

Abstract
Dystrophin deficiency results in lethal Duchenne muscular dystrophy (DMD). Substituting missing dystrophin with abbreviated microdystrophin has dramatically alleviated disease in mouse DMD models. Unfortunately, translation of microdystrophin therapy has been unsuccessful in dystrophic dogs, the only large mammalian model. Approximately 70% of the dystrophin-coding sequence is removed in microdystrophin. Intriguingly, loss of ≥50% dystrophin frequently results in severe disease in patients. To test whether the small gene size constitutes a fundamental design error for large mammalian muscle, we performed a comprehensive study using 22 dogs (8 normal and 14 dystrophic). We delivered the ΔR2-15/ΔR18-19/ΔR20-23/ΔC microdystrophin gene to eight extensor carpi ulnaris (ECU) muscles in six dystrophic dogs using Y713F tyrosine mutant adeno-associated virus (AAV)-9 (2.6 × 1013 viral genome (vg) particles/muscle). Robust expression was observed 2 months later despite T-cell infiltration. Major components of the dystrophin-associated glycoprotein complex (DGC) were restored by microdystrophin. Treated muscle showed less inflammation, fibrosis, and calcification. Importantly, therapy significantly preserved muscle force under the stress of repeated cycles of eccentric contraction. Our results have established the proof-of-concept for microdystrophin therapy in dystrophic muscles of large mammals and set the stage for clinical trial in human patients.
Introduction
Duchenne muscular dystrophy (DMD) is caused by dystrophin deficiency. Several thousands of disease-causing mutations have been identified in the dystrophin gene. Replacing the defective gene with a functional copy offers a mutation-independent therapy for all patients. Excessively truncated microdystrophin has been pursued for over a decade as a substitute of the full-length protein.1,2 The microgene is developed to overcome the 5 kb packaging constrain of adeno-associated virus (AAV), the only viral vector with the potential of transducing all striated muscles in the body through vasculature delivery. It is hypothesized that rational deletion of ~70% of the 427 kD full-length dystrophin protein may yield novel synthetic microdystrophins that still protect muscle. More than a dozen, ~4 kb microgenes have been tested in various mouse models including severely affected strains such as aged dystrophin null mdx, dystrophin/utrophin double null, and dystrophin/myoD double null mice.3,4,5 Collectively, these studies suggest that microdystrophin can ameliorate cellular and biochemical defects, increase muscle force, and prolong life span.
Failure to translate therapeutic efficacy from mice to human patients has been a major obstacle in the field of DMD research. As a matter of fact, many promising therapies (such as myoblast transfer and myostatin-neutralizing antibody) are stalled at this stage.6,7 Considering the inherent anatomic, biochemical, and physiological differences in muscles of small and large mammals, it becomes crucial to conduct tests in large animal models before human trial. Dystrophin-deficient dog is the only large mammalian model for DMD. Affected dogs share clinical and genetic similarity to human patients (reviewed in ref. 1). To further develop the promising microdystrophin therapy, many laboratories tried microdystrophin in the canine model (reviewed in ref. 1). Unfortunately, most studies were exclusively on gene expression and only a few papers examined histological and/or physiological improvement. In contrast to the unequivocal muscle-strengthening effect in mice, these limited dog studies have yielded conflicting results. A study based on a single dog suggests that microdystrophin may reduce central nucleation.8 However, results from two other studies that included more dogs demonstrated that microdystrophin did not protect muscles in large mammals.9,10 Specifically, Sampaolesi et al. found that the performance of four microdystrophin-treated dogs declined rather than improved.10 Kornegay et al. delivered AAV microdystrophin to three newborn dystrophic puppies. Instead of disease amelioration, surprisingly, treatment resulted in growth delay, limb muscle atrophy, and contracture.9
The initial idea of dystrophin minimization came from a study reporting very mild symptoms in an old patient who missed 46% of dystrophin due to in-frame deletion.11 However, patients who have lost ≥50% dystrophin are almost invariably associated with a severe phenotype even if the truncated protein is expressed at a fairly high level.12,13,14,15,16,17,18,19 Clinical reports from human patients and the results of dog studies seem to suggest that excessively abbreviated microdystrophin may not meet the need of large species such as dogs and humans. To address this fundamental question, we expressed the ΔR2-15/ΔR18-19/ΔR20-23/ΔC microdystrophin gene in a forelimb muscle of dystrophic dogs using tyrosine mutant AAV-9. We report here that microdystrophin gene therapy reduced macrophage infiltration, fibrosis, and calcification. When challenged with eccentric contraction, AAV-injected muscle significantly outperformed untreated muscle. Our results establish the premise for microdystrophin therapy in muscles of large mammals.
Results
Y731F tyrosine mutant AAV-9 resulted in robust microdystrophin expression in dystrophic dogs
To determine whether excessive truncation of dystrophin compromises muscle protection in large mammals, we packaged a flag-tagged codon-optimized canine ΔR2-15/ΔR18-19/ΔR20-23/ΔC microgene using Y731F tyrosine mutant AAV-9.20 We first tested this vector in mdx mice. Tail vein injection of 6.5 × 1012 viral genome (vg) particles of AAV significantly reduced central nucleation and fibrosis, increased specific muscle force, and minimized eccentric contraction-induced force drop (Figure 1).
Figure 1.

Evaluation of the flag-tagged codon-optimized canine ΔR2-15/ΔR18-19/ΔR20-23/ΔC microgene by systemic Y731F tyrosine mutant AAV-9 gene transfer in mdx mice. 6.5 × 1012 vg particles of the AAV vector were injected to 5-week-old male mdx mice via the tail vein. Microdystrophin expression, muscle pathology, and force were evaluated at 2 months after gene transfer. (a) Representative immunofluorescence staining photomicrographs showing microdystrophin expression. Serial muscle sections were stained with antibodies specific for the indicated epitopes. Microdystrophin carries epitopes for the flag tag, dystrophin H1 region and dystrophin R16 but not dystrophin R6-8. Microdystrophin expression also restored sarcolemmal nNOS expression. *, a revertant fiber that was transduced by the AAV microgene vector. This fiber is not only positive for the flag tag and the H1 antibody but was also stained by R6-8 antibody; †, an area that was not transduced by AAV microdystrophin vector. (b) Quantitative evaluation of muscle pathology in the anterior tibialis muscle. AAV microdystrophin therapy significantly reduced degeneration/regeneration and fibrosis as reflected by the reduction of central nucleation (CN) and the hydroxyproline (HP) content, respectively. (c) Canine microdystrophin significantly increased the contractility of the extensor digitorum longus muscle and prevented eccentric contraction-induced force drop in mdx mice. Left panel, cross-sectional area normalized specific muscle force; right panel, relative force changes during 10 rounds of eccentric contraction. The sample size for b and c are n = 7 for untreated, and n = 10 for AAV treated. *, significantly different from the other group. AAV, adeno-associated virus; nNOS, neuronal nitric oxide synthase; vg, viral genome.
Next, we delivered 2.6 × 1013 vg particles/muscle of virus to the extensor carpi ulnaris (ECU) muscles of six dystrophic dogs (Table 1). Four dogs (Caesar, Chipotle, Jack, and Lanai) received AAV microdystrophin treatment in one ECU muscle. Two dogs (Honey Dijon and Sonic) received treatment in both ECU muscles (Table 1). To minimize viral capsid and/or microdystrophin-induced immune rejection, we applied a transient immune suppression protocol using cyclosporine and mycophenolate mofetil.21 At 2 months after gene transfer, AAV transduction, histopathology, and muscle force were examined. Saturated microdystrophin expression was confirmed in every treated ECU muscle by immunostaining and western blot (Figure 2, Supplementary Figure S2). Consistently, abundant vector genomes were detected in injected ECU muscles (Figure 2). T-cell response was not detected to either AAV capsids or microdystrophin by interferon-γ enzyme-linked immunosorbent spot (ELISPOT) assay (Figure 3a). However, compared with that of uninjected muscles, AAV-injected ECU muscles showed much more infiltration of CD4+ and CD8+ T cells (Figure 3b).
Table 1. Summary of AAV microdystrophin-infected dogs.
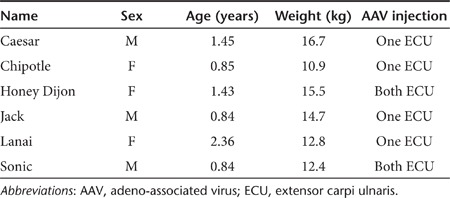
Figure 2.

AAV gene transfer leads to robust microdystrophin expression in the ECU muscle of dystrophic dogs. (a) A representative montage immunofluorescence staining photomicrograph showing widespread microdystrophin expression in the AAV-injected ECU muscle from Chipotle. (b) A representative western blot showing microdystrophin expression at the expected size. Two AAV-injected dystrophic ECU muscles were from Jack and Lanai. Untreated dystrophic muscle was the non-injected ECU muscle from Lanai. Dys, dystrophic dog; WT, normal dog. Open arrowhead, full-length dystrophin; filled arrowhead, microdystrophin. (c) Quantification of AAV genome copy. EDL, extensor digitorum longus muscle; one side, only one ECU muscle received AAV injection. The contralateral ECU muscle served as the uninjected control; two sides, both ECU muscles were injected with AAV. *, significantly higher than that of non-injected EDL and contralateral ECU muscles. AAV, adeno-associated virus; ECU, extensor carpi ulnaris.
Figure 3.

AAV microdystrophin expression accompanies T-cell infiltration in dystrophic dog muscle but without a positive ELISPOT response. (a) Interferon-γ ELISPOT responses to pools of AAV-9 capsid peptides or microdystrophin peptides. Lymphocytes were isolated from the spleen at the time of necropsy (2 months after AAV injection) and stimulated with each of three pools (A,B,C) of AAV-9 capsid peptides, each of five pools (A,B,C,D,E) of microdystrophin peptides, or phorbol 12-myristate 13-acetate (PMA)/ionomycin (positive control). Medium alone served as the no stimulation negative control. Results are presented as spot-forming units (SFU) per 106 splenocytes for experimental groups and as SFU per 2 × 104 splenocytes for PMA/ION control. Neither AAV-9 capsid nor microdystrophin induced the ELISPOT positive T-cell response. (b) Representative immunohistochemical staining montage photomicrographs from normal, dystrophic, and AAV-treated dystrophic dog ECU muscles. Top panel, CD4+ T cells; bottom panel, CD8+ T cells. Robust CD4+ and CD8+ T-cell infiltration were seen only in dystrophic dog ECU muscles that received AAV injection. AAV, adeno-associated virus; ECU, extensor carpi ulnaris; ELISPOT, enzyme-linked immunosorbent spot; ION, ionomycin.
Microdystrophin restored the dystrophin-associated glycoprotein complex, ameliorated histopathology, and mitigated eccentric contraction-induced injury in dystrophic dogs
We examined major components of the dystrophin-associated glycoprotein complex (DGC) by immunofluorescence staining (Figure 4). Expression of β-dystroglycan, β-sarcoglycan, syntrophin, and dystrobrevin was substantially enhanced in microdystrophin-transduced myofibers (Figure 4). We also examined utrophin, an autosomal paralog of dystrophin. Staining intensity for utrophin was clearly reduced in microdystrophin-positive myofibers compared with that of the neighboring untransduced myofibers (Figure 4).
Figure 4.

AAV-9 microdystrophin gene transfer restores the major components of the dystrophin-associated glycoprotein complex (DGC). (a) Immunofluorescence staining was performed with epitope-specific antibodies on serial muscle sections. The canine microdystrophin gene used in this study carries the N-terminal domain, spectrin-like repeat 16 (R16), an engineered Dys2 epitope at the C-terminal end, and a flag tag at the N-terminal end. However, it does not contain R4-6 and R11. The results confirmed flag-tagged canine microdystrophin expression in AAV-treated ECU muscle. Top panel, normal ECU muscle; middle panel, untreated dystrophic ECU muscle; bottom panel, AAV microdystrophin-treated ECU muscle. *, the same myofiber in serial sections. (b) Representative photomicrographs of serial immunofluorescence staining for laminin, major DGC components including β-dystroglycan (β-DG), β-sarcoglycan (β-SG), syntrophin (Syn), dystrobrevin (Dbr), and utrophin. These stainings were on the same serial tissue sections as in a. *, the same myofiber in serial sections. Laminin staining outlines individual myofiber. Expression of the major DGC components (β-DG, β-SG, Syn, and Dbr) was greatly reduced in untreated dystrophic muscle. AAV microdystrophin therapy increased expression of these DGC components. AAV, adeno-associated virus; ECU, extensor carpi ulnaris.
Untreated ECU muscles showed typical dystrophic pathology such as inflammation, degeneration/regeneration, fibrosis, and calcification (Figure 5a).22 AAV microdystrophin therapy attenuated most but not all morphological lesions. Treated muscles had much less myofiber calcification (Figure 5a). Macrophage infiltration and fibrosis were also substantially reduced (Figure 5a, Supplementary Figure S1). Further, myofiber size distribution showed a trend towards normalization (Figure 5b). Interestingly, treatment did not alter cellular markers of regeneration such as the percentage of centrally nucleated myofiber and the quantity of embryonic myosin heavy chain (eMHC)-positive myofibers (Figure 5c).
Figure 5.

AAV microdystrophin therapy ameliorates histological lesions in dystrophic dogs. (a) Representative photomicrographs of histopathology in untreated and AAV microdystrophin-treated ECU muscles from Caesar. Treated muscle had much less macrophage infiltration, minimal calcification, and substantially reduced fibrosis. Dys, dystrophin immunostaining; Mφ, nonspecific esterase staining for macrophage (insert is the high power magnification image of the boxed area in the same image). Macrophages show deep, dark brown staining; Cal, Alizarin red staining for calcification, calcified myofibers stained in red. (Macrophage immunostaining and low magnification view of Alizarin red staining are shown in Supplementary Figure S3); MT, Masson trichrome staining for fibrosis (fibrotic tissue is in blue color). *, the same myofiber in serial sections. Arrow, calcified myofiber in untreated ECU muscle. (b) Myofiber size distribution in normal (8,844 myofibers from five normal dogs), untreated (5,055 myofibers from four untreated dogs) and AAV microdystrophin-treated ECU muscles (8,374 myofibers from six AAV-injected dogs). (c) Quantification of central nucleation (top panel) and embryonic myofibers (bottom panel). Central nucleation shows percentage of myofibers containing centrally localized nuclei. N = 448 myofibers for normal dogs; N = 3,964 myofibers for dystrophic dogs (from four dystrophic dogs); and N = 8,530 myofibers for AAV-treated dystrophic dogs (from eight ECU muscles of six treated dystrophic dogs). The results of embryonic myofiber quantification are obtained from ECU muscles of two normal dogs and four dogs that received AAV injection in one side of the ECU muscle. *, significantly lower than other two groups. There was no significant difference between treated and untreated ECU muscles. AAV, adeno-associated virus; ECU, extensor carpi ulnaris; HE, hematoxylin and eosin.
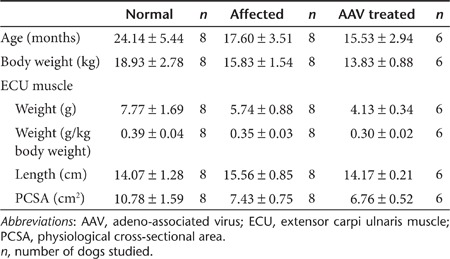
To determine whether microdystrophin therapy improved muscle physiology, we measured ECU force in situ (Figure 6).22 AAV injection did not affected anatomic properties of the ECU muscle (Table 2). Despite clear histology improvement, specific tetanic muscle force was not significantly changed following microdystrophin therapy (Figure 6a). Heightened susceptibility to repeated eccentric contraction-induced injury is a hallmark of dystrophin-deficient muscle.22,23 We thus applied five rounds of eccentric contraction. Strenuous lengthening contraction resulted in negligible force loss in the normal ECU muscle (Figure 6b).22 Untreated dystrophic muscles showed dramatic force reduction when challenged by eccentric contraction (Figure 6b).22 AAV microdystrophin therapy significantly protected the dystrophic ECU muscle from eccentric contraction-induced force decline (Figure 6b).
Figure 6.

AAV microdystrophin therapy protects dystrophic dog muscle from eccentric contraction-induced injury although it does not increase specific muscle force. (a) Specific tetanic muscle force in normal, dystrophic, and AAV-treated dystrophic ECU muscles. PCSA, physiological cross-sectional area. *, the value from normal dogs is significantly higher than dystrophic dogs (treated or untreated). (b) Relative change of the tetanic force during five cycles of eccentric contraction in normal, untreated, and microdystrophin-treated ECU muscles. AAV-treated muscles were significantly more resistant to eccentric contraction-induced force loss than untreated muscles. *, significantly different from those of normal and untreated. For both a and b, N = 8 dogs for normal, N = 8 dogs for dystrophic, and N = 6 dogs for AAV treated. AAV, adeno-associated virus; ECU, extensor carpi ulnaris.
Table 2. Dog age, body weight, and anatomic properties of the ECU muscle.
Discussion
Despite remarkable success in the murine models of DMD, microdystrophin therapy has encountered great challenges when tested in dogs and humans.9,10,24 Untoward cellular immune response has been considered as the primary barrier because such immune rejection eliminates therapeutic microdystrophin expression in treated muscles.24 While overcoming immunological hurdle remains an important research area (especially for human trials), several effective regimes have been developed to allow robust microdystrophin delivery in dystrophic dogs.9,10,21,25 Surprisingly, successful microgene expression has so far not resulted in convincing physiological improvement in the canine DMD model.9,10,21,25 Validating therapeutic efficacy in dystrophic dogs is thus a top priority for microdystrophin-based DMD therapy. With this in mind, we delivered a codon-optimized canine microdystrophin gene to the ECU muscle in young adult dystrophic dogs. Consistent with our earlier publications,22,26 untreated muscles displayed severe histological lesions and force deficit characteristic of a DMD phenotype. AAV gene transfer resulted in widespread expression of microdystrophin and restoration of major components of the DGC (Figures 2 and 4, Supplementary Figure S2).21,25 Morphology studies revealed appreciable reduction of macrophage invasion, fibrosis, and myofiber calcification in treated muscles (Figures 5, Supplementary Figure S3). Importantly, microdystrophin therapy significantly prevented lengthening contraction-induced force drop, a cardinal feature of dystrophin-deficient muscle (Figure 6).
While our results are very encouraging, it is important to exclude possible “contribution ” from immune suppressive drugs used in the study.27 Conflicting data have been reported from small, uncontrolled clinical studies on the therapeutic effect of immunosuppressants in DMD. One research group concluded that cyclosporine increased muscle strength in DMD patients.28 However, Mendell and colleagues found that immune suppressive drugs (such as cyclosporine and azathioprine) had no clinical benefit in DMD.6 Similarly, results from mdx mice are also inconsistent.29,30 To accurately determine therapeutic benefits of microdystrophin in our study, we intentionally delivered AAV to only oneside of the ECU muscle in four dogs. The results from the contralateral non-AAV–injected ECU muscle were compared with those of dystrophic dogs that have never received immunosuppressants.22 We did not see any apparent difference in muscle pathology. The eccentric contraction profiles were identical with or without immune suppression. In support of our results, Cerletti et al. found that cyclosporine therapy in newborn dystrophic dogs did not reduce fibrosis.31 Dell'Agnola et al. reported that cyclosporine did not improve clinical course in dystrophic dogs.32 Rouger et al. compared dystrophic dogs that either received or did not receive immune suppressive drugs. They found that immune suppression did not improve clinical condition neither did it reduce fibrosis nor calcification.33 Recently, a placebo-controlled, double blind multicentered clinical trial has finally settled the controversy.34 This randomized study suggests that cyclosporine has no beneficial effect for DMD.34 Taken together, we believe histological and physiological improvement observed in this study is due to AAV microdystrophin therapy.
The microdystrophin vector used in this study effectively reduced muscle disease in both mdx mice and dystrophic dogs (Figures 1,4,5 and 6). However, we found important differences between the murine and canine model. Central nucleation was significantly reduced in mdx mice but remained unchanged in dystrophic dogs (Figures 1 and 5). The frequency of embryonic myofiber, another marker of degeneration/regeneration was not altered in dystrophic dogs either (Figure 5). In physiological assay, treatment significantly increased specific muscle force in mdx mice but not in dystrophic dogs (Figures 1 and 6). It is currently unclear why the exact same vector has yielded different results in these aspects of muscle disease in mice and dogs. There are several possibilities. First, the inherent differences between mouse muscle and dog muscle may have contributed to our findings. In this regard, mouse muscle shows a much higher regenerative capacity than that of dog muscle. In the absence of treatment, centrally nucleated myofibers reached ~80% in mdx mice but only ~20% in dystrophic dogs (Figures 1 and 5). Second, there may exist a species-related difference in the structure-function relationship of dystrophin. C-terminal domain truncation has minimal impact on muscle histology and function in mice.35 Interestingly, both mild and severe cases have been reported in patients whose C-terminal domain is partially or completely deleted in humans.36,37,38,39,40 While it is clear that C-terminal truncation may have compromised mRNA and/or protein stability, it is also possible that other yet unknown species-related factors may have played a role. Future studies are needed to clarify these intriguing observations.
A recent study examined microdystrophin therapy in a single dystrophic dog.8 At 2 months after gene transfer in the cranial tibialis muscle, the authors quantified central nucleation in four random fields and found a fivefold reduction in the treated muscle.8 We counted ~4,000 myofibers from four untreated ECU muscles and ~8,500 fibers from eight AAV-injected ECU muscles (Figure 5). No significant difference was detected. Using eMHC immunostaining, another marker of regeneration, we also did not see a significant difference between treated and untreated ECU muscles (Figure 5). It is currently unclear whether the difference between our results and that of Koo et al. is due to small versus large sample size or the cranial tibialis muscle versus the ECU muscle.
Another interesting finding is persistent microdystrophin expression despite infiltration of CD4+ and CD8+ T cells (Figure 3). We have previously shown that the immune suppression protocol used in the current study resulted in long-term microdystrophin expression for at least half year in dystrophic dogs.21 However, cellular immune reaction was not examined in the previous study. Here, we performed ELISPOT assay and immunohistochemical staining for CD4+ and CD8+ T cells (Figure 3). Using pooled peptide libraries, we did not detect interferon-γ response to either viral capsids or microdystrophin (Figure 3a). However, CD4+ and CD8+ T cells were detected on immunostaining in microdystrophin-transduced muscles (Figure 3b). Future mechanistic studies are needed to clarify the atypical immune reaction results seen in our study (negative ELISPOT in the presence of T-cell infiltration). A T-cell–mediated response has been shown to erase AAV-transduced hepatocytes within 2 months of gene transfer in a hemophilia trial.41 It is puzzling that T-cell infiltration did not eliminate AAV transduction in our study. Nevertheless, our results are in line with another muscle-directed AAV trial in which Flotte and colleagues found that the T-cell response did not compromise AAV transduction following direct muscle injection.42
In contrast to the disappointing outcome in previous dog studies by Sampaolesi et al. and Kornegay et al.,9,10 our results suggest that microdystrophins can at least partially protect skeletal muscle in large mammals. While precise reasons remain to be determined, we suspect it may relate to the experimental design/protocol such as the differences in immune suppression regimen, gene delivery method (stem cell versus AAV, AAV-9 versus tyrosine mutant AAV-9), and dog age (newborn versus adult). Another important factor is the microdystrophin construct, especially the origin (human gene versus canine gene) and configuration (specific repeats and hinges used in the synthetic microgene, number of repeats and hinges etc.). Different regions of the dystrophin rod domain exhibit great disparities in their biophysical properties and/or function.3,43,44 Further, the arrangement of non-native protein junction also influences the folding and stability of the synthetic dystrophin proteins.43,45,46,47
In summary, our results have cleared uncertainty on microdystrophin therapy arisen from other dog studies.9,10 However, compared with what was reported in the mouse model, the improvement we saw in dystrophic dogs remained suboptimal. Future studies are needed to optimize the microgene configuration, expression cassette, AAV serotype, and immune suppression protocol.
Materials and Methods
AAV microdystrophin vector. The codon-optimized canine microgene ΔR2-15/ΔR18-19/ΔR20-23/ΔC was synthesized by GenScript (Piscataway, NJ). It contains the N-terminal domain, three hinges (H1, H3, and H4), four spectrin-like repeats (R1, R16, R17, and R24), and the cysteine-rich domain. To unambiguously detect microdystrophin, we engineered a flag tag at the N-terminus and a Dys-2 epitope at the C-terminus. The expression cassette was under transcriptional regulation of the cytomegalovirus promoter and the SV40 pA. The AAV plasmid was called pSJ46. Endotoxin-free Y731F tyrosine mutant AAV-9 was generated and titrated using our published protocol.48 The stock virus titer was 1.625 × 1013 vg particles/ml. The biological activity and the safety of the virus were evaluated by systemic injection of stock virus to mdx mice (Figure 1).3 We did not observe any gross abnormality in injected mice. Histological examination of the liver and kidney was not remarkable (data not shown).
Experimental mice and gene transfer. All mouse experiments were approved by the Animal Care and Use Committee of the University of Missouri and were performed in accordance with National Institutes of Health guidelines. Experimental dystrophin null mdx mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were housed in a specific pathogen-free animal facility. Only male mice were used in the study. For systemic gene transfer, 6.5 × 1012 vg particles of codon-optimized canine ΔR2-15/ΔR18-19/ΔR20-23/ΔC microgene Y731F tyrosine mutant AAV-9 viruses were injected to 5-week-old male mdx mice in a total volume of 100 µl via the tail vein. At 2 months after AAV gene transfer, muscles were harvested and muscle pathology and force were examined. Age- and sex-matched untreated mdx mice were used as the control.
Experimental dogs and gene transfer. All dog experiments were approved by the Animal Care and Use Committee of the University of Missouri and were performed in accordance with National Institutes of Health guidelines. Experimental dogs were produced and genotyped as described before.49 A total of 22 dogs were recruited for the study (Table 2). Six affected dogs received AAV microdystrophin injection under a 5-week transient immune suppression using cyclosporine (10–20 mg/kg/day, Novartis, East Hanover, NJ) and mycophenolate mofetil (40 mg/kg/day; Genentech, South San Francisco, CA) (Table 1).21 For AAV injection, dogs were gently restrained on the surgery table. The ECU muscle was identified by palpating the anatomical markers on the body surface. The proximal end of the ECU muscle was located at the lateral epicondyle of the humerus at the elbow. The distal end of the ECU muscle was located at the styloid process of the ulna at the wrist. A total of 2.6 × 1013 vg particles were delivered to each ECU muscle in four separate injections in a total volume of 1.6 ml.
Mouse muscle function assay. Experimental mice were anesthetized via intraperitoneal injection of a cocktail containing 25 mg/ml ketamine, 2.5 mg/ml xylazine, and 0.5 mg/ml acepromazine at 2.5 µl/g body weight. The extensor digitorum longus muscle was gently dissected and mounted to an intact muscle test system (Aurora Scientific, Aurora, Ontario, Canada).50 The maximum isometric tetanic force was measured at 150 Hz. Specific muscle force was determined by dividing the maximum isometric tetanic force with the muscle cross-sectional area as we described before.50 The eccentric contraction protocol was performed according to our published protocol.50
Dog muscle function assay. Dogs were sedated with ketamine (15 mg/kg body weight) and acepromazine (0.12 mg/kg body weight). Anesthesia was induced with 4% isoflurane and maintained with 2% isoflurane. Dogs were intubated and the breath rate was set at 15–18 per minute and the tidal volume at 10–15 ml/minute/kg body weight. A catheter was placed in the thoracic aorta to monitor blood pressure. Another catheter was placed in the jugular vein for lactated saline infusion. Core body temperature was maintained at 37 °C during the assay. The subject was placed in the supine position on the in situ muscle function assay force transducer plate. The entire ECU muscle was exposed and the muscle length was measured. The distal ECU tendon was cut at its insertion and fastened onto the force transducer (SM-250-38; Interface, Scottsdale, AZ). The forearm was subsequently fixed with two bone pins to align the ECU muscle with the force transducer. The radial nerve was carefully dissected and mounted on a bipolar electrode. Muscle force was measured using 8 V and 0.2 ms pulse duration electric stimulation (Grass S48 Stimulator; Grass Instruments, Quincy, MA). The optimal muscle length (Lo) was determined using single twitch stimulation. The tetanic force was determined by applying 200 ms tetanic stimulation at the frequency of 100 Hz. The physiological cross-sectional area was calculated according to the equation physiological cross-sectional area = (muscle weight in gram × cos10.03)/(1.056 g/cm3 × optimal fiber length in cm). 1.0597 g/cm3 is the muscle density. 10.03 is the pennation angle of the ECU muscle.22 The optimal fiber length (Lf) was determined by multiplying the measured Lo of the muscle by the ECU muscle Lf/Lo ratio of 0.0448.22 The specific muscle force was calculated by normalizing the tetanic muscle force by the physiological cross-sectional area.
For eccentric contraction, the ECU muscle was stimulated for a total of 1,200 ms. In the last 1,100 ms, the ECU muscle was stretched by ~5% of its original length. After a 2-minute rest, a second cycle of eccentric contraction was applied. A sequence of eccentric contraction was conducted in each ECU muscle. The percentage of force drop after each cycle of eccentric contraction was calculated.
At the end of study, the subject was euthanized with Euthasol (Virbac Animal Health, Fort Worth, TX). Tissues were collected for subsequent studies.
Morphology studies. General muscle histopathology was revealed with hematoxylin and eosin staining. Nonspecific esterase staining, Alizarin red staining, and Masson trichrome staining were used to reveal macrophage infiltration, myofiber calcification, and muscle fibrosis according to our published protocols.26 Macrophage infiltration was further confirmed by immunostaining with the canine macrophage-specific antibody (1:2,000; AbD Serotec, Raleigh, NC). CD4+ and CD8+ T cells were detected by immunohistochemical staining using the canine-specific antibodies at the dilution of 1:1,000 and 1:200, respectively (AbD Serotec). The cross-sectional area of individual myofiber was determined from the digitized images using the quantitative image analysis module of the extended version of the Photoshop CS5.5 software (Adobe Systems, San Jose, CA). Dystrophin expression was detected using seven dystrophin-specific antibodies (two polyclonal and five monoclonal). The N-terminal domain of dystrophin was detected with a rabbit polyclonal antibody (1:600) (a gift from Dr Jeffrey Chamberlain at the University of Washington). Spectrin-like repeats 4-6 (R4-6) was detected with the H-300 rabbit polyclonal antibody (1:400; Santa Cruz Biotechnology, Santa Cruz, CA). Hinge 1 (H1) was detected with DysB (1:80; Novocastra, Newcastle, UK). R6-8 was detected with Dys1 (1:100; Vector Laboratories, Burlingame, CA). R11 was detected with Mandys8 (1:200; Sigma, St Louis, MO). R16 was detected with Mandys103 (1:20; a gift from Dr Glenn Morris at the Robert Jones and Agnes Hunt Orthopaedic Hospital, Oswestry, UK). Dys-2 epitope at the C-terminal end was detected with Dys2 (1:20; Novocastra). The flag tag was revealed with the anti-FLAG M2 antibody (1:400; Sigma). The DGC components were evaluated with monoclonal antibodies against β-dystroglycan (1:50; Novocastra), β-sarcoglycan (1:50; Novocastra), dystrobrevin (1:200; BD Biosciences, San Diego, CA), and syntrophin (1:200; Abcam, Cambridge, MA). Laminin was revealed with a rabbit polyclonal antibody (1:200; Sigma). Utrophin was examined with a mouse monoclonal antibody against the utrophin N-terminal domain (1:20; Vector Laboratories). Embryonic myofiber was detected with a monoclonal antibody against eMHC (1:250; Developmental Studies Hybridoma Bank, Iowa City, IA). The quantity of the eMHC-positive myofiber was determined by dividing the eMHC-staining positive area with the total cross-sectional area of the entire muscle section.
Western blot. Whole muscle lysate (100 µg/lane) was loaded on an 8% sodium dodecyl sulfate-polyacrylamide gel and protein was transferred to a polyvinylidene difluoride membrane. Dystrophin was detected with an antibody against the Dys2 epitope (1:100; Novocastra). As a loading control, membrane was also probed with an anti-α-tubulin antibody (1:3,000; Sigma). The full-length dystrophin protein migrated at 427 kD. The canine ΔR2-15/ΔR18-19/ΔR20-23/ΔC microdystrophin protein migrated at 140 kD. α-Tubulin migrated at 50 kD.
AAV genome copy determination. The AAV genome copy in muscle was determined by quantitative PCR in an ABI 7900 HT QPCR machine (Applied Biosystems, Foster City, CA).48 The primers are DL560 (TTACGGTAAACTGCCCACTTG) and DL561 (CATAAGGTCAT GTACTGGGCATAA). The threshold cycle value was converted to copy number by measuring against the copy number standard curve of known amount of pSJ46 plasmid. The copy number was normalized by DNA concentration.
Interferon-γ ELISPOT assay. Splenocytes were isolated from the spleen strips (1 × 2 × 3 cm) freshly harvested at the time of necropsy. The 15-mer peptide library was made for codon-optimized canine microdystrophin and the AAV-9 VP1 capsid protein (Mimotopes, Clayton, Australia). In the library, each peptide shared 10 amino acids overlap with the neighboring peptides. The microdystrophin peptide library was comprised of 237 peptides and these peptides were grouped into five pools with pool A containing peptide 1 to 47, pool B containing peptide 48 to 95, pool C containing peptide 96 to 142, pool D containing peptide 143 to 188, and pool E containing peptide 189 to 237. A total of 146 peptides were made for the AAV-9 capsid library and they were divided into three pools with pool A containing peptide 1 to 50, pool B containing peptide 51 to 100, and pool C containing peptide 101 to 146. The entire lyophilized library was reconstituted with dimethyl sulfoxide to reach a concentration of 100 mg/ml for each peptide. In the peptide pool, the concentration of each peptide was 2 mg/ml. To test the T-cell response, splenocytes were incubated with the peptide pools at the individual peptide concentration of 2 µg/ml. No peptide stimulation (medium containing dimethyl sulfoxide at the same concentration used for peptide stimulation) was used as the negative control. Phorbol 12-myristate 13-acetate (50 ng/ml, Sigma) and ionomycin (1 µg/ml; Sigma) were used as the positive control. Interferon-γ ELISPOT was set up using the canine ELISPOT EL781 kit following instructions from the manufacturer (R&D Systems, Minneapolis, MN). Spot formation was quantified with an immunospot reader (CTL, Cleveland, OH). A positive T-cell response was defined as a value at least three times higher than the background (medium alone control). Results are presented as spot-forming units per 106 splenocytes for experimental groups and as spot-forming units per 2 × 104 splenocytes for the phorbol 12-myristate 13-acetate/ionomycin control.
Statistical analysis. Data are presented as mean ± SEM. For multiple group comparison, we analyzed data by one-way analysis of variance using the IBM SPSS software (IBM, Armonk, NY). Statistical difference between treated and untreated muscle of the same dog was assessed with paired Student's t-test. A P < 0.05 was considered statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. Schematic outline of the experimental timeline. Figure S2. AAV gene transfer results in widespread microdystrophin expression in dystrophic dog ECU muscle. Figure S3. Microdystrophin gene therapy reduces macrophage infiltration in dystrophic dog muscle.
Acknowledgments
This work was supported by grants from the National Institutes of Health AR-49419 (D.D.), HL-91883 (D.D.), Jessey's Journey-The Foundation for Cell and Gene Therapy (D.D.), and the Muscular Dystrophy Association (D.D.). We acknowledge the Gene Therapy Resource Program of the National Heart, Lung, and Blood Institute, National Institutes of Health, and the Immunology Core at the Gene Therapy Program of the University of Pennsylvania. The authors thank Bruce Smith, Dawna Voelkl, Scott Korte, Lonny Dixon, Arun Srivastava, Jeffrey Chamberlain, Glenn Morris, and Marianne Hakim for providing experimental reagents and/or technical support. The authors declared no conflict of interest.
Supplementary Material
REFERENCES
- Duan D. Duchenne muscular dystrophy gene therapy: Lost in translation. Res Rep Biol. 2011;2011:31–42. doi: 10.2147/RRB.S13463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain JS. Gene therapy of muscular dystrophy. Hum Mol Genet. 2002;11:2355–2362. doi: 10.1093/hmg/11.20.2355. [DOI] [PubMed] [Google Scholar]
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C.et al. (2009Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy J Clin Invest 119624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu M., and, Duan D. C-terminal-truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double-knockout mice. Mol Ther. 2006;14:79–87. doi: 10.1016/j.ymthe.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L.et al. (2006rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice Nat Med 12787–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Kissel JT, Amato AA, King W, Signore L, Prior TW.et al. (1995Myoblast transfer in the treatment of Duchenne's muscular dystrophy N Engl J Med 333832–838. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM.et al. (2008A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy Ann Neurol 63561–571. [DOI] [PubMed] [Google Scholar]
- Koo T, Okada T, Athanasopoulos T, Foster H, Takeda S., and, Dickson G. Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. J Gene Med. 2011;13:497–506. doi: 10.1002/jgm.1602. [DOI] [PubMed] [Google Scholar]
- Kornegay JN, Li J, Bogan JR, Bogan DJ, Chen C, Zheng H.et al. (2010Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs Mol Ther 181501–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaolesi M, Blot S, D'Antona G, Granger N, Tonlorenzi R, Innocenzi A.et al. (2006Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs Nature 444574–579. [DOI] [PubMed] [Google Scholar]
- England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE.et al. (1990Very mild muscular dystrophy associated with the deletion of 46% of dystrophin Nature 343180–182. [DOI] [PubMed] [Google Scholar]
- Arikawa-Hirasawa E, Koga R, Tsukahara T, Nonaka I, Mitsudome A, Goto K.et al. (1995A severe muscular dystrophy patient with an internally deleted very short (110 kD) dystrophin: presence of the binding site for dystrophin-associated glycoprotein (DAG) may not be enough for physiological function of dystrophin Neuromuscul Disord 5429–438. [DOI] [PubMed] [Google Scholar]
- Den Dunnen JT, Grootscholten PM, Bakker E, Blonden LA, Ginjaar HB, Wapenaar MC.et al. (1989Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications Am J Hum Genet 45835–847. [PMC free article] [PubMed] [Google Scholar]
- Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T.et al. (1989The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion Am J Hum Genet 45498–506. [PMC free article] [PubMed] [Google Scholar]
- Fanin M, Freda MP, Vitiello L, Danieli GA, Pegoraro E., and, Angelini C. Duchenne phenotype with in-frame deletion removing major portion of dystrophin rod: threshold effect for deletion size. Muscle Nerve. 1996;19:1154–1160. doi: 10.1002/mus.880190902. [DOI] [PubMed] [Google Scholar]
- Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, United Dystrophinopathy Project Consortium et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–1666. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo Y, Muntoni F, Sewry C, Legum C, Kutai M, Harel S.et al. (2003Large in-frame deletions of the rod-shaped domain of the dystrophin gene resulting in severe phenotype Isr Med Assoc J 594–97. [PubMed] [Google Scholar]
- Winnard AV, Klein CJ, Coovert DD, Prior T, Papp A, Snyder P.et al. (1993Characterization of translational frame exception patients in Duchenne/Becker muscular dystrophy Hum Mol Genet 2737–744. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Burghes AH, Mora M, Tomé FM, Morandi L, Cornello F.et al. (1994Immunohistochemical analysis of dystrophin-associated proteins in Becker/Duchenne muscular dystrophy with huge in-frame deletions in the NH2-terminal and rod domains of dystrophin J Clin Invest 9399–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M.et al. (2008Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses Proc Natl Acad Sci USA 1057827–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Yue Y, Srivastava A, Smith B, Lai Y., and, Duan D. A simplified immune suppression scheme leads to persistent micro-dystrophin expression in Duchenne muscular dystrophy dogs. Hum Gene Ther. 2012;23:202–209. doi: 10.1089/hum.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HT, Shin JH, Hakim CH, Pan X, Terjung RL., and, Duan D. Dystrophin deficiency compromises force production of the extensor carpi ulnaris muscle in the canine model of Duchenne muscular dystrophy. PLoS ONE. 2012;7:e44438. doi: 10.1371/journal.pone.0044438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrof BJ, Shrager JB, Stedman HH, Kelly AM., and, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S.et al. (2010Dystrophin immunity in Duchenne's muscular dystrophy N Engl J Med 3631429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Storb R, Halbert CL, Banks GB, Butts TM, Finn EE.et al. (2012Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies Mol Ther 201501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BF, Yue Y, Woods PR, Kornegay JN, Shin JH, Williams RR.et al. (2011An intronic LINE-1 element insertion in the dystrophin gene aborts dystrophin expression and results in Duchenne-like muscular dystrophy in the corgi breed Lab Invest 91216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KE., and, Grounds MD. Treating muscular dystrophy with stem cells. Cell. 2006;127:1304–1306. doi: 10.1016/j.cell.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Sharma KR, Mynhier MA., and, Miller RG. Cyclosporine increases muscular force generation in Duchenne muscular dystrophy. Neurology. 1993;43 3 Pt 1:527–532. doi: 10.1212/wnl.43.3_part_1.527. [DOI] [PubMed] [Google Scholar]
- Weller B, Massa R, Karpati G., and, Carpenter S. Glucocorticoids and immunosuppressants do not change the prevalence of necrosis and regeneration in mdx skeletal muscles. Muscle Nerve. 1991;14:771–774. doi: 10.1002/mus.880140812. [DOI] [PubMed] [Google Scholar]
- De Luca A, Nico B, Liantonio A, Didonna MP, Fraysse B, Pierno S.et al. (2005A multidisciplinary evaluation of the effectiveness of cyclosporine a in dystrophic mdx mice Am J Pathol 166477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerletti M, Negri T, Cozzi F, Colpo R, Andreetta F, Croci D.et al. (2003Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer Gene Ther 10750–757. [DOI] [PubMed] [Google Scholar]
- Dell'Agnola C, Wang Z, Storb R, Tapscott SJ, Kuhr CS, Hauschka SD.et al. (2004Hematopoietic stem cell transplantation does not restore dystrophin expression in Duchenne muscular dystrophy dogs Blood 1044311–4318. [DOI] [PubMed] [Google Scholar]
- Rouger K, Larcher T, Dubreil L, Deschamps JY, Le Guiner C, Jouvion G.et al. (2011Systemic delivery of allogenic muscle stem cells induces long-term muscle repair and clinical efficacy in Duchenne muscular dystrophy dogs Am J Pathol 1792501–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner J, Schessl J, Schara U, Reitter B, Stettner GM, Hobbiebrunken E.et al. (2010Treatment of Duchenne muscular dystrophy with ciclosporin A: a randomised, double-blind, placebo-controlled multicentre trial Lancet Neurol 91053–1059. [DOI] [PubMed] [Google Scholar]
- Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC., and, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol. 2000;150:1399–1410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe ER, Towbin J, Chamberlain J, Baumbach L, Witkowski J, van Ommen GJ.et al. (1989Complementary DNA probes for the Duchenne muscular dystrophy locus demonstrate a previously undetectable deletion in a patient with dystrophic myopathy, glycerol kinase deficiency, and congenital adrenal hypoplasia J Clin Invest 8395–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ., and, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34:135–144. doi: 10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- Tuffery-Giraud S, Béroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L.et al. (2009Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase Hum Mutat 30934–945. [DOI] [PubMed] [Google Scholar]
- Prior TW, Bartolo C, Pearl DK, Papp AC, Snyder PJ, Sedra MS.et al. (1995Spectrum of small mutations in the dystrophin coding region Am J Hum Genet 5722–33. [PMC free article] [PubMed] [Google Scholar]
- Deburgrave N, Daoud F, Llense S, Barbot JC, Récan D, Peccate C.et al. (2007Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene Hum Mutat 28183–195. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, Maus MV, Hui DJ, Sabatino DE, Murphy SL, Rasko JE.et al. (2007CD8(+) T-cell responses to adeno-associated virus capsid in humans Nat Med 13419–422. [DOI] [PubMed] [Google Scholar]
- Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT.et al. (2009Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy Proc Natl Acad Sci USA 10616363–16368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza A, Sagathevan M, Sahni N, Choi L., and, Menhart N. A biophysical map of the dystrophin rod. Biochim Biophys Acta. 2010;1804:1796–1809. doi: 10.1016/j.bbapap.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Banks GB, Judge LM, Allen JM., and, Chamberlain JS. The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet. 2010;6:e1000958. doi: 10.1371/journal.pgen.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni N, Mangat K, Le Rumeur E., and, Menhart N. Exon edited dystrophin rods in the hinge 3 region. Biochim Biophys Acta. 2012;1824:1080–1089. doi: 10.1016/j.bbapap.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Henderson DM, Belanto JJ, Li B, Heun-Johnson H., and, Ervasti JM. Internal deletion compromises the stability of dystrophin. Hum Mol Genet. 2011;20:2955–2963. doi: 10.1093/hmg/ddr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruszczak C, Mirza A., and, Menhart N. Differential stabilities of alternative exon-skipped rod motifs of dystrophin. Biochim Biophys Acta. 2009;1794:921–928. doi: 10.1016/j.bbapap.2009.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Yue Y., and, Duan D. Recombinant adeno-associated viral vector production and purification. Methods Mol Biol. 2012;798:267–284. doi: 10.1007/978-1-61779-343-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine DM, Shin JH, Yue Y, Volkmann D, Leach SB, Smith BF.et al. (2011Age-matched comparison reveals early electrocardiography and echocardiography changes in dystrophin-deficient dogs Neuromuscul Disord 21453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim CH, Li D., and, Duan D. Monitoring murine skeletal muscle function for muscle gene therapy. Methods Mol Biol. 2011;709:75–89. doi: 10.1007/978-1-61737-982-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.