this percentage to approximate the random integration rate in these cells. Therefore, we predicted that approximately 27.4% of the cells in the ZFN samples were targeted at the CCR5 locus. The same percentage of these cells remained stably GFP positive for more than 4 months in culture, confirming that there was no appreciable silencing of the Ubc promoter when targeted to the CCR5 locus (data not shown). Negative selection with ganciclovir led to a 2.5-fold enrichment of targeted cells compared with the random integrants, consistent with targeting at the same genomic locus (Supplementary Figure S2).
To confirm that targeting occurred at the CCR5 locus, we designed a polymerase chain reaction strategy with a forward primer that binds a sequence in the CCR5-GFP targeting vector and a reverse primer recognizing a site in the CCR5 locus (Figure 1e). A band indicative of targeting was detected in samples treated with both the CCR5-GFP targeting vector and the ZFNs, but not in those transfected only with the targeting vector (Figure 1f). Clonal molecular analysis demonstrated that 89% of the GFP positive cells (25/28 clones) were the result of targeting the GFP expression cassette to the CCR5 locus (Figure 1g and Supplementary Figure S3a) compared with 0% of the samples without ZFNs (Supplementary Figure S3b), thus showing an overall targeting rate of 27% in the unsorted population.
Generation of HIV-resistant cell lines by targeting restriction factors to CCR5
We next investigated whether targeting an anti-HIV restriction factor to the CCR5 locus by homologous recombination would confer greater restriction to HIV infection compared with CCR5 disruption alone. To do so, we modified the CCR5-GFP targeting vector to replace the GFP expression cassette with a TRIM5α-IRES-puro cassette (CCR5-TRIM5α targeting vector' Figure 1b). Three versions of this targeting vector were constructed in which the TRIM5α gene is either human (h), which confers very little restriction against HIV infection; rhesus (rh), which confers robust resistance25;25 or a human-rhesus hybrid (hrh), which has been shown to confer intermediate resistance to HIV.6 A fourth version of the targeting vector was created to include only the puromycin resistance gene, allowing for the selection of CCR5 disrupted cells (“CCR5-hTRIM5α,” “CCR5-rhTRIM5α,” “CCR5-hrhTRIM5α,” “CCR5-IRES-puro” targeting vectors; Supplementary Figure S4). ZFN-mediated integration of the CCR5-rhTRIM5α targeting vector at the CCR5 locus was detected at greater than 50% of the alleles in K562 cells following selection with puromycin (Supplementary Figure S3b).
For HIV challenge experiments, we used the JLTRG-R5 line, a human Jurkat T-cell reporter line.37 JLTRG-R5 cells express CD4 and CXCR4 at levels similar to primary CD4+ T-cells and express CCR5 at levels similar to peripheral blood mononuclear cells.37 In addition, they have an integrated long terminal repeat (LTR) -GFP reporter that expresses GFP upon HIV infection. We used the CCR5 ZFNs to integrate the CCR5-hrhTRIM5α and CCR5-rhTRIM5α targeting vectors into the CCR5 locus in JLTRG-R5 cells. Again, we showed targeting of up to 50% of the alleles in the puromycin-selected population (Figure 2a). Similarly, we targeted CCR5-hTRIM5α and CCR5-IRES-puro to CCR5 in JLTRG-R5 (data not shown). Protein levels of each of the targeted TRIM5α variants were detected (Figure 2b, lanes 2,3,4), quantified by band intensities, and normalized to the CCR5-hrhTRIM lane (Figure 2c). A separate membrane with the same samples was stained with Coomassie blue as a loading control. These cells maintained expression of the transgenes by western blot for at least 6 months in culture without any evidence of locus silencing (data not shown). To check the status of the untargeted CCR5 allele, we stained the targeted cells for CCR5 and analyzed by fluorescence-activated cell sorting. Notably, in the puromycin-selected CCR5-IRES-puro population, greater than 99% of the untargeted CCR5 alleles had been disrupted and did not produce a functional gene product as evidenced by the absence of CCR5 on the cell surface (Figure 2d). Similar rates of disruption (93–99%) of the untargeted CCR5 allele were seen in the other CCR5-targeted samples (Supplementary Figure S5). In this way, we efficiently created a population of cells, most of which that are disrupted at both CCR5 alleles, one by gene targeting and the second by mutagenic NHEJ.
Figure 2.

Establishment of JLTRG-R5 T-cell line derived cells expressing anti-HIV genes. (a) JLTRG-R5 cells were transfected with the rhesus or human-rhesus hybrid CCR5-TRIM5α targeting vector and CCR5 ZFNs, and were analyzed for targeting by Southern blot. (b) Protein expression of restriction factors targeted to CCR5, probed by α-HA (TRIM5α), α-myc (APOBEC3G D128K) and α-Rev (Rev M10). The Coomassie stained blot is a loading control. (c) Quantification of bands from western blots in panel b using ImageJ software and normalized to hTRIM, rev-APO and rev lanes, respectively. Error bars represent the variation between quantification from two gels. (d) FACS plots showing ZFN-mediated disruption of the untargeted CCR5 allele in CCR5-IRES-puro cells. See also Supplementary Figure S5. a, APOBEC3G D128K-myc; FACS, fluorescence-activated cell sorting; h, human TRIM5α HA; hrh, human-rhesus hybrid TRIM5α-HA; r, Rev M10; rh, rhesus TRIM5α-HA; WT, wild type; ZFN, zinc finger nuclease.
Targeted single-factor cell lines are significantly protected against R5-tropic and X4-tropic HIV
To quantitate the level of resistance conferred by targeting these TRIM5α variants to CCR5, we challenged the targeted cells with R5-tropic HIV-1BaL-1. Samples were analyzed by flow cytometry for GFP expression from the integrated LTR-GFP reporter, and ratio to cell negative (RTCN) values were calculated as previously described.38 Briefly, RTCN is the cross product of percent GFP positive cells times mean fluorescence intensity, normalized to uninfected samples and was used to quantify level of infection (the higher the RTCN value, the greater the level of infection). Infection was followed over the course of 14 days, and is reported as RTCN values or cumulative RTCN values calculated from the area under the RTCN curve. Wild-type JLTRG-R5 cells were susceptible to infection by R5-tropic virus with a maximum RTCN value of 660 (Figure 3a, blue line, day 11) and a cumulative RTCN of 2,438 (Table 1). CCR5-IRES-puro cells, which mimic previously reported CCR5 disrupted cells, reached a maximum infection of 42 with R5-tropic HIV and a cumulative RTCN of 155, a protection of 16-fold (Figure 3a and Table 1). Similarly, CCR5-hTRIM5α cells showed a fivefold reduction in cumulative RTCN following R5-tropic HIV infection, consistent with simply knocking out CCR5. In contrast, CCR5-hrhTRIM5α cells and CCR5-rhTRIM5α cells demonstrated 1,642-fold (cumulative RTCN = 1.5) and 2,365-fold (cumulative RTCN = 1.0) resistance to R5-tropic HIV, respectively as measured through day 14 (Figure 3a and Table 1). Thus, compared with the level of protection achieved by CCR5 disruption alone through the targeting of IRES-puro, combining CCR5 disruption with the targeted integration of an anti-HIV factor increases the resistance of these cells an additional 100- to 150-fold, providing virtually complete inhibition of R5-tropic infection through day 14.
Figure 3.

Targeting a single anti-HIV restriction factor to CCR5 confers significant resistance to R5-tropic and X4-tropic HIV. Infection time course of CCR5-TRIM5α cells and CCR5-IRES-puro cells with (a) R5-tropic and (b) X4-tropic HIV as measured by RTCN values. Cumulative RTCN values were determined by integrating the area under the RTCN curve. Infection time course of CCR5-APO and CCR5-rev cells with (c) R5-tropic and (d) X4-tropic HIV. (*p < 0.05 cumulative RTCN compared with the wild type. Error bars represent standard deviation of three independent biological replicates.) h, human; hrh, human-rhesus hybrid; rh, rhesus; RTCN, ratio to cell negative; WT, wild type.
Table 1. Infection and fold protection of CCR5-targeted cells after 14 daysa.
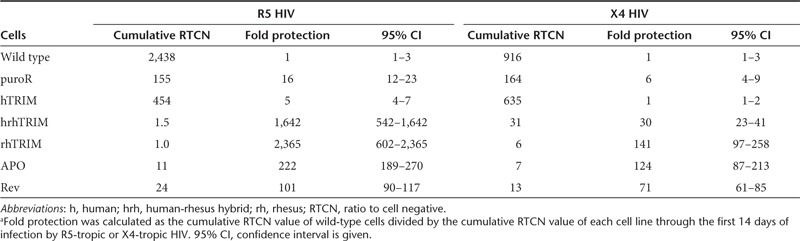
As TRIM5α acts as a postentry restriction factor, we hypothesized that the CCR5-hrhTRIM5α and CCR5-rhTRIM5α cells would also be resistant to infection by X4-tropic HIV, whereas the CCR5-IRES-puro cells would not. Each of the targeted cell lines was challenged with X4-tropic HIV-1NL4-3. By day 14, wild-type JLTRG-R5 cells had a cumulative RTCN value of 915, and the CCR5-IRES-puro cells and the CCR5-hTRIM5α cells were not significantly protected from X4-tropic infection (Figure 3b and Table 1), supporting the hypothesis that the resistance these cells displayed against R5-tropic virus was due solely to disruption of CCR5. Importantly, the CCR5-hrhTRIM5α cells and the CCR5-rhTRIM5α cells displayed 30-fold (cumulative RTCN = 31) and 141-fold (cumulative RTCN = 6.5) resistance to X4-tropic HIV, respectively (Figure 3b and Table 1). In this way, we showed that compared with CCR5 disruption alone, targeting rhesus- or human-rhesus hybrid TRIM5α to CCR5 both increases the protection of a T-cell line against R5-tropic infection and confers significant resistance to X4-tropic infection.
To investigate whether other postentry restriction factors would also serve to protect against both viral tropisms, we modified the targeting vectors to include the Vif-resistant D128K mutant of APOBEC3G or dominant negative Rev M10 (“CCR5-APO” and “CCR5-Rev” targeting vectors, Supplementary Figure S4). Following targeting, we detected expression of the myc-tagged APOBEC3G D128K and Rev M10 (Figure 2b, lanes 5 and 7, and Figure 2c). We showed that the CCR5-APO cells and the CCR5-Rev cells were protected 222-fold (cumulative RTCN = 11) and 101-fold (cumulative RTCN = 24) against R5-tropic HIV, respectively (Figure 3c and Table 1). Similarly, against X4-tropic HIV, the CCR5-APO and CCR5-Rev cells were protected 124-fold (cumulative RTCN = 7) and 71-fold (cumulative RTCN = 13) through day 14, respectively (Figure 3d and Table 1).
Expression of TRIM5α inhibits the initial round of infection
To further investigate the mechanism of HIV resistance in each of these single-factor cell lines, we used a single-round infectivity assay.39,40 First, we sought to determine whether this assay was sensitive enough to detect differences in the susceptibility of our targeted cell lines. To do this, we generated replication incompetent CCR5-tropic pseudovirions containing red fluorescent protein in place of the viral envelope protein by cotransfection of HEK 293T cells with the NL4-3-δE-RFP plasmid and the CCR5-tropic envelope SF162 plasmid. Culture supernatants were collected and used to infect wild-type JLTRG-R5 cells. Infection was assessed by quantifying RFP positive cells at 2 days postinfection. Importantly, wild-type cells had measurable infection at day 2, while cell lines targeted at CCR5 were significantly protected against infection due to the disruption of CCR5 (Figure 4a). Slightly variable levels of CCR5 disruption in the targeted cell lines (Supplementary Figure 5) accounted for the subtle but nonsignificant differences in the level of protection against single-round R5-tropic virus.
Figure 4.

TRIM5α blocks the initial round of infection by HIV. Infection of CCR5-targeted cell lines with single-round HIV pseudotyped with (a) R5 envelope or (b) VSVG envelope. (error bars represent standard deviation of three biological replicates. *p < 0.05, **p < 0.005, ***p < 0.0005.) hrh, human-rhesus hybrid; rh, rhesus; VSVG, vesicular stomatitis virus-G.
To quantify the contribution the targeted anti-HIV restriction factors have independent of the effects from CCR5 disruption, we made vesicular stomatitis virus-G (VSVG) pseudotyped single-round RFP virus (HIV-1NL4-3-δE-RFP-VSVG) and infected cells as above. Because TRIM5α is a postentry, preintegration restriction factor, CCR5-hrhTRIM5α cells and CCR5-rhTRIM5α cells should still be protected against single-round VSVG pseudotyped HIV infection. In fact, when challenged in the single-round assay, CCR5-hrhTRIM5α cells became significantly less infected, reaching only 59% the level of infection compared with the wild-type cells. Similarly, CCR5-rhTRIM5α cells were infected at 32% of wild type (Figure 4b). These data indicate that in the single-round infectivity assay, blocking viral uncoating through the expression of rh- or hrhTRIM5α confers significant, but less robust resistance against the initial round of infection compared with the inhibition of viral entry, presumably because some of the viral particles are able to overwhelm the TRIM5α resistance and become effectively uncoated. Furthermore, because Rev M10 and APOBEC3G D128K are postintegration restriction factors, they should not inhibit the initial cycle of infection, but only subsequent rounds. As predicted, the cells targeted with CCR5-APO and CCR5-Rev showed no protection in the single-round infectivity assay (Figure 4b).
Targeted stacking of restriction factors confers complete resistance to R5-tropic and X4-tropic infection
Finally, as the combination of CCR5 disruption with TRIM5α expression was more protective than CCR5 disruption alone against R5-tropic HIV in the multiround infection experiments, we sought to determine if stacking resistance factors together in combination as a cassette of anti-HIV genes would confer greater resistance to both R5-tropic and X4-tropic variants. To do this we again modified the targeting vectors to include all combinations of Rev M10, APOBEC3G D128K and hrhTRIM5α (“CCR5-Rev-hrh,” “CCR5-APO-hrh,” “CCR5-Rev-APO,” and “CCR5-hrh-triple,” Supplementary Figure S4) and targeted them to CCR5 in JLTRG-R5 cells. We chose to include the hrhTRIM5α variant instead of the rhTRIM5α version in the combination vectors because of its effectiveness alone, and the potentially lower antigenicity associated with the hybrid protein compared with the rhesus protein. Protein expression was analyzed by western blot (Figures 2b,c). Notably, we were unable to detect TRIM5α expression from the CCR5-Rev-hrh cells, making it difficult to accurately compare the efficacy of this combination of factors in conferring resistance to infection by HIV.
Because of the robust protection provided by each of the factors alone (cumulative RTCN values between 1.0 and 31), there was no significant benefit of adding additional factors through day 14 of infection (data not shown). To investigate the protection conferred by stacking restriction factors during prolonged infection, we measured the cumulative RTCN through day 25. Against R5-tropic HIV, all of the one-, two-, and three-factor cell lines had a significantly lower cumulative level of infection compared with the wild-type cells (Figure 5a, *). Notably, when compared with the cumulative RTCN of the CCR5-hrh-triple cells, all of the one- and two-factor cell lines showed significantly higher levels of infection, demonstrating the added benefit of stacking restriction factors (Figure 5a, # and Table 2).
Figure 5.

Stacking genetic resistance provides complete protection from infection by R5-tropic and X4-tropic HIV. Cumulative level of infection (area under RTCN curve) of cell lines targeted at CCR5 with zero (red bars), one (yellow), two (blue) or three (green) restriction factors by day 25 of infection with (a) R5-tropic HIV or (b) X4-tropic HIV. (Error bars represent standard deviation of three independent biological replicates, *p < 0.05 compared with the wild type, #p < 0.05 compared with CCR5-hrh-triple.) h, human; hrh, human-rhesus hybrid; RTCN, ratio to cell negative.
Table 2. Infection and fold protection of CCR5-targeted cells after prolonged exposure to HIVa.
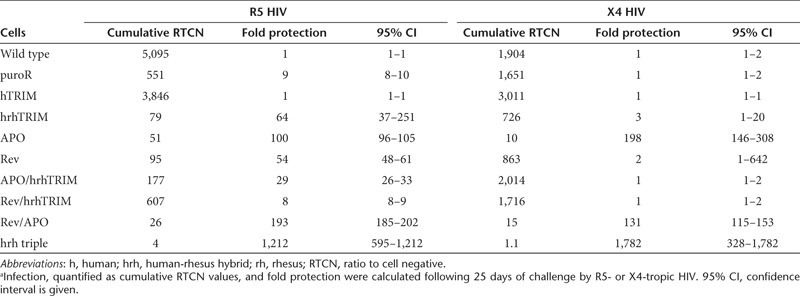
Against X4-tropic HIV, of the one- and two-factor lines, the CCR5-hrhTRIM, CCR5-APO, and CCR5-Rev-APO cells showed statistically lower levels of infection than wild-type cells, while the other lines became infected at near wild-type levels during prolonged HIV challenge. Importantly, the CCR5-hrh triple line remained uninfected during prolonged infection, again highlighting the efficacy of our trait-stacking approach (Figure 5b and Table 2).
Discussion
Using ZFN-mediated homologous recombination to target a cassette of anti-HIV restriction factors (hrhTRIM5α, APOBEC3G D128K and Rev M10) to the CCR5 locus, we have created robust resistance to infection by both X4- and R5-tropic HIV and have established a framework to test the relative fold protection genetic restriction factors provide against HIV infection (Table 1 and Table 2). This is the first demonstration of targeted trait stacking against HIV infection using homologous recombination to precisely integrate the restriction factors into the genome. A recent report41 describes ZFN-mediated insertion of GFP into CCR5 at targeting rates of up to 10% using a baculoviral delivery method in Ghost-CCR5 cells. A second study42 used integration-defective lentivirus to deliver CCR5 ZFNs and targeting vector and achieved targeting rates of 0.1% in human CD34+ hematopoietic stem/progenitor cells and 5% in human embryonic stem cells, but neither group infected the targeted cells with HIV. Untargeted trait-stacking approaches against HIV have also been previously reported and show significant restriction of infection,32,33 but they rely on the random integration of the anti-HIV factors in the genome. Here, we achieve precise gene targeting to the CCR5 locus of a cassette of anti-HIV restriction factors and demonstrate effective resistance to infection by HIV.
As targeting a puromycin resistance gene alone to CCR5 is functionally equivalent to CCR5 disruption by ZFN-induced NHEJ, we were able to compare the efficiency of our ZFN-mediated homologous recombination trait-stacking strategy with the published ZFN-mediated CCR5 disruption approach currently in clinical trials.43,44 In fact, our strategy overestimates the effectiveness of CCR5 disruption alone because we selected for targeted cells, most of which have undergone mutagenic NHEJ at the untargeted CCR5 allele (Figure 2d), resulting in a population of cells with most showing bi-allelic CCR5 disruption. Notably, even though only 1–7% of the targeted cells have detectable CCR5 expression by fluorescence-activated cell sorting, there is sufficient expression to establish infection in the cells without additional anti-HIV factors (the CCR5-IRES-puro and CCR5-hTRIM5α lines). Because of the small differences across samples in CCR5 expression, it is difficult to absolutely contrast the effect of targeting particular restriction factors in protecting against R5-tropic infection, but the CCR5-IRES-puro cells had among the lowest expression of CCR5 from the untargeted allele (Figure 2d and Supplementary Figure S5). Even in this context, against R5-tropic HIV, targeted integration of an anti-HIV gene to CCR5 provides up to 150-fold increased protection compared with CCR5 disruption alone in the CCR5-IRES-puro cells.
In an effort to restrict infection by X4-tropic virus, CXCR4 ZFNs have been recently described,16,17 and when used in combination with CCR5 ZFNs could confer resistance to both viral tropisms. However, the possible implementation of this approach is limited to the mature postthymic CD4+ T-cell compartment and would not be feasible in HSCs because of the requirement of functional CXCR4 in B-cell development and HSC homing.45,46 The use of ZFNs against CXCR4 and CCR5 to confer dual-tropic HIV resistance would also require bi-allelic gene disruption at two distinct sites in the same cell, a relatively rare occurrence even with highly active ZFN pairs. Finally, the simultaneous creation of two double-strand breaks would likely lead to increased genomic instabilities as simultaneous double-strand breaks can lead to chromosomal translocations47,48 or deletions.49
Our results suggest an alternative strategy to generate cells that are significantly protected from infection by either R5- or X4-tropic HIV. Infection was quantified using cumulative RTCN, calculated as the area under the RTCN time course curve. Cumulative RTCN describes the total viral burden over time and was used to compare the susceptibility to infection of our targeted cell lines. Our single-factor cell lines showed significant protection through the first two weeks of infection by either R5-tropic or X4-tropic HIV. By day 25 of the R5-tropic HIV challenge, the one- and two-factor cell lines, still had significantly lower cumulative RTCN values compared with the wild-type cells, but these cells did show modest levels of infection. In contrast, the CCR5-hrh-triple cell line showed statistically lower levels of infection than the one- and two-factor cell lines, remaining virtually uninfected. Against X4-tropic virus, several of the one- and two-factor lines (specifically, the CCR5-hrhTRIM, CCR5-APO, and CCR5-rev-APO cells) maintained significantly lower levels of infection compared with the wild-type cells, whereas the others became infected at near wild-type levels. Importantly, all but one of the one- and two-factor lines were infected at significantly higher levels than were the CCR5-hrh-triple cells, showing the benefit of stacking at least three layers of restriction against both R5-tropic and X4-tropic HIV. The one cell line that was not statistically different from the CCR5-hrh-triple cells was the CCR5-Rev line in which each of the three replicates was infected at higher levels than the CCR5-hrh-triple cells, but the variability among samples was high because one of the replicates became highly infected earlier in the time course than the other two replicates (data not shown). In addition, the fact that the double-factor CCR5-Rev-hrhTRIM cells showed considerably higher susceptibility to infection compared with the single-factor CCR5-Rev or CCR5-hrhTRIM cells is explained by the lower level of Rev expression and the virtually undetectable level of TRIM5α expression from the double-factor line. Because of this variable protein expression, it is difficult to directly evaluate the data from the CCR5-Rev-hrhTRIM cells. In contrast, the presence of low levels of three anti-HIV factors in the CCR5-hrh-triple cells is sufficient to provide robust protection against prolonged infection.
In summary, targeted trait stacking is an improvement over previous studies that solely relied on CCR511,12,50 or CXCR416,17 disruption to prevent HIV infection for three reasons. First, unique to our system, monoallelically modified cells confer effective resistance to infection. Second, just as pharmacological therapies against HIV infection inhibit multiple stages of the HIV lifecycle, our system provides multiple genetic blockades against infection (Supplementary Figure S1). In the first 14 days following infection, all combinations of anti-HIV factors provided resistance and there were only slight differences in the effectiveness of some of the single-, double- and triple-factor cassettes. However, by day 25 of infection with R5-tropic HIV, many of the cell lines targeted with single-factor cassettes had become infected. Notably, the CCR5-hrh-triple cells remained uninfected throughout the course of the experiment. The third major improvement of this strategy is that targeted trait stacking at the CCR5 locus is effective in preventing infection by both R5- and X4-tropic HIV. Cumulative RTCN values for CCR5-hrh-triple cells were less than 4 after prolonged infection by both viral tropisms. Combining the targeted trait-stacking strategy described here with recent advances in the ZFN modification of human hematopoietic stem and progenitor cells is the next preclinical hurdle in developing robust, long-term genetic protection against infection by HIV.
Materials and Methods
Cell lines and cell culture. K562s (ATCC, Manassas, VA) and JLTRG-R5 (AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH from Dr Olaf Kutsch, Germantown, MD) were maintained in RPMI 1640 (Hyclone, Logan, UT) supplemented with 10% bovine growth serum, 100 units/ml penicillin, 100 µg/ml streptomycin, and 2 mmol/l L-glutamine.
Plasmid construction. Flag-tagged CCR5 ZFNs11 were synthesized and cloned into pcDNA6 (Invitrogen, Grand Island, NY). Targeting vectors were constructed by polymerase chain reaction amplifying CCR5 arms of homology centered on the ZFN cut site using the following primers: 5′fwd, 5′-TTCCTGCCTCATAAGGTTGC-3′, 5′ rev 5′-AGGA TGAGGATGACCAGCAT-3′, 3′fwd 5′-GATAAACTGCAAAAGGCTG AAGAG-3′, 3′ rev 5′-AGACCCTCTATAACAGTAACTTCCT-3′. pcRev (AIDS Research and Reference Reagent Program from Dr Bryan R. Cullen, Germantown, MD) and pcDNA-APO3G (AIDS Research and Reference Reagent Program from Drs Klaus Strebel and Sandra Kao, Germantown, MD) were subjected to site-directed mutagenesis to create the rev M1031 and APOBEC3G D128K7 mutants. HA-tagged TRIM5α constructs were used as previously described.6 The three anti-HIV genes were cloned in all combinations and followed by IRES-puromycin acetyltransferase between the CCR5 arms of homology. Included outside the homology arms was HSV-TK domain for negative selection.
Virus preparation and titer. Plasmid pNL4-3 (generous gift of Dr Beth Levine) was transfected into HEK293T cells to make replication competent CXCR4-tropic HIV. Supernatant was collected on day 2 and viral titer was measured by p24 ELISA (Cell Biolabs, San Diego, CA) following the manufacturer's protocol. Cell-free viral supernatant of CCR5-tropic HIV-1Ba-L (AIDS Research and Reference Reagent Program from Dr Suzanne Gartner, Dr Mikulas Popovic, and Dr Robert Gallo, Germantown, MD) was propagated in wild-type JLTRG-R5 cells and viral supernatant was collected, titered, and used in subsequent experiments. Single-round HIV was produced as previously described.39 Briefly pNL4-3δE-RFP was cotransfected into HEK293T cells with either pVSVG (VSVG envelope) or pSF162 (R5 envelope) and viral supernatant was collected on day 2 after transfection.
Cell transfection and selection. K562 cells were nucleofected (Lonza, Basel, Switzerland) with 10 µg targeting vector and 1 µg of each ZFN plasmid or mRNA using program T-016 and nucleofection buffer containing 100 mmol/l KH2PO4, 15 mmol/l NaHCO3, 12 mmol/l MgCl2 • 6 H20, 8 mmol/l adenosine 5′-triphosphate, 2 mmol/l glucose, pH 7.4. JLTRG-R5 cells were nucleofected with the same conditions except with program I-010. Targeted cells were selected with 0.5 µg/ml puromycin. Negative selection was performed with 5 µmol/l ganciclovir.
Gene targeting. Genomic polymerase chain reaction was performed to show targeting to CCR5 using a forward primer within GFP (5′-TTCAAGATCCGCCACAACATCG-3′) and a reverse primer outside the 3′ arm of homology (5′-ACAGATGCCAAATAAATGGATG-3′). A Southern blot probe was generated upstream of the 5′ arm of homology by polymerase chain reaction amplification of genomic DNA using the following primers: fwd 5′-GGCCAGAAGAGCTGAGACATCCG-3′, rev 5′-CGTCTG CCACCACAGATGAATGTC-3′. Radioactive Southern blotting was performed using standard techniques.
Western blot. Expression of targeted anti-HIV genes was detected by western blot using the following primary antibodies: 1:500 mouse α-rev (Thermo, Rockford, IL), 1:5,000 mouse α-myc (Roche, Indianapolis, IN), 1:300 rabbit α-HA (Santa Cruz Biotechnology, Santa Cruz, CA); and secondary antibodies: 1:10,000 goat α-mouse-HRP (Santa Cruz Biotechnology) and 1:10,000 goat α-rabbit-HRP (Santa Cruz Biotechnology). Band intensities were quantified using ImageJ (NIH, Bethesda, MD).
Immunostaining for CCR5. Surface expression of CCR5 was measured by staining 0.5 million JLTRG-R5 cells with 10 µl APC-conjugated α-CCR5 antibody (BD Biosciences, San Jose, CA). Incubation was performed in 100 µl phosphate-buffered saline/2% serum for 30 minutes at 4°C and flow cytometry was performed using an Accuri C6 cytometer (Accuri, Ann Arbor, MI).
Quantitation of infection. In multiround infection experiments, 105 wild-type or targeted JLTRG-R5 cells were incubated with either 1 ng p24 X4-tropic or 10 ng p24 R5-tropic HIV. Optimal viral amount were determined by titration of virus on wild-type JLTRG-R5 cells, and the 10-fold difference in amounts of R5 and X4 virus is attributable to the variable susceptibility of these cells to infection by R5- or X4-tropic virus. Because JLTRG-R5 cells contain an integrated LTR-GFP,37 the level of infection was determined using GFP fluorescence and was calculated as the RTCN (RTCN = (% GFP)sample × (MFI)sample/(% GFP)uninfected × (MFI)uninfected).38 Cumulative RTCN values were calculated as the area under the RTCN curve using the trapezoid method of integration, minus the area under the curve of uninfected cells (which by definition is one time the number of days). Fluorescence was measured by flow cytometry using an Accuri C6 every 2–3 days. At every time point, an aliquot of each sample was pelleted and fixed by resuspension in either 4% paraformaldehyde or 2% formaldehyde, and incubated 30 minutes at 4°C before fluorescence-activated cell sorting. In single-round infection experiments, 105 wild-type or targeted cells were spin infected with 100 µl viral supernatant at 1,200g for 2 hours. On day 2, cells were prepared for fluorescence-activated cell sorting analysis as above.
Statistical analysis. All infections were performed in biological triplicate and statistical significance was calculated by Student's t test and 95% confidence intervals.
SUPPLEMENTARY MATERIAL Figure S1. Disruption of the HIV lifecycle by targeted gene therapy. Figure S2. Negative selection with ganciclovir enriches for targeted cells. Figure S3. Clonal analysis of K562 cells at targeted CCR5. Figure S4. CCR5 targeting vectors. Figure S5. CCR5 expression in targeted cell lines. ZFN-mediated disruption of CCR5 measured by antibody staining and FACS.
Acknowledgments
All data are tabulated in the main paper and supplementary materials. R.A.V., M.H.P., and S.S. conceived the project. R.A.V., M.M., and M.H.P. designed experiments performed by R.A.V. R.A.V. wrote the manuscript and all authors approved its content. We thank David Katzenstein for access to a BL-2+ facility and Shaina Porter for review of the manuscript. R.A.V. is supported by the U.T. Southwestern MSTP and Pharmacology Training Grant. This work was funded by an award from amFar (107776). The authors declare no conflict of interest.
Supplementary Material
References
- Cohen J. HIV. Escape artist par excellence. Science. 2003;299:1505–1508. doi: 10.1126/science.299.5612.1505. [DOI] [PubMed] [Google Scholar]
- Podsakoff GM, Engel BC, Carbonaro DA, Choi C, Smogorzewska EM, Bauer G.et al. (2005Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34(+) cells Mol Ther 1277–86. [DOI] [PubMed] [Google Scholar]
- Ranga U, Woffendin C, Verma S, Xu L, June CH, Bishop DK.et al. (1998Enhanced T cell engraftment after retroviral delivery of an antiviral gene in HIV-infected individuals Proc Natl Acad Sci USA 951201–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi JJ, June CH., and, Kohn DB. Genetic therapies against HIV. Nat Biotechnol. 2007;25:1444–1454. doi: 10.1038/nbt1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Böhnlein S, Hauber J., and, Cullen BR. Functional dissection of the HIV-1 Rev trans-activator–derivation of a trans-dominant repressor of Rev function. Cell. 1989;58:205–214. doi: 10.1016/0092-8674(89)90416-9. [DOI] [PubMed] [Google Scholar]
- Sawyer SL, Wu LI, Emerman M., and, Malik HS. Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA. 2005;102:2832–2837. doi: 10.1073/pnas.0409853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Svarovskaia ES, Barr R, Zhang Y, Khan MA, Strebel K.et al. (2004A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion Proc Natl Acad Sci USA 1015652–5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuitemaker H, van 't Wout AB., and, Lusso P. Clinical significance of HIV-1 coreceptor usage. J Transl Med. 2011;9 Suppl 1:S5. doi: 10.1186/1479-5876-9-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R.et al. (1996Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection Cell 86367–377. [DOI] [PubMed] [Google Scholar]
- Chen W, Zhan P, De Clercq E., and, Liu X. Recent progress in small molecule CCR5 antagonists as potential HIV-1 entry inhibitors. Curr Pharm Des. 2012;18:100–112. doi: 10.2174/138161212798919084. [DOI] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O.et al. (2008Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases Nat Biotechnol 26808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V.et al. (2010Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo Nat Biotechnol 28839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allers K, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E.et al. (2011Evidence for the cure of HIV infection by CCR5?32/?32 stem cell transplantation Blood 1172791–2799. [DOI] [PubMed] [Google Scholar]
- Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K.et al. (2009Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation N Engl J Med 360692–698. [DOI] [PubMed] [Google Scholar]
- Porteus MH., and, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- Wilen CB, Wang J, Tilton JC, Miller JC, Kim KA, Rebar EJ.et al. (2011Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases PLoS Pathog 7e1002020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Wang J, Crain K, Fearns C, Kim KA, Hua KL.et al. (2012Zinc-finger Nuclease Editing of Human cxcr4 Promotes HIV-1 CD4(+) T Cell Resistance and Enrichment Mol Ther 20849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P.et al. (2003LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1 Science 302415–419. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F.et al. (2010Transfusion independence and HMGA2 activation after gene therapy of human ß-thalassaemia Nature 467318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U.et al. (2006Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1 Nat Med 12401–409. [DOI] [PubMed] [Google Scholar]
- Schmit JC., and, Weber B. Recent advances in antiretroviral therapy and HIV infection monitoring. Intervirology. 1997;40:304–321. doi: 10.1159/000150564. [DOI] [PubMed] [Google Scholar]
- Baumann JG. Intracellular restriction factors in mammalian cells–An ancient defense system finds a modern foe. Curr HIV Res. 2006;4:141–168. doi: 10.2174/157016206776055093. [DOI] [PubMed] [Google Scholar]
- Grütter MG., and, Luban J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr Opin Virol. 2012;2:142–150. doi: 10.1016/j.coviro.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertel T, Hausmann S, Morger D, Züger S, Guerra J, Lascano J.et al. (2011TRIM5 is an innate immune sensor for the retrovirus capsid lattice Nature 472361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P., and, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- Towers GJ. The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology. 2007;4:40. doi: 10.1186/1742-4690-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MR, Sacha JB, Weiler AM, Borchardt GJ, Glidden CE, Sheppard NC.et al. (2011The TRIM5{alpha} genotype of rhesus macaques affects acquisition of simian immunodeficiency virus SIVsmE660 infection after repeated limiting-dose intrarectal challenge J Virol 859637–9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santa-Marta M, da Silva FA, Fonseca AM., and, Goncalves J. HIV-1 Vif can directly inhibit apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G-mediated cytidine deamination by using a single amino acid interaction and without protein degradation. J Biol Chem. 2005;280:8765–8775. doi: 10.1074/jbc.M409309200. [DOI] [PubMed] [Google Scholar]
- Harris RS, Petersen-Mahrt SK., and, Neuberger MS. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol Cell. 2002;10:1247–1253. doi: 10.1016/s1097-2765(02)00742-6. [DOI] [PubMed] [Google Scholar]
- Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES.et al. (2007Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration J Virol 817099–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, McCarn DF, Tiley LS., and, Cullen BR. Mutational definition of the human immunodeficiency virus type 1 Rev activation domain. J Virol. 1991;65:4248–4254. doi: 10.1128/jvi.65.8.4248-4254.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JS, Javien J, Nolta JA., and, Bauer G. Preintegration HIV-1 inhibition by a combination lentiviral vector containing a chimeric TRIM5 alpha protein, a CCR5 shRNA, and a TAR decoy. Mol Ther. 2009;17:2103–2114. doi: 10.1038/mt.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JE, Chen RX, McGee J, Nacey C, Pollard RB, Abedi M.et al. (2012Generation of an HIV-1-resistant immune system with CD34(+) hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector J Virol 865719–5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S.et al. (2005Highly efficient endogenous human gene correction using designed zinc-finger nucleases Nature 435646–651. [DOI] [PubMed] [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M.et al. (2008Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification Mol Cell 31294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I.et al. (2007An improved zinc-finger nuclease architecture for highly specific genome editing Nat Biotechnol 25778–785. [DOI] [PubMed] [Google Scholar]
- Ochsenbauer-Jambor C, Jones J, Heil M, Zammit KP., and, Kutsch O. T-cell line for HIV drug screening using EGFP as a quantitative marker of HIV-1 replication. BioTechniques. 2006;40:91–100. doi: 10.2144/000112072. [DOI] [PubMed] [Google Scholar]
- Vödrös D., and, Fenyö EM. Quantitative evaluation of HIV and SIV co-receptor use with GHOST(3) cell assay. Methods Mol Biol. 2005;304:333–342. doi: 10.1385/1-59259-907-9:333. [DOI] [PubMed] [Google Scholar]
- McMahon MA, Jilek BL, Brennan TP, Shen L, Zhou Y, Wind-Rotolo M.et al. (2007The HBV drug entecavir - effects on HIV-1 replication and resistance N Engl J Med 3562614–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon MA, Parsons TL, Shen L, Siliciano JD., and, Siliciano RF. Consistent inhibition of HIV-1 replication in CD4+ T cells by acyclovir without detection of human herpesviruses. J Virol. 2011;85:4618–4622. doi: 10.1128/JVI.02423-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Lee CL, Joo KI, Zarzar J, Liu Y, Dai B.et al. (2011Gene Editing of Human Embryonic Stem Cells via an Engineered Baculoviral Vector Carrying Zinc-finger Nucleases Mol Ther 19942–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA.et al. (2007Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery Nat Biotechnol 251298–1306. [DOI] [PubMed] [Google Scholar]
- Ando D, Tebas P, Stein D, Wang S, Lee G, Holmes MC.et al. (2011HAART Treatment Interruption Following Adoptive Transfer of Zinc Finger Nuclease (ZFN) CCR5 Modified AutologousCD4 T-cells (SB-728-T) to HIV-infected Subjects Demonstrates Durable Engraftment and Suppression of Viral LoadIn: 51st ICAAC, Chicago, IL.
- Mitsuyasu R, Lalezari J, Deeks S, Wang S, Lee G, Holmes MC.et al. (2011Adoptive Transfer of Zinc Finger Nuclease CCR5 Modified Autologous CD4 T-cells (SB-728-T) to Aviremic HIV-infected Subjects with Suboptimal CD4 Counts (200–500 cells/mm3)In: 51st ICAAC, Chicago, IL.
- Dar A, Kollet O., and, Lapidot T. Mutual, reciprocal SDF-1/CXCR4 interactions between hematopoietic and bone marrow stromal cells regulate human stem cell migration and development in NOD/SCID chimeric mice. Exp Hematol. 2006;34:967–975. doi: 10.1016/j.exphem.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y.et al. (1996Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1 Nature 382635–638. [DOI] [PubMed] [Google Scholar]
- Brunet E, Simsek D, Tomishima M, DeKelver R, Choi VM, Gregory P.et al. (2009Chromosomal translocations induced at specified loci in human stem cells Proc Natl Acad Sci USA 10610620–10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ.et al. (2011Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells Cell 147107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Kim E., and, Kim JS. Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res. 2010;20:81–89. doi: 10.1101/gr.099747.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Lee HJ, Kim H, Cho SW., and, Kim JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19:1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.