

Abstract
L-dopa-induced dyskinesia in Parkinson’s disease (PD) is a major clinical problem. The prevailing view is that in PD patients and animal PD models dyskinesia develops after repeated L-dopa use or priming, independent of L-dopa’s anti-PD therapeutic effect that occurs immediately. Here we show that in mice with severe and consistent dopamine (DA) loss in the dorsal striatum, rendered by transcription factor Pitx3 null mutation, the very first injection of L-dopa or D1-like agonist SKF81297 induced both normal ambulatory and dyskinetic movements. Furthermore, the robust stimulating effects on normal and dyskinetic movements had an identical time course and parallel dose-response curves. In contrast, D2-like agonist ropinirole stimulated normal and dyskinetic movements relatively modestly. These results demonstrate that severe DA loss in the dorsal striatum sets the stage for dyskinesia to occur on the first exposure to L-dopa or a D1 agonist without any priming. These results also indicate that L-dopa stimulated both normal and dyskinetic movements primarily via D1 receptor activation and that proper D1 agonism is potentially an efficacious therapy for PD motor deficits.
Keywords: basal ganglia; L-3,4-dihydroxyphenylalanine (L-dopa); dopamine D1 receptor; dyskinesia; medium spiny neuron; Parkinson’s disease
Introduction
In PD, the degeneration of the nigrostriatal DA system causes motor control dysfunction (Hornykiewicz, 2001). Since its introduction more than 40 years ago, the DA precursor L-dopa has been the most effective treatment for the motor symptoms of PD (Cotzias et al., 1969; Standaert and Young, 2006; LeWitt, 2009). L-dopa’s therapeutic anti-PD effect occurs immediately upon the first dose of L-dopa and is mediated by activating DA receptors. However, precisely which DA receptor type is primarily responsible for L-dopa’s anti-PD effect is usually not defined in the literature (Fahn and Przedborki, 2005; Standaert and Young, 2006; LeWitt, 2009; Olanow et al., 2009). The use of D2-like agonists such as ropinirole but not D1-like agonists (Jenner, 2003) to treat PD motor symptoms gives an impression that DA D2 type receptors (D2Rs) in the indirect pathway are the primary mediator of the therapeutic effects of L-dopa. Experimental studies, however, have demonstrated an overwhelming importance of the D1R-expressing medium spiny neurons (D1-MSNs) in the direct pathway to promoting motor activity and designated the direct pathway as the Go pathway (Chevalier and Deniau, 1990; DeLong, 1990; Hikosaka et al., 2000; Bateup et al., 2010; Kravitz et al., 2010; Berthet et al., 2012). Therefore, we reason that D1Rs may be primarily responsible for L-dopa’s anti-PD effect.
Another important question is about the mechanisms underlying the motor side effects of L-dopa. In advanced stage PD patients, L-dopa often triggers abnormal involuntary movements, known as L-dopa-induced dyskinesias (LID) (Ahlskog and Muenter, 2001; Fahn, 2008), thus limiting the use of an otherwise efficacious drug (Olanow et al., 2009). Although D1Rs are known to be critically involved, the mechanisms underlying LID are not fully understood (Cenci et al., 2010; LeWitt, 2010; Iravani and Jenner 2011; Smith et al., 2012). Because LID often occurs after long-term, repeated use of L-dopa, it is commonly thought that L-dopa’s therapeutic effect and dyskinetic side effect are mediated by separate mechanisms with the therapeutic effect occurring immediately whereas the dyskinetic effect developing only after repeated L-dopa use or priming (Jenner, 2008). This prevailing view advocates delaying L-dopa use, allowing the DA-depleted basal ganglia to undergo compensatory and also detrimental maladaptive changes (Schapira and Obeso, 2006). It also leads to a considerable research effort seeking to either stimulate or suppress one effect without affecting the other (Bezard et al., 2003; Santini et al., 2009; Gottwald and Aminoff, 2011; Huot et al., 2011). However, since L-dopa/DA likely activates the same DA receptors in the basal ganglia, the normal and dyskinetic motor control signals may be generated by at least partially shared mechanisms and thus intrinsically linked and difficult to separate. Indeed, clinical studies indicate that measures that reduce LID often worsen PD symptoms, i.e., dyskinetic and normal movements change in the same direction (Katzenschlager et al., 2008; Nutt, 2008). Further, it has been reported that after LID’s first appearance, L-dopa’s anti-PD effect and dyskinetic effect occurred with identical temporal profile (Nutt et al., 2010). These clinical observations suggest a common pharmacological mechanism that stimulates both normal and dyskinetic movements. However, because pharmacological manipulation is not practical in PD patients, the underlying pharmacological mechanism was not investigated. In this study, we seek to determine this shared pharmacological mechanism. Based on literature data and our reasoning in the preceding section that D1Rs may be primarily responsible for L-dopa’s therapeutic effect, we now further hypothesize that D1Rs are the primary mediator of both LID and the therapeutic effect such that these two effects have identical time courses and dose-response curves. We will perform our experiments in transcription factor Pitx3 mutant mice, taking advantage of their consistent DA loss in the dorsal striatum and their robust L-dopa-induced motor effects.
Experimental procedures
Animals
Pitx3 mutant mice were used for the following reasons. In the brain, the transcription factor Pitx3 gene is expressed only in midbrain DA neurons (Smits et al., 2006). Loss of Pitx3 gene function leads to the death of the vast majority of nigral DA neurons shortly after their birth, whereas many DA neurons in the ventral tegmental area survive (Nunes et al., 2003; van den Munckhof et al., 2003; Smidt et al., 2004; Smits et al., 2006). Consequently, these mice have a severe and consistent DA deficiency in the dorsal striatum (Fig. 1), leading to robust and consistent L-dopa responses including LID (Hwang et al., 2005; Ding et al., 2007, 2011). Thus, these mice are suitable for our questions.
Fig. 1.

Severe DA loss in the dorsal striatum in PixtHomo mice. (A) A confocal image of 3 μm optical section showing the typical intense DA axons as labeled by tyrosine hydroxylase (TH) immunostain in the entire striatum in a PitxWT mouse. The box area is expanded and displayed in A1. (B) A confocal image, obtained under identical conditions as in A, evidenced by the identical background signal in the non-striatal areas, showing the typical gradient pattern of DA axon loss in the striatum in PitxHomo mice. The dorsal striatum is largely void of DA axons, the middle striatum retains a significant number of DA axons, whereas the ventral striatum or the nucleus accumbens retains substantial amounts of DA axons. The box area is expanded and displayed in B1. (C) The intensity of DA axons in the striatal subregions was quantified by calculating the fractional area occupied by DA axons in 4 PitxWT mice (16 sections) and 4 PitxHomo mice (16 sections). All sections were from the middle part of the striatum on the anterior-posterior axis. AC, anterior commissure. OT, olfactory tubercle. **, p<0.01, t-test.
Two breeding pairs of heterozygous Pitx3+/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME), resulting in a small colony of homozygous Pitx3−/− (PitxHomo), heterozygous Pitx3+/−, and wild-type Pitx3+/+ (PitxWT) mice. The genotypes were determined by PCR-based genotyping to identify WT, homozygotes, and heterozygotes. PitxHomo mice are also aphakia and thus clearly identified. PitxHomo mice are highly viable and fertile. Mice had free access to food and water. Male mice of 11–12 weeks old were used for testing. All procedures were approved by The Institutional Animal Care and Use Committee of The University of Tennessee Health Science Center in Memphis, Tennessee.
Treatment regimen
L-3,4-dihydroxyphenylalanine methyl ester hydrochloride (L-dopa), benserazide hydrochloride, D1-like agonist SKF81297 hydrobromide, tyrosine hydroxylase inhibitor α-methyl-DL-tyrosine methyl ester hydrochloride (AMPT) and ropinirole hydrochloride were either purchased from Sigma-Aldrich (St. Louis, MO) or supplied by the Drug Supply Program of the National Institute of Mental Health. These drugs were dissolved in 0.9% saline and delivered to the mice via intraperitoneal (IP) injection. L-dopa was always injected together with 5 mg/kg benserazide to inhibit peripheral dopa decarboxylase. L-dopa, SKF81297 and ropinirole were injected twice a day (the first injection was in the morning and the second one in the late afternoon). To avoid potential dopaminergic sensitization effects, each of these drugs was injected into a fresh and separate group of mice. For dose-response experiments, each dose was also tested in a fresh and separate group of mice. The number of mice in each group was noted where the treatment was described. AMPT treatment (30 mg/kg, 1 injection/day in the morning) started 5 days before L-dopa to allow the mice to acclimatize. Peak AMPT effect in PitxHomo mice was reached 1 hour after injection and the peak effect lasted for ~ 2.5 hours, and then recovered gradually. Experiments were performed during the peak effect time window. Behavioral tests were performed between 9 AM and 1 PM.
Monitoring and quantification of motor activity
Ambulatory normal motor activity was monitored and quantified by an Activity Monitor system (Med Associates, St. Albans, VT). Horizontal movements are the most common natural motor activity in rodents and are equivalent to walking in humans (Kuoppamaki et al., 2007; Fox and Lang, 2008; Stockwell et al., 2008; Taylor et al., 2010; Moretti et al., 2011). Horizontal movement speed was used as a measure for normal movements. The speed was calculated by dividing the horizontal travel distance by the time the mouse was in horizontal positions, thus excluding the interference from the time used for dyskinesia. The abnormal involuntary movements or dyskinesias consisted mainly of abnormal stereotypic paw movements combined with abnormal stereotypic vertical trunk movements (Movie 1, video still 1). These events are qualitatively identical to the dyskinetic events described by Ding et al. (2007). Quantification of dyskinetic events was performed manually, following the published method of Ding et al. (2007). We placed individual mice in home cages and video-recorded their motor activity before and after injection of L-dopa or other drugs. A 10-min video 30 min after L-dopa injection was manually analyzed off-line to calculate the total dyskinesia duration that included one-paw, two-paw, and three-paw and occasional 4-paw dyskinetic events. When necessary, videos were played and viewed at slow motion or frame-by-frame to identify the start and end of the abnormal paw movements. The abnormal trunk movements were not used because it can not be clearly separated from the normal rearings. Also, we did not use the dyskinesia rating system because it was developed for unilateral DA-lesioned rodent PD models (Lundblad et al. 2005) and is not suitable for animals with bilateral DA loss such as PitxHomo mice. As indicated by Ding et al. (2007), dyskinesia duration provides a direct measure of the dyskinesia in PitxHomo mice.
For the challenging beam test, a 106 cm long beam was precision custom-made using the design and materials described by Fleming et al. (2004). Baseline PitxHomo mice were trained to traverse the beam in 5 sessions per day for 2 days for a total of 10 sessions, starting at the widest end (3.5 cm in width) and finishing at the narrowest end (0.5 cm in width). In each session, the mouse was required to traverse the beam 5 times. The traversal time was counted with a hand-held timer. By 6th or 7th training session, in different mice, the traversal time had plateaued. Thus, the baseline beam traversal time was measured on 11th session on day 3. Then the AMPT treatment and dopaminergic (L-dopa or SKF81297 or ropinirole) treatment were started, as described in the Treatment Regimen section, while a daily maintenance session of challenging beam traversal was performed. Since dopaminergic responses stabilized within 4 days after starting treatment, the challenging beam test was performed on treatment day 5.
Immunohistochemistry
Conventional immunofluorescence methods were used to detect DA axons in the striatum (Zhou et al., 2009). The brains were fixed in 4% paraformaldehyde dissolved in a phosphate buffer at 4°C overnight and then sectioned on a vibratome. The free-floating sections (50 μm in thickness) were incubated with 2% fat-free milk, 1% bovine serum albumin, and 0.4% Triton X-100 in a phosphate buffered saline (PBS) for 1 h at room temperature to block nonspecific binding and permeate the cell membrane, respectively. After thorough rinsing, the free-floating sections were incubated for 48 h at 4°C with the primary antibody, a polyclonal tyrosine hydroxylase antibody raised in rabbit (diluted at 1:1000; Novus Biologicals, Littleton, CO), and then rinsed in the PBS, followed by incubating with a donkey anti-rabbit secondary antibody conjugated with Alexa Fluor 488 (diluted at 1:200; Invitrogen), for 3 h at room temperature. Fluorescence images were acquired on a Zeiss 710 confocal laser scanning microscope. Images were analyzed using ImageJ (http://rsbweb.nih.gov/ij/).
Fast cyclic voltammetry (FCV) in striatal brain slices
Coronal striatal brain slices (400 μm in thickness) were cut on a vibratome (VT1200S, Leica Microsystems, Wetzlar, Germany). FCV was performed at 30 °C, according to published methods (Zhou et al., 2001, 2005). Carbon fiber electrodes were home-made from P55S carbon fibers of 10 μm diameter (Amoco Polymers, Greenville, SC). A Multiclamp 700B patch clamp amplifier, pClamp 9 software and Digidata 1320A interface (Axon Instruments) were used to acquire and analyze data. The peaks of the voltammograms were plotted over time and converted to concentrations by post-experiment calibrating with 0.1–5 μM DA standards. Local electrical stimulation (0.2 ms, 0.05–0.15 mA, single pulse every 2 min) in the striatum was delivered through bipolar tungsten electrodes (Microprobes, Gaithersburg, MD). Stimuli of increasing intensity were used to evoked the maximal DA signal.
Statistics
Descriptive statistics was used to obtain mean and standard error (SE). Unpaired t test and one way ANOVA combined with post hoc Turkey test were used to compare measurements in different groups of mice. Correlation was determined by the Pearson correlation test. p < 0.05 was considered statistically significant. Calculation was performed using Origin (OriginLab, Northampton, MA) and StatMost (Dataxiom Software, Los Angeles, CA).
Results
Mild inhibition of DA synthesis renders PitxHomo mice hypokinetic
The DA innervation in the striatum in PitxHomo mice is lost in a gradient fashion with the loss in the upper dorsal striatum being virtually complete and with considerable residual DA axons in the middle and more ventral parts of the striatum (Fig. 1A–C). However, these mice do not show overt deficits in their natural locomotor activity (Beeler et al., 2009; Hwang et al., 2005), thereby complicating the observation and analysis of anti-PD effects and motor side effects produced by pharmacological agents. We reasoned that the significant residual DA innervation in the middle and ventral striatal subregions (Fig. 1) may release sufficient DA that may be able to support the basic normal motor function in these mice. Therefore, a further fine-graded reduction in the residual DA level may render these mice hypokinetic. Indeed, IP injection of 30 mg/kg tyrosine hydroxylase inhibitor AMPT, a low dose relative to the doses reported in the literature (Sotnikova et al. 2005), caused a clear hypokinesia in PitxHomo mice, reducing normal horizontal locomotion to about 25% of that in PitxWT littermates 1 hour after AMPT injection (Fig. 2A). The same 30 mg/kg AMPT dosing did not cause any significant motor effect in PitxWT mice (Fig. 2A). The commonly used dose of 200–250 mg/kg (e.g., Sotnikova et al., 2005) led to a complete immobility in PitxHomo mice and was lethal to these mice, whereas these same high doses of AMPT only caused a recoverable hypokinesia in PitxWT mice.
Fig. 2.

A small dose of AMPT renders PitxHomo mice hypokinetic. (A) IP injection of 30 mg/kg AMPT had no overt motor effect in PitxWT mice but induced a clear hypokinesia in PitxHomo mice. Measurements were taken 1 hour after saline or AMPT injection. Five mice in each group. **, p<0.01, t-test. (B) Example FCV records (B1) and summary FCV data (B2) showing the moderate reduction in DA release caused by 30 mg/kg AMPT pretreatment in 4 PitxWT (3 slices from each mouse) and 4 PitxHomo mice (3 slices from each mouse). All slices were from the middle part of the striatum on the anterior-posterior axis. The residual DA signal was still substantial in the striatal subregions in PitxWT mice. In contrast, in the striatal subregions in PitxHomo mice, the already diminished DA signal was further reduced by AMPT such the residual DA signal became very small. *, p<0.05, t-test.
Next, we used fast cyclic voltammetry (FCV) at the carbon fiber microelectrode to estimate the effect of AMPT on DA release in striatal brain slices. Mice received an IP injection of saline or 30 mg/kg AMPT and were sacrificed 1 hour later to make brain slices containing the striatum. FCV measurements detected a maximal DA release signal of 3.12±0.27 μM, 3.15±0.32 μM, and 2.98±0.31 μM in the dorsal and middle striatum and nucleus accumbens core (NAc), respectively, in 4 saline-treated PitxWT mice. In 4 saline-treated PitxHomo mice, FCV detected a maximal DA release signal of 0 μM, 0.22±0.02 μM, 1.22±0.13 μM DA in the dorsal and middle striatum and in NAc core, respectively (Fig. 2B1,2). As shown in Fig. 2B1,2, AMPT (30 mg/kg, IP) pretreatment reduced the DA signal by ~ 40% in the middle striatum and NAc in both PitxWT and PitxHomo mice. However, the absolute amplitude of the DA signal was still substantial (2 μM in the middle striatum) in PitxWT mice but very small (0.09 μM in the middle striatum) in PitxHomo mice. These results indicate that a low dose of AMPT further reduced DA availability in the already DA deficient middle and ventral striatal subregions such that the normal motor activity can no longer be maintained, leading to substantially reduced locomotor activity or hypokinesia (Fig. 2A). In contrast, in WT mice, after treatment with the same low dose (30 mg/kg) AMPT, the striatum still had sufficient DA to maintain normal motor function. These results also indicate that a small dose of AMPT adds a clear hypokinetic feature or face validity to PitxHomo mice, making these mice a more useful model to study the behavioral neuropharmacology of L-dopa-induced motor activity. For the convenience of description, hereafter we will refer to these 30 mg/kg AMPT-treated PitxHomo mice as AMPT-PitxHomo mice.
First injection of L-dopa or D1-like agonist SKF81297 triggers dyskinesia
For this set of experiments, we injected 6 mg/kg L-dopa (IP), a clinically relevant dose (Standaert and Young, 2006), together with 5 mg/kg benserazide, into L-dopa-naive mice. In pilot experiments, we visually observed (without quantification) that the very first injection of L-dopa stimulated normal and also dyskinetic movements in every AMPT-PitxHomo mice (n=20) but showed no overt effect in PitxWT mice (n=10). First L-dopa injection-induced LID was also observed in baseline PitxHomo mice by Ding et al. (2007). Next, we set out to quantify L-dopa’s motor effects. Pilot experiments indicated that L-dopa effects were at their peak around 30 min after injection. So we used a locomotor activity monitor to quantify L-dopa-induced normal horizontal movements during 30th and 39th min after injection. We found that the first injection of 6 mg/kg L-dopa increased the normal horizontal movement speed from 1.7 ± 0.2 cm/10 s during the 10 min before injection to 11.5 ± 1.3 cm/10 s during 30th and 39th min after injection (Fig. 3A). During the first 3 treatment days, L-dopa’s normal motor-stimulating effect increased with each injection and then was stable during our 2-week treatment period.
Fig. 3.
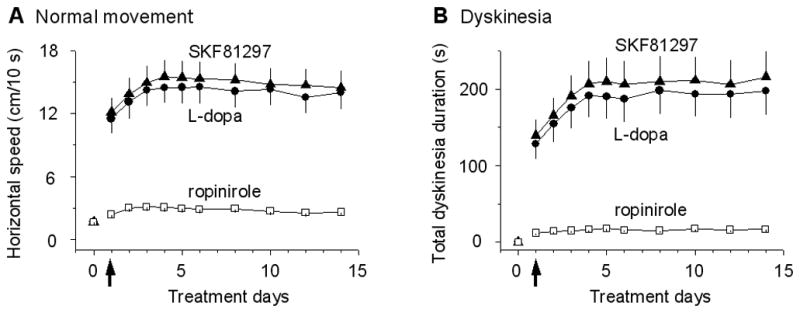
Motor effects of the first and subsequent injections of L-dopa in AMPT-PitxHomo mice. (A) The first IP injection of 6 mg/kg L-dopa with 5 mg/kg benserazide or 1 mg/kg SKF81297 induced strong normal horizontal movements in AMPT-PitxHomo mice. The first IP injection of 0.1 mg/kg ropinirole induced only weak responses in AMPT-PitxHomo mice. Note the increase in the responses during the first 3 days. Horizontal speed was measured during 30th and 39th min after injection. The horizontal speeds under L-dopa and SKF81297 were strongly correlated with a correlation coefficient of 1, but were weakly correlated with the horizontal speeds under ropinirole with a correlation coefficient of 0.61, Pearson test. The weak correlation is likely due to the coincidental drug injection and coincidental D1 and D2 activation and also indicates that L-dopa’s stimulating effect on horizontal motor activity was primarily mediated by D1Rs, not D2Rs. (B) In separate, fresh groups of AMPT-PitxHomo mice, the first IP injection of 6 mg/kg L-dopa with 5 mg/kg benserazide or 1 mg/kg SKF81297 induced dyskinetic movements. The first IP injection of 0.1 mg/kg ropinirole induced relatively weak responses in AMPT-PitxHomo mice. Dyskinesia duration was manually measured during 30th and 39th min after injection. N=5 mice for each data point. The 2 arrows indicate the first injections. Note dyskinetic movements increased during the first 3 days. The dyskinesia durations under L-dopa and SKF81297 were correlated with a strong correlation coefficient of 1, indicating a common D1R activation besides coincidental drug injection during these days. However, the dyskinesia duration under ropinirole was weakly correlated with the dyskinesia durations under L-dopa and SKF81297 with a correlation coefficient of 0.66, Pearson test. The weak correlation was likely due to coincidental drug injection during these days and also indicates that L-dopa’s effect was primarily mediated by D1Rs, not D2Rs.
Equally important, the very first IP injection of 6 mg/kg L-dopa also consistently induced dyskinetic movements with qualitative characteristics identical to those induced by repeated L-dopa described in Ding et al. 2007 (see also Movie 1). To quantify the dyskinetic movements induced by the first injection of L-dopa, we video-recorded the motor activity of individual AMPT-PitxHomo mice in their home cages for 10 min between 30th and 39th min after injection. We then reviewed the video recordings offline and manually counted and tallied the duration of dyskinetic events according to the method of Ding et al. (2007). As shown in Fig. 3B, under our experimental condition, upon the first IP injection of 6 mg/kg L-dopa, these mice displayed clear dyskinetic movements with a total dyskinetic duration of about 2 min during the 10 min recording period (Fig. 3B). During the first 3 days, the dyskinesia duration increased with each injection and plateaued on 4th day of treatment (Fig. 3B), but the qualitative characteristics of the dyskinetic events were constant during the entire treatment period. No dyskinetic event was seen in PitxWT mice upon L-dopa injection (1, 2, 4, 6, 10 and 20 mg/kg with 5 mg/kg benserazide, IP).
Because of the established movement-promoting function of the direct pathway D1R-MSNs that express the vast majority of brain’s D1Rs (Bateup et al., 2010; Kravitz et al., 2010), we reasoned that D1Rs in these D1-MSNs may contribute critically to the generation of the first L-dopa injection-stimulated normal and dyskinetic movements observed here. If this is correct, then a D1 agonist should mimic L-dopa’s effects. Indeed, as illustrated in Fig. 3A,B, IP injection of the D1-like agonist SKF81297 at 1 mg/kg clearly stimulated both normal horizontal movements and dyskinetic movements (Movie 1, video still 1) in AMPT-PitxHomo mice, almost fully mimicking the effects of 6 mg/kg L-dopa. In contrast, IP injection of D2-like agonist ropinirole (0.1 mg/kg) induced relatively modest normal movements and dyskinetic movements in AMPT-PitxHomo mice (Fig. 3A,B) (higher doses of ropinirole induced motor inhibition, see Movie 2 and also next section), suggesting a more limited role for D2 receptors in stimulating motor activity. Taken together, these results clearly show that the very first injection of L-dopa and a D1-like agonist SKF81297 can stimulate normal movements and trigger dyskinesia without any priming. The strong effects of SKF81297 and the weak effects of ropinirole indicate a primary role for D1Rs in stimulating normal and dyskinetic movements.
Identical time courses of L-dopa- and D1R agonism-stimulated normal and dyskinetic movements
Next, we set out to determine if the effects of L-dopa and SKF81297 on normal and dyskinetic movements are triggered by common pharmacological mechanisms. If they are, then these two effects should have similar time courses, although it is only a necessary but not a sufficient evidence. To test this possibility, we measured the time course of L-dopa’s effect on the normal horizontal and dyskinetic movements in AMPT-PitxHomo mice. Since L-dopa effects reached their peak and stabilized on treatment day 4 (Fig. 3), we performed our experiment during the 9th injection of L-dopa on treatment day 5. As shown in Fig. 4A1, the horizontal movement speed and dyskinesia duration started to increase simultaneously around 5 min after IP injection of 6 mg/kg L-dopa, reached their peak effects around 25 min after injection, and started to decline at about 60 min after injection. D1 agonist SKF81297 at 1 mg/kg IP injection also stimulated both normal and dyskinetic movements with a similar time course in AMPT-PitxHomo mice (Fig. 4B1). The weak motor-stimulating effects of ropinirole (0.1 mg/kg) also had similar time courses for normal and dyskinetic movements (Fig. 4C1). Correlation analysis indicated that the two time courses in each of the three time course pairs (Fig. 4A1, B1, C1) were correlated with correlation coefficients being between 0.87 to 1 (Pearson test). These result show that the time courses of normal and dyskinetic movements induced by L-dopa, SKF81297 and ropinirole were similar for each drug, consistent with, though not proving, the idea that normal and dyskinetic movements are triggered by pharmacological mechanisms with shared components. The similarly strong effects of L-dopa and D1 agonist SKF81297 and the weak effects of D2 agonist ropinirole indicate that D1Rs are the primary mediator of L-dopa’s stimulating effects on normal and dyskinetic movements.
Fig. 4.

Identical time course and parallel dose-response curves of L-dopa-stimulated normal and dyskinetic movements in AMPT-PitxHomo mice. (A1, B1, C1) L-dopa (6 mg/kg, A1), SKF81297 (1 mg/kg, B1) and ropinirole (0.1 mg/kg, C1) stimulated normal and dyskinetic movements with identical time courses for each drug. The normal horizontal speed was measured, using a Locomotor Monitor, during the 9th injection of L-dopa on treatment day 5 when the responses had plateaued and were stable. Dyskinesia duration was measured manually in separate groups of mice. N=5–6 mice for each data point. The correlation coefficient for the 3 pairs of curves was 1, 1 and 0.87, respectively (Pearson test). (A2, B2, C2) L-dopa (A2), SKF81297 (B2) and ropinirole (C2) stimulated normal and dyskinetic movements with parallel dose-response curves. The horizontal movement speed and the dyskinesia duration were measured during the 9th injection of L-dopa on treatment day 5 in separate groups of PitxHomo mice. N=4–6 mice for each data point. The correlation coefficient for the 3 pairs of curves was 0.98, 0.98 and 0.93, respectively, Pearson test.
Parallel dose-response curves of L-dopa- and D1R agonism-stimulated normal and dyskinetic movements
A similar time course of two different pharmacological effects may be due to the same pharmacokinetics of the drug activating different receptors. Therefore, to further determine if L-dopa- and SKF81297-induced normal and dyskinetic movements are mediated by the same D1R-dependent pharmacological mechanism, we next compared the dose-response relationships of the normal horizontal movements and dyskinetic movements in AMPT-PitxHomo mice. We first constructed doses-response curves for L-dopa. L-dopa stimulated both normal and dyskinetic movements at 0.5 mg/kg (injected with 5 mg/kg benserazide), the lowest dose we tested (Fig. 4A2). L-dopa’s stimulating effects on both normal and dyskinetic movements increased dose-dependently, reaching the peak effects at 20 mg/kg. As shown in Fig. 4A2, 40 mg/kg L-dopa did not further increase its motor-stimulating effects. Instead, an inhibitory component started to appear with both normal and dyskinetic movements becoming slow or sticky. Therefore, L-dopa had a biphasic dose-response relationship. However, the dose-response curves for L-dopa-stimulated normal horizontal movement speed and total dyskinesia duration were in parallel for the entire dose range tested, and when normalized, these two curves overlap.
We also constructed dose-response curves for D1-like agonist SKF81297- and D2-like agonist ropinirole-induced normal and dyskinetic movements. As illustrated in Fig. 4B2, SKF81297 robustly stimulated both normal horizontal movements and dyskinetic movements with parallel dose-response curves. Unlike L-dopa’s biphasic responses, SKF81297’s stimulating effects were monophasic (Fig. 4B2). In contrast to the robust motor-stimulating effects of L-dopa and SKF81297, D2-like agonist ropinirole at doses of 0.1 to 0.2 mg/kg stimulated normal and dyskinetic movements only modestly (Fig. 4C2). Higher (≥0.5 mg/kg) doses of ropinirole induced a slowness or stickiness in both normal and dyskinetic movements. At doses of ≥1 mg/kg, ropinirole often induced a complete stop or freezing of normal and dyskinetic movements (Movie 2, video still 2). A consistent observation was that when the normal horizontal movements were slowed or frozen, the dyskinetic movements were also slowed or frozen. Correlation analysis showed that the two dose-response courses in each of the three dose-response curve pairs (Fig. 4A2, B2, C2) were correlated with correlation coefficients being between 0.93 to 0.98 (Pearson test). These results indicate that the therapeutic effect and dyskinetic side effect induced by each of these drugs were highly correlated and likely to be intrinsically linked.
Given that ropinirole is a current anti-PD drug (Olanow et al., 2009; Standaert and Young, 2006), the relatively modest stimulating effect of ropinirole on ambulatory horizontal movements prompted us to examine its effect on the challenging beam traversal, another common parameter for normal motor function and anti-PD effect (Ding et al., 2007; Fleming et al., 2004; Hwang et al., 2005). As shown in Fig. 5A and B, 6 mg/kg and 20 mg/kg L-dopa and 0.5 mg/kg and 2 mg/kg SKF81297 substantially increased the walking speed and thus shortened the time for AMPT-PitxHomo mice to traverse the challenging beam. In contrast, ropinirole exerted biphasic effects: at 0.05 mg/kg, 0.1 mg/kg and 0.2 mg/kg, it modestly but significantly reduced the beam traversal time, while 0.8 mg/kg ropinirole increased the time for the mice to traverse the challenging beam (Fig. 5C). Additionally, as illustrated in Fig. 5B* and C*, AMPT-PitxHomo mice injected with ≥1 mg/kg ropinirole were often unable to stand on the narrowest segment of the beam, and instead, they often straddled the beam, either sliding forward or becoming stuck. These results further indicate that D1Rs may play a larger role in stimulating normal motor function than D2Rs and that strong D2R agonism may also inhibit movements.
Fig. 5.

Effects of L-dopa, SKF81297 and ropinirole on challenging beam traversal time. Each data point is the average of the measurements from 5 male AMPT-PitxHomo mice, while the measurement from an individual mouse was the average of 5 consecutive runs to minimize variation. The baseline traversal time in 11–12 week old male PitxHomo mice before any drug treatment (i.e. before AMPT treatment) was around 22 s. (A and B) L-dopa and SKF81297 substantially shortened the time to traverse the beam. One-way ANOVA for L-dopa effects: F(2, 12) = 91.2, p < 0.01. Post hoc Turkey test: p < 0.01 for 0 mg/kg L-dopa vs. 6 mg/kg L-dopa and 0 mg/kg L-dopa vs. 20 mg/kg L-dopa, p < 0.05 for 6 mg/kg L-dopa vs. 20 mg/kg L-dopa. One-way ANOVA for SKF81297 effects: F(2, 12) = 185.5, p < 0.01. Post hoc Turkey test: p < 0.01 for 0 mg/kg SKF81297 vs. 0.5 mg/kg SKF81297 and 0 mg/kg SKF81297 vs. 2 mg/kg SKF81297, p < 0.01 for 0.5 mg/kg SKF81297 vs. 2 mg/kg SKF81297. The dose of 0 mg/kg was saline control. (B*) AMPT-PitxHomo mice injected with 2 mg/kg SKF81297 were able to stand and walk on the narrowest part of the beam. The beam does not show well since it is made of transparent plastic. (C) Ropinirole at 0.05 mg/kg, 0.1 mg/kg and 0.2 mg/kg (IP) reduced the beam traversal time while a higher dose (0.8 mg/kg) prolonged the traversal time. One-way ANOVA: F(4, 20) = 65.6, p < 0.01. Post hoc Turkey test: p < 0.05 for all pairs except p > 0.05 for 0.05 mg/kg and 0.2 mg/kg Ropinirole. (C*) AMPT-PitxHomo mice injected with 1 mg/kg ropinirole often straddled the narrowest part of the beam. The beam shows better here than in B* because the mouse moved slowly in C* and the camera was set for the best picture while it was set for speed in B* for the fast moving mouse.
Discussion
Our key observations are that in mice with severe DA loss in the dorsal striatum, the very first dose of L-dopa induced both normal and dyskinetic movements with an identical time course and parallel dose-response curves, and D1R but not D2R agonism mimicked L-dopa’s stimulation of normal and dyskinetic movements. These observations have important implications to our understanding and treatment of PD. In the following sections, we will first address the concerns on the validity of using PitxHomo mice to study the consequences of DA loss in PD and then discuss our main findings.
Concerns on PitxHomo mice as a model of DA loss in PD
One concern is that early loss of the nigrostriatal DA projection (Nunes et al. 2003; Smidt et al. 2004) may induce developmental compensatory changes. For example, the generally normal locomotor activity in PitxHomo mice may be due to a developmental compensation, thus confounding data interpretation. Our data indicate that this is not the case. Instead, our data show that a further moderate depletion of DA by a small dose of TH inhibitor AMPT, not effective in PitxWT mice, rendered these PitxHomo mice hypokinetic. These results suggest that the motor function in PitxHomo mice is fragilely maintained by the residual DA in the middle striatum and the upper ventral striatal region that have motor function (Voorn et al., 2004). Another concern is the relevance of our conclusions on L-dopa pharmacology and LID made in PitxHomo mice to PD. We argue that since DA receptor pharmacology and other properties such as supersensitization are similar in rodents and primates (Corvol et al., 2004; Nadjar et al., 2009), our conclusions made in PitxHomo mice are relevant to PD. Thus, PitxHomo mice are a valid and highly convenient mouse model to study the consequences of DA loss in PD.
L-dopa induces dyskinesia without any priming
In this study, we observed that the very first injection of L-dopa consistently induced dyskinetic movements, without any priming, in PitxHomo mice that have a severe DA loss in the dorsal striatum. In contrast, in WT mice, L-dopa, even at high doses, had no motor effect. Ding et al. (2007) also described first L-dopa-induced dyskinetic events. These results suggest that severe chronic DA loss may cause pathological changes in the striatum that set the stage for dyskinesia upon the first exposure to L-dopa. We propose that severe long-term DA loss induces a latent dyskinetic state. Once the latent dyskinetic state is established, dyskinesia is inescapable whenever L-dopa is administered. Our data also indicate that repeated L-dopa use or priming modestly increased dyskinesia duration. However, this increase in dyskinesia or priming was fully dependent on severe DA loss and thus secondary, because L-dopa, as a precursor to DA, does not induce dyskinesia in normal animals or animals with moderate toxin-induced DA loss (Jenner, 2008; Nadjar et al., 2009; unpublished data of Li and Zhou).
Since our conclusion is that the severely DA deficient dorsal striatum is ready to produce abnormal motor signals upon the first L-dopa stimulation, why is dyskinesia often absent or mild during the first few years’ L-dopa treatment in PD patients? We suggest that this is likely due to the significant (30–10%) residual DA axons in the dorsal striatum during early to mid-stages of PD (Hornykiewicz, 2001). The residual DA system may compensate in benign ways for up to 80% DA loss and that D1R supersensitivity, a key factor in dyskinesia pathogenesis, develops only after DA loss is ≥90% (Schwarting and Huston, 1996; Cenci et al., 2010). Therefore, it is likely that while the PD motor symptoms are treated with L-dopa, the residual DA neurons continue to degenerate (Morrish et al., 1996; Lee et al. 2004; Hilker et al., 2005; Cheng et al., 2010). A few years later, the DA loss becomes severe (≥90%) such that dyskinesia occurs readily upon DA stimulation. Thus, the temporal lag between the start of L-dopa use and the first appearance of LID probably reflects mainly the disease progression and the severity of DA loss. Our results and interpretation are consistent with clinical observations that LID occurs readily in untreated late stage PD patients with severe nigrostriatal DA degeneration (Nadjar et al., 2009). The accidentally DA neuron-lesioned patients developed LID quickly (Ballard et al., 1985; Langston, 1985), providing additional strong support for our conclusion. Furthermore, experimental studies show that animals with severe DA lesion develop LID readily, even on first injection of L-dopa (Putterman et al., 2007; Nadjar et al., 2009). In contrast, when DA loss is moderate, L-dopa does not induce dyskinesia. Taken together, our results and literature evidence indicate that severe nigrostriatal DA loss sets the stage for LID, and priming is not required although it may occur and contribute to the total intensity of LID. Additionally, non-DA lesion factors, currently not well defined, may also contribute (Guigoni et al., 2005).
L-dopa stimulates both normal and dyskinetic movements
The prevailing view is that L-dopa’s anti-PD and dyskinetic effects are mediated by separate mechanisms (Olanow et al., 2009; Santini et al., 2009; Gottwald and Aminoff, 2011; see LeWitt, 2010 and Nutt et al., 2010 for discussion). The key evidence for this view is the apparent temporal lag between the start of L-dopa treatment and the first appearance of LID. As discussed in the preceding section, however, the temporal lag is likely due to the progressive DA neuron degeneration, not truly separate mechanisms. Thus, evidence for the prevailing view is largely lacking, raising the possibility that L-dopa’s anti-PD and dyskinetic effects are mediated by shared mechanisms. Indeed, we observed that L-dopa at all doses tested always stimulated normal and dyskinetic movements simultaneously and with parallel dose-response curves. These observations support our idea that L-dopa triggered the same DA receptors in the basal ganglia that stimulate both normal and dyskinetic movements. Certainly, these observations, when viewed in isolation, are not sufficient to prove our idea, because the identical time courses may be due to L-dopa dosing and the dose-response curves may be similar coincidently. However, our data also show that L-dopa’s motor stimulating effect was predominantly mediated by D1Rs (discussed in the next section) that are expressed mainly in the D1-MSNs in the striatum. D1-MSNs are known to promote both normal and dyskinetic movements (Darmopil et al., 2009; Bateup et al., 2010; Kravitz et al., 2010). Therefore, in aggregate, our data make a strong case that via at least partially shared mechanisms, L-dopa stimulates both normal and dyskinetic movements. Consequently, it may be difficult to separate L-dopa’s anti-PD effect from its dyskinetic effect after LID first appears when DA loss is severe enough that creates the conditions for dyskinesia to occur. This conclusion is consistent with the clinical observation that measures that reduce dyskinesia usually worsen parkinsonism (Fabbrini et al., 2007; Nutt, 2008; Olanow et al., 2009).
D1 agonism but not D2 agonism underlies L-dopa’s motor-stimulating effects
We observed that relatively low doses (< 4 mg/kg) of SKF81297 potently stimulated both normal and dyskinetic movements, mimicking the motor-stimulating effects of L-dopa. In contrast, the maximal stimulatory responses of ropinirole were relatively weak, although the normal movements and dyskinetic movements induced by ropinirole still had identical time courses and parallel dose-response curves. The only plausible explanation for these observations is that normal and dyskinetic movements induced by L-dopa and SKF81297 were generated by shared D1R-dependent pharmacological mechanisms in the D1-MSN direct pathway. Additional support for this interpretation comes from the following observations. SKF81297’s motor-stimulating effects were monophasic and plateaued at 2 mg/kg. In contrast, ropinirole had biphasic effects: while being stimulatory at relatively low doses, high doses of ropinirole and, to a less extent, L-dopa, induced a slowness or stickiness in normal and dyskinetic movements. High doses of ropinirole may even cause a complete movement freezing. These results indicate that D1R activity is the key common component of the mechanisms stimulating normal and dyskinetic movements. Because of the heavy expression of D1Rs in the direct pathway MSNs in the striatum (Levey et al. 1993; Yung et al. 1995) and the known motor-promoting function of these D1-MSNs (Bateup et al. 2010; Kravitz et al. 2010), D1Rs in the direct pathway MSNs are likely responsible for the normal and dyskinetic motor-stimulating effects of L-dopa and SKF81297. Consequently, the anti-PD effect and the dyskinetic side effect are two intrinsically linked components of D1R agonism-triggered movements. This interpretation is consistent with recent studies indicating that D1-MSNs generate the Go signals required for both normal and abnormal movements (Darmopil et al., 2009; Bateup et al., 2010; Kravitz et al., 2010; Berthet et al., 2012). The low stimulating effects of ropinirole on normal movements and dyskinesia observed here are consistent with the clinical observation that D2R agonists induce less dyskinesia but also weaker anti-PD effects than L-dopa such that PD patients eventually need L-dopa when the disease is severe (Katzenschlager et al., 2008; Nutt, 2008). The mechanisms for high dose ropinirole-induced motor freezing observed here and in PD patients are unknown (Fahn and Przedborki, 2005; Fox and Lang, 2008; Olanow et al., 2009). One possibility is that a high dose of ropinirole may inhibit DA release from the residual DA neurons and also disrupt the function of neuronal circuits that expressing D2Rs.
The following question is also important: How can D1R activation trigger both normal and dyskinetic movements? This question can only be answered by future studies. Here we propose that the same D1Rs and the signaling pathways, activated at different intensity and duration, may produce both normal and dyskinetic motor control signal. Additionally, there are different types of LID, ranging from being dystonic to being choreic. If normal and dyskinetic movements require D1Rs with different downstream signaling mechanisms, then these different types of LID should also require different downstream signaling mechanisms. This is unlikely because there are only few D1R signaling pathways but more types of LID.
Finally, we need to note that clinical data suggest that D2R agonism may have stronger motor-stimulating effects in PD patients (Olanow et al. 2009) than in mice (Darmopil et al., 2009; Bateup et al., 2010; present study), although it is certain that L-dopa, likely via D1R agonism, has a stronger anti-PD effect than D2R agonists. Given the structural differences in the basal ganglia in primates and rodents, it is possible that even though the direct pathway may still be the dominant force in promoting both normal and abnormal movements in primates, the indirect pathway and hence D2R agonism may play a relatively larger role in humans than in mice.
Clinical implications
Our results indicate that severe DA loss may cause sufficient abnormalities or a latent dyskinetic state, in the basal ganglia, such that the first dose of L-dopa or a D1 agonist may induce dyskinesia without any priming. Our results also indicate that L-dopa’s anti-PD effect and dyskinetic side effect are primarily mediated by D1Rs, whereas D2Rs play a smaller role. These findings have important implications to future research and treatment strategies for PD and LID. First, it may be difficult, if not impossible, to separate the anti-PD effect from the dyskinetic effect after chronic severe DA loss. Second, a better strategy is perhaps to manage or prevent the detrimental maladaptive functional and structural changes in the basal ganglia following DA loss. Third, compared with the current D2R agonism-based therapy, proper D1R agonism may be a more efficacious, unused route to treat the motor deficits in PD while keeping dyskinesia at acceptable levels because PD patients prefer dyskinesia over akinesia (Hung et al., 2010).
Supplementary Material
First injection of 2 mg/kg SKF81297 induced dyskinetic movements characterized by abnormal stereotypic paw movements accompanied by vertical trunk movements in an AMPT-PitxHomo mouse (left). This movie is intended to show the qualitative aspects of dyskinetic movements in PitxHomo mice. Lower doses of SKF81297 induced qualitatively identical dyskinetic movements of lower intensity and shorter duration. L-dopa also induced qualitatively identical dyskinetic movements in a dose-dependent manner. While every PitxHomo mouse generated this type of dyskinetic movements upon L-dopa or SKF81297 stimulation, it was never observed in PitxWT mice. These dyskinetic movements are also qualitatively identical to those induced by repeated 25 mg/kg L-dopa described in Ding et al. (2007). The mouse in the right compartment was an AMPT-PitxHomo mouse that received a saline injection as control.
First injection of 1 mg/kg ropinirole induced a complete motor freezing in an AMPT-PitxHomo mouse (left). Note the mouse was frozen in the middle of a forward movement. The mouse in the right compartment was an AMPT-PitxHomo mouse that received a saline injection instead of ropinirole.
Highlights.
DA D1 receptors are the primary mediator of L-dopa’s anti-PD effect
Severe DA loss in the dorsal striatum sets the stage for L-dopa-induced dyskinesia
L-dopa can induce dyskinesia without any priming
D1 receptors are the shared mechanism for L-dopa’s anti-PD and dyskinetic effects
L-dopa’s anti-PD and dyskinetic effects are intrinsically linked
Acknowledgments
This work was supported by NIH grant R01NS058850.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16:448–458. doi: 10.1002/mds.1090. [DOI] [PubMed] [Google Scholar]
- Ballard PA, Tetrud JW, Langston JW. Permanent human parkinsonism due to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): seven cases. Neurology. 1985;35:949–956. doi: 10.1212/wnl.35.7.949. [DOI] [PubMed] [Google Scholar]
- Bateup HS, Santini E, Shen W, Birnbaum S, Valjent E, Surmeier DJ, Fisone G, Nestler EJ, Greengard P. Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc Natl Acad Sci USA. 2010;107:14845–14850. doi: 10.1073/pnas.1009874107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Cao ZF, Kheirbek MA, Zhuang X. Loss of cocaine locomotor response in Pitx3-deficient mice lacking a nigrostriatal pathway. Neuropsychopharmacology. 2009;34:1149–1161. doi: 10.1038/npp.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med. 2003;9:762–767. doi: 10.1038/nm875. [DOI] [PubMed] [Google Scholar]
- Berthet P, Hellgren-Kotaleski J, Lansner A. Action selection performance of a reconfigurable basal ganglia inspired model with Hebbian-Bayesian Go-NoGo connectivity. Front Behav Neurosci. 2012;6:65. doi: 10.3389/fnbeh.2012.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci MA. Molecular mechanisms of L-dopa-induced dyskinesia. In: Steiner H, Tseng KY, editors. Handbook of Basal Ganglia Structure and Function. San Diego: Academic Press; 2010. pp. 625–640. [Google Scholar]
- Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 2010;67:715–725. doi: 10.1002/ana.21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier G, Deniau JM. Disinhibition as a basic process in the expression of striatal functions. Trends Neurosci. 1990;13:277–280. doi: 10.1016/0166-2236(90)90109-n. [DOI] [PubMed] [Google Scholar]
- Corvol JC, Muriel MP, Valjent E, Féger J, Hanoun N, Girault JA, Hirsch EC, Hervé D. Persistent increase in olfactory type G-protein alpha subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson disease. J Neurosci. 2004;24:7007–7014. doi: 10.1523/JNEUROSCI.0676-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotzias GC, Papavasiliou PS, Gellene R. Modification of Parkinsonism--chronic treatment with L-dopa. N Engl J Med. 1969;280:337–345. doi: 10.1056/NEJM196902132800701. [DOI] [PubMed] [Google Scholar]
- Darmopil S, Martín AB, De Diego IR, Ares S, Moratalla R. Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol Psychiatry. 2009;66:603–613. doi: 10.1016/j.biopsych.2009.04.025. [DOI] [PubMed] [Google Scholar]
- Delong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–2855. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- Ding Y, Restrepo J, Won L, Hwang DY, Kim KS, Kang UJ. Chronic 3,4-dihydroxyphenylalanine treatment induces dyskinesia in aphakia mice, a novel genetic model of Parkinson’s disease. Neurobiol Dis. 2007;27:11–23. doi: 10.1016/j.nbd.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Won L, Britt JP, Lim SA, McGehee DS, Kang UJ. Enhanced striatal cholinergic neuronal activity mediates L-DOPA-induced dyskinesia in parkinsonian mice. Proc Natl Acad Sci U S A. 2011;108:840–845. doi: 10.1073/pnas.1006511108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG. Levodopa-induced dyskinesias. Mov Disord. 2007;22:1379–1389. doi: 10.1002/mds.21475. [DOI] [PubMed] [Google Scholar]
- Fahn S. How do you treat motor complications in Parkinson’s disease: Medicine, surgery, or both? Ann Neurol. 2008;64(Suppl 2):S56–64. doi: 10.1002/ana.21453. [DOI] [PubMed] [Google Scholar]
- Fahn S, Przedborki S. Parkinsonism. In: Rowland LP, editor. Merritt’s Neurology. New York: Lippincott Williams & Wilkins; 2005. pp. 828–846. [Google Scholar]
- Fleming SM, Salcedo J, Fernagut PO, Rockenstein E, Masliah E, Levine MS, Chesselet MF. Early and progressive sensorimotor anomalies in mice overexpressing wild-type human alpha-synuclein. J Neurosci. 2004;24:9434–9440. doi: 10.1523/JNEUROSCI.3080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox SH, Lang AE. Levodopa-related motor complications--phenomenology. Mov Disord. 2008;23(Suppl 3):S509–514. doi: 10.1002/mds.22021. [DOI] [PubMed] [Google Scholar]
- Gottwald MD, Aminoff MJ. Therapies for dopaminergic-induced dyskinesias in Parkinson disease. Ann Neurol. 2011;69:919–927. doi: 10.1002/ana.22423. [DOI] [PubMed] [Google Scholar]
- Guigoni C, Dovero S, Aubert I, Li Q, Bioulac BH, Bloch B, Gurevich EV, Gross CE, Bezard E. Levodopa-induced dyskinesia in MPTP-treated macaques is not dependent on the extent and pattern of nigrostrial lesioning. Eur J Neurosci. 2005;22:283–287. doi: 10.1111/j.1460-9568.2005.04196.x. [DOI] [PubMed] [Google Scholar]
- Hikosaka O, Takikawa Y, Kawagoe R. Role of the basal ganglia in the control of purposive saccadic eye movements. Physiol Rev. 2000;80:953–978. doi: 10.1152/physrev.2000.80.3.953. [DOI] [PubMed] [Google Scholar]
- Hilker R, Schweitzer K, Coburger S, Ghaemi M, Weisenbach S, Jacobs AH, Rudolf J, Herholz K, Heiss WD. Nonlinear progression of Parkinson disease as determined by serial positron emission tomographic imaging of striatal fluorodopa F18 activity. Arch Neurol. 2005;62:378–382. doi: 10.1001/archneur.62.3.378. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Chemical neuroanatomy of the basal ganglia — normal and in Parkinson’s disease. J Chem Neuroanat. 2001;22:3–12. doi: 10.1016/s0891-0618(01)00100-4. [DOI] [PubMed] [Google Scholar]
- Hung SW, Adeli GM, Arenovich T, Fox SH, Lang AE. Patient perception of dyskinesia in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2010;81:1112–1115. doi: 10.1136/jnnp.2009.173286. [DOI] [PubMed] [Google Scholar]
- Huot P, Fox SH, Newman-Tancredi A, Brotchie JM. Anatomically selective serotonergic type 1A and serotonergic type 2A therapies for Parkinson’s disease: an approach to reducing dyskinesia without exacerbating parkinsonism? J Pharmacol Exp Ther. 2011;339:2–8. doi: 10.1124/jpet.111.184093. [DOI] [PubMed] [Google Scholar]
- Hwang DY, Fleming SM, Ardayfio P, Moran-Gates T, Kim H, Tarazi FI, Chesselet MF, Kim KS. 3,4-dihydroxyphenylalanine reverses the motor deficits in Pitx3-deficient aphakia mice: behavioral characterization of a novel genetic model of Parkinson’s disease. J Neurosci. 2005;25:2132–2137. doi: 10.1523/JNEUROSCI.3718-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iravani MM, Jenner P. Mechanisms underlying the onset and expression of levodopa-induced dyskinesia and their pharmacological manipulation. J Neural Transm. 2011;118:1661–1690. doi: 10.1007/s00702-011-0698-2. [DOI] [PubMed] [Google Scholar]
- Jenner P. Dopamine agonists, receptor selectivity and dyskinesia induction in Parkinson’s disease. Curr Opin Neurol. 2003;16(Suppl 1):S3–7. doi: 10.1097/00019052-200312001-00002. [DOI] [PubMed] [Google Scholar]
- Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- Katzenschlager R, Head J, Schrag A, Ben-Shlomo Y, Evans A, Lees AJ. Parkinson’s Disease Research Group of the United Kingdom. Fourteen-year final report of the randomized PDRG-UK trial comparing three initial treatments in PD. Neurology. 2008;71:474–480. doi: 10.1212/01.wnl.0000310812.43352.66. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuoppamaki M, Al-Barghouthy G, Jackson MJ, Smith LA, Quinn N, Jenner P. L-dopa dose and the duration and severity of dyskinesia in primed MPTP-treated primates. J Neural Transm. 2007;114:1147–1153. doi: 10.1007/s00702-007-0727-3. [DOI] [PubMed] [Google Scholar]
- Langston JW. MPTP neurotoxicity: an overview and characterization of phases of toxicity. Life Sci. 1985;36:201–206. doi: 10.1016/0024-3205(85)90059-1. [DOI] [PubMed] [Google Scholar]
- Lee CS, Schulzer M, de la Fuente-Fernández R, Mak E, Kuramoto L, Sossi V, Ruth TJ, Calne DB, Stoessl AJ. Lack of regional selectivity during the progression of Parkinson disease: implications for pathogenesis. Arch Neurol. 2004;61:1920–1925. doi: 10.1001/archneur.61.12.1920. [DOI] [PubMed] [Google Scholar]
- Levey AI, Hersch SM, Rye DB, Sunahara RK, Niznik HB, Kitt CA, Price DL, Maggio R, Brann MR, Ciliax BJ. Localization of D1 and D2 dopamine receptors in brain with subtype-specific antibodies. Proc Natl Acad Sci USA. 1993;90:8861–8865. doi: 10.1073/pnas.90.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewitt PA. Levodopa for the treatment of Parkinson’s disease. N Engl J Med. 2009;359:2468–2476. doi: 10.1056/NEJMct0800326. [DOI] [PubMed] [Google Scholar]
- Lewitt PA. Relief of parkinsonism and dyskinesia: one and the same dopaminergic mechanism? Neurology. 2010;74:1169–1170. doi: 10.1212/WNL.0b013e3181d90076. [DOI] [PubMed] [Google Scholar]
- Lundblad M, Usiello A, Carta M, Håkansson K, Fisone G, Cenci MA. Pharmacological validation of a mouse model of l-DOPA-induced dyskinesia. Exp Neurol. 2005;194:66–75. doi: 10.1016/j.expneurol.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Sonsalla PK, Chesselet MF. Animal models of Parkinson’s disease progression. Acta Neuropathol. 2008;115:385–398. doi: 10.1007/s00401-008-0350-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti R, Torre P, Antonello RM, Esposito F, Bellini G. The on-freezing phenomenon: cognitive and behavioral aspects. Parkinsons Dis. 2011;2011:746303. doi: 10.4061/2011/746303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrish PK, Sawle GV, Brooks DJ. An [18F]dopa-PET and clinical study of the rate of progression in Parkinson’s disease. Brain. 1996;119:585–591. doi: 10.1093/brain/119.2.585. [DOI] [PubMed] [Google Scholar]
- Nadjar A, Gerfen CR, Bezard E. Priming for l-dopa-induced dyskinesia in Parkinson’s disease: a feature inherent to the treatment or the disease? Prog Neurobiol. 2009;87:1–9. doi: 10.1016/j.pneurobio.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes I, Tovmasian LT, Silva RM, Burke RE, Goff SP. Pitx3 is required for development of substantia nigra dopaminergic neurons. Proc Natl Acad Sci U S A. 2003;100:4245–4250. doi: 10.1073/pnas.0230529100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt JG. Pharmacokinetics and pharmacodynamics of levodopa. Mov Disord. 2008;23(Suppl 3):S580–584. doi: 10.1002/mds.22037. [DOI] [PubMed] [Google Scholar]
- Nutt JG, Chung KA, Holford NH. Dyskinesia and the antiparkinsonian response always temporally coincide: a retrospective study. Neurology. 2010;74:1191–1197. doi: 10.1212/WNL.0b013e3181d90050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease (2009) Neurology. 2009;72(21 Suppl 4):S1–136. doi: 10.1212/WNL.0b013e3181a1d44c. [DOI] [PubMed] [Google Scholar]
- Putterman DB, Munhall AC, Kozell LB, Belknap JK, Johnson SW. Evaluation of levodopa dose and magnitude of dopamine depletion as risk factors for levodopa-induced dyskinesia in a rat model of Parkinson’s disease. J Pharmacol Exp Ther. 2007;323:277–284. doi: 10.1124/jpet.107.126219. [DOI] [PubMed] [Google Scholar]
- Santini E, Heiman M, Greengard P, Valjent E, Fisone G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci Signal. 2009;2(80):ra36. doi: 10.1126/scisignal.2000308. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Obeso J. Timing of treatment initiation in Parkinson’s disease: a need for reappraisal? Ann Neurol. 2006;59:559–562. doi: 10.1002/ana.20789. [DOI] [PubMed] [Google Scholar]
- Schwarting RK, Huston JP. The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol. 1996;50:275–331. doi: 10.1016/s0301-0082(96)00040-8. [DOI] [PubMed] [Google Scholar]
- Smidt MP, Smits SM, Bouwmeester H, Hamers FP, van der Linden AJ, Hellemons AJ, Graw J, Burbach JP. Early developmental failure of substantia nigra dopamine neurons in mice lacking the homeodomain gene Pitx3. Development. 2004;131:1145–1155. doi: 10.1242/dev.01022. [DOI] [PubMed] [Google Scholar]
- Smith Y, Wichmann T, Factor SA, DeLong MR. Parkinson’s disease therapeutics: new developments and challenges since the introduction of levodopa. Neuropsychopharmacology. 2012;37:213–246. doi: 10.1038/npp.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits SM, Burbach JP, Smidt MP. Developmental origin and fate of meso-diencephalic dopamine neurons. Prog Neurobiol. 2006;78:1–16. doi: 10.1016/j.pneurobio.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Sotnikova TD, Beaulieu JM, Barak LS, Wetsel WC, Caron MG, Gainetdinov RR. Dopamine-independent locomotor actions of amphetamines in a novel acute mouse model of Parkinson disease. PLoS Biol. 2005;3:e271. doi: 10.1371/journal.pbio.0030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert DG, Young AB. Treatment of central nervous system degenerative disorders. In: Brunton L, Lazo J, Parker K, editors. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw-Hill; 2006. pp. 527–545. [Google Scholar]
- Stockwell KA, Virley DJ, Perren M, Iravani MM, Jackson MJ, Rose S, Jenner P. Continuous delivery of ropinirole reverses motor deficits without dyskinesia induction in MPTP-treated common marmosets. Exp Neurol. 2008;211:172–179. doi: 10.1016/j.expneurol.2008.01.019. [DOI] [PubMed] [Google Scholar]
- Taylor TN, Greene JG, Miller GW. Behavioral phenotyping of mouse models of Parkinson’s disease. Behav Brain Res. 2010;211:1–10. doi: 10.1016/j.bbr.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Munckhof P, Luk KC, Ste-Marie L, Montgomery J, Blanchet PJ, Sadikot AF, Drouin J. Pitx3 is required for motor activity and for survival of a subset of midbrain dopaminergic neurons. Development. 2003;130:2535–2542. doi: 10.1242/dev.00464. [DOI] [PubMed] [Google Scholar]
- Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM. Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci. 2004;27:468–474. doi: 10.1016/j.tins.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Yung KK, Bolam JP, Smith AD, Hersch SM, Ciliax BJ, Levey AI. Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience. 1995;65:709–730. doi: 10.1016/0306-4522(94)00536-e. [DOI] [PubMed] [Google Scholar]
- Zhou F-M, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- Zhou F-M, Liang Y, Salas R, Zhang L, De Biasi M, Dani JA. Co-release of dopamine and serotonin from striatal dopamine terminals. Neuron. 2005;46:65–74. doi: 10.1016/j.neuron.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Zhou FW, Jin Y, Matta SG, Xu M, Zhou F-M. An ultra-short dopamine pathway regulates basal ganglia output neurons. J Neurosci. 2009;29:10424–1035. doi: 10.1523/JNEUROSCI.4402-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
First injection of 2 mg/kg SKF81297 induced dyskinetic movements characterized by abnormal stereotypic paw movements accompanied by vertical trunk movements in an AMPT-PitxHomo mouse (left). This movie is intended to show the qualitative aspects of dyskinetic movements in PitxHomo mice. Lower doses of SKF81297 induced qualitatively identical dyskinetic movements of lower intensity and shorter duration. L-dopa also induced qualitatively identical dyskinetic movements in a dose-dependent manner. While every PitxHomo mouse generated this type of dyskinetic movements upon L-dopa or SKF81297 stimulation, it was never observed in PitxWT mice. These dyskinetic movements are also qualitatively identical to those induced by repeated 25 mg/kg L-dopa described in Ding et al. (2007). The mouse in the right compartment was an AMPT-PitxHomo mouse that received a saline injection as control.
First injection of 1 mg/kg ropinirole induced a complete motor freezing in an AMPT-PitxHomo mouse (left). Note the mouse was frozen in the middle of a forward movement. The mouse in the right compartment was an AMPT-PitxHomo mouse that received a saline injection instead of ropinirole.