

Abstract
Endoplasmic reticulum–associated degradation (ERAD) is a cellular protein quality-control process that disposes of proteasomal substrates from the early secretory pathway. Recent work shows that the endoplasmic reticulum–resident rhomboid protease RHBDL4 facilitates ERAD by recognizing and cleaving integral membrane substrates. The work indicates that intramembrane proteolysis may have a general role in the extraction of misfolded membrane proteins from the endoplasmic reticulum.
Secreted and transmembrane proteins account for approximately one-third of the eukaryotic proteome, and the endoplasmic reticulum (ER) provides a specialized environment for their oxidative folding, modification and oligomerization. In addition, the ER must constantly combat competing off-pathway folding reactions culminating in non-native, aggregation-prone conformers that can result not only in nonfunctional proteins but also in proteotoxic cell stress. The ER has therefore evolved a robust quality-control system composed of two branches: the unfolded-protein response (UPR)1, which transmits stress signals to the cytosol and nucleus to effect global changes in transcription and translation, and ERAD2,3, which targets misfolded secretory proteins for destruction.
The ERAD machinery consists of luminal, integral-membrane and cytosolic factors that are organized around membrane-embedded E3 ubiquitin ligases to mediate the recognition, dislocation and ubiquitination of terminally misfolded proteins and their delivery from the ER to cytosolic proteasomes2,4. The underlying mechanism(s) by which ERAD substrates are dislocated into the cytosol remains unknown, but it is clear that in most cases, this process is coupled to the energy derived from ATP hydrolysis by p97/VCP2,3, a highly abundant cytosolic AAA ATPase. Many of the proteins involved in ERAD contain p97/VCP–binding sites, perhaps reflecting the need to adapt p97/VCP function toward substrate-specific ERAD pathways in order to accommodate the diverse set of proteins degraded by ERAD.
Membrane proteins represent an especially complex challenge for the dislocation machinery, as they may contain a combination of luminal and membrane domains. The energy required to extract a membrane domain is likely to largely depend on factors such as the hydrophobicity of the component transmembrane segments and the presence of exoplasmic disulfide bonds. For example, increasing the hydrophobicity of a transmembrane segment of the ERAD substrate cystic fibrosis transmembrane conductance regulator (CFTR) resulted in an increased dependence on p97/VCP for CFTR degradation in a semireconstituted system5. In the case of CFTR, dislocation is highly coupled to proteolysis by the proteasome, and CFTR is released from the ER in fragments6. In theory, proteolytic cleavages of integral membrane substrates within regions inaccessible to the proteasome—the membrane or ER lumen—could facilitate dislocation by destabilizing membrane segments and releasing them from luminal domains, thus reducing what is likely a substantial energy barrier.
Although full-length intermediates of ERAD substrates have been observed in the cytosol, which suggests that the dislocation machinery is able to dislocate entire proteins3, fragments of ERAD substrates have been detected with proteasome inhibition7–9, which indicates that another protease may be involved. Intriguingly, recent data demonstrated that several ERAD factors (Fig. 1), Derlins10, UBAC2 (refs. 4,10) and iRhoms11 are inactive homologs of intramembrane proteases known as rhomboids, which have key roles in controlling epidermal growth factor receptor (EGFR) signaling and in regulating mitochondrial fusion dynamics12. These data suggest that common mechanistic features underlie dislocation and rhomboid-mediated intramembrane proteo lysis. The aspartyl intramembrane protease signal peptide peptidase (SPP) has also been suggested to participate in ERAD13, but its precise role is unclear, and SPP-mediated cleavage outside of signal-sequence cleavage sites has yet to be reported for an ERAD substrate. Therefore, questions of whether a protease besides the proteasome is involved in dislocation, whether observed fragments of membrane substrates represent true degradation inter mediates or are simply experimental artifacts and what the exact role of ER-localized inactive rhomboid-protease homologs is in dislocation remain unanswered.
Figure 1.
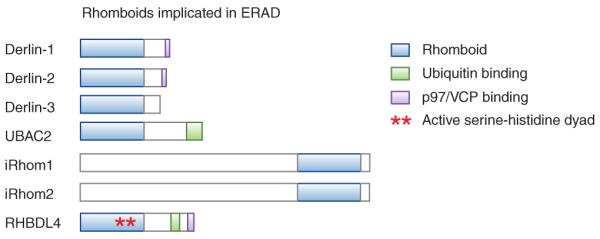
Domain structure of ERAD-implicated rhomboid family members.
In a recent paper14, Fleig et al. provide a tantalizing clue to this puzzle by suggesting a proteolytic role for the rhomboid RHBDL4 in the ERAD of unstable membrane proteins. The authors found that RHBDL4 localizes to the ER and is upregulated upon chemical induction of ER stress, which suggests a role for RHBDL4 in maintaining ER homeostasis. Unlike Derlins and iRhoms, RHBDL4 has a conserved serine-histidine pair homologous to active site residues of known rhomboid proteases, consistent with RHBDL4 being a bona fide protease.
To test whether RHBDL4 proteolytic activity is involved in an ERAD-like process, the authors examined the effect of coexpressing RHBDL4 with a constitutively degraded orphan α subunit of the pre–T-cell receptor (pTα). The overexpression of wild-type RHBDL4 but not a catalytic-site mutant resulted in the accumulation of fragments of pTα. These fragments were localized to the cytosol and stabilized by proteasome inhibition, which implies that overexpressed RHBDL4 is capable of cleaving an ERAD substrate into fragments that are subsequently dislocated and degraded. Knocking down RHBDL4 or overexpressing the dominant-negative mutant resulted in not only impaired pTα degradation but also activation of the UPR, which suggests that RHBDL4 activity is required for ERAD of at least some endogenous substrates. These data do not preclude the possibility that RHBDL4 itself functions in the UPR directly or indirectly, for example by regulating anterograde trafficking of ER proteins, as has been demonstrated for iRhom2 (refs. 15,16).
How general is the function of RHBDL4 in ERAD? Fleig et al. tested whether RHBDL4 is capable of cleaving an assortment of structurally unrelated ERAD membrane substrates. pTα and an orphan chain of the T-cell receptor, TCRα, each contain basic amino acids, a feature that is used for substrate recognition by ERAD. By inserting destabilizing positively charged amino acids into the transmembrane regions of the single-pass membrane protein myelin protein zero (MPZ) and the multiple-pass membrane proteins opsin and polycystin-1, the authors were able to convert these proteins into ERAD substrates as well. RHBDL4 overexpression resulted in the accumulation of fragments of each of these proteins. Interestingly, ectodomain cleavages were observed for pTα as well as for the secreted hormone prolactin fused to the transmembrane and cytosolic domains of pTα, consistent with prior studies showing that the rhomboid cleavage site is accessible from outside of the membrane. In addition, knockdown of RHBDL4 resulted in a stabilization of mutant opsin, which suggests that its dislocation is promoted by endogenous RHBDL4 activity. Together, these data indicate that RHBDL4 is able to cleave topologically diverse membrane substrates, both within and outside of the membrane, consistent with a rather general role in ERAD.
Promiscuous RHBDL4 activity could potentially result in the deleterious cleavage of nascent membrane proteins that have not had the opportunity to fold. Fleig et al. describe an elegant mechanism for specifying RHBDL4 activity toward ERAD substrates. The authors identified a ubiquitin-binding UIM domain in the cytosolic C terminus of RHBDL4 and demonstrated that it is required for the association of RHBDL4 with ubiquitinated proteins. Mutation of the RHBDL4 UIM domain substantially reduced cleavage of pTα, which suggests that the commitment of pTα to RHBDL4 cleavage is mediated at least in part through ubiquitination. Consistent with this hypothesis, overexpression of a dominant-negative version of the E3 ligase gp78 also blocked RHBDL4-dependent cleavage of pTα.
The identification of an interaction between RHBDL4 and p97/VCP suggests a straightforward model of RHBDL4 function in ERAD (Fig. 2a): membrane ERAD substrates accumulate ubiquitin chains, thereby specifying RHBDL4 activity toward these proteins. p97/VCP is subsequently recruited directly by RHBDL4 to cytosolically exposed substrate fragments, which are then dislocated, whereas luminal fragments are presumably disposed of by an alternate pathway. Several other ERAD components contain ubiquitin-binding domains, including the rhomboid UBAC2, which is also involved in ERAD and is indirectly coupled to p97/VCP through its interaction with UBXD8 (ref. 4), which suggests that ubiquitin binding may represent a common mechanism for increasing the specificity of the dislocation machinery toward substrates.
Figure 2.
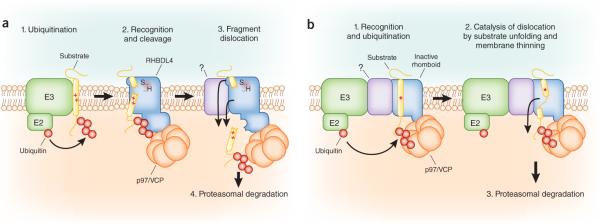
Roles for the rhomboid family in ERAD. (a) Model of the RHBDL4 ERAD pathway. RHBDL4 recognizes integral membrane substrates bearing transmembrane helices destabilized by an exposed positively charged residue(s) (red + symbols). Ubiquitination by an upstream E3 ligase provides a critical signal that is recognized by the RHBDL4 UIM and commits the substrate for RHBDL4-mediated proteolytic cleavage. The resulting substrate fragments are subsequently dislocated in a p97/VCP-dependent manner and targeted to the proteasome for degradation. (b) Model of a potential mechanism for inactive rhomboids in ERAD. Inactive rhomboids may function in the recognition and recruitment of misfolded substrates (red * symbols) to dislocation complexes. Structural features within inactive rhomboids22 may catalyze p97/VCP-dependent substrate dislocation by destabilizing substrates through a combination of a-helical transmembrane-domain unwinding and local membrane thinning.
Further studies will determine how general RHBDL4 function is in ERAD and the rules for substrate selection by RHBDL4. In the case of pTα but not TCRα, RHBDL4-generated fragments were dislocated and were degraded at a more rapid rate than the full-length protein. This raises an important question: Why does pTα but not TCRα require RHBDL4 for degradation? One difference between these ERAD substrates is that the transmembrane domain of pTα is considerably more hydrophobic than that of TCRα, with a predicted ΔG of insertion into the bilayer of −1.73 kcal mol−1 versus −0.34 kcal mol−1, respectively, as determined using the TOPCONS prediction server (http://www.topcons.net)17. Thus, RHBDL4 may be required for the dislocation of only membrane substrates whose extraction cannot be driven energetically by p97/VCP and proteasome-mediated ATP hydrolysis alone.
The highly irregular shape of rhomboid proteases is thought to alter their local bilayer environment to facilitate intramembrane cleavage, in part by thinning the membrane18. Perhaps the requirement for inactive rhomboids such as Derlins and iRhoms for some ERAD substrates reflects a similar role—destabilizing ERAD substrates or associated factors to facilitate their exposure to cytosolic ERAD factors (Fig. 2b). The recent identification of rhomboid family members in ERAD, together with the observed promiscuity of bacterial and eukaryotic rhomboid proteases toward transmembrane substrates containing helix-breaking residues12,19,20 and the demonstration of the cleavage of an aberrant membrane protein by the bacterial rhomboid GlpG (ref. 21), supports a unified role for rhomboids in bacterial and eukaryotic membrane-protein quality control.
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Ron D, Walter P. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 2.Vembar SS, Brodsky JL. Nat. Rev. Mol. Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagola K, Mehnert M, Jarosch E, Sommer T. Biochim. Biophys. Acta. 2011;1808:925–936. doi: 10.1016/j.bbamem.2010.06.025. [DOI] [PubMed] [Google Scholar]
- 4.Christianson JC, et al. Nat. Cell Biol. 2012;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlson EJ, Pitonzo D, Skach WR. EMBO J. 2006;25:4557–4566. doi: 10.1038/sj.emboj.7601307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiong X, Chong E, Skach WR. J. Biol. Chem. 1999;274:2616–2624. doi: 10.1074/jbc.274.5.2616. [DOI] [PubMed] [Google Scholar]
- 7.Loo TW, Clarke DM. J. Biol. Chem. 1998;273:32373–32376. doi: 10.1074/jbc.273.49.32373. [DOI] [PubMed] [Google Scholar]
- 8.Huppa JB, Ploegh HL. Immunity. 1997;7:113–122. doi: 10.1016/s1074-7613(00)80514-2. [DOI] [PubMed] [Google Scholar]
- 9.Moriyama T, Sather SK, McGee TP, Simoni RD. J. Biol. Chem. 1998;273:22037–22043. doi: 10.1074/jbc.273.34.22037. [DOI] [PubMed] [Google Scholar]
- 10.Greenblatt EJ, Olzmann JA, Kopito RR. Nat. Struct. Mol. Biol. 2011;18:1147–1152. doi: 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zettl M, Adrain C, Strisovsky K, Lastun V, Freeman M. Cell. 2011;145:79–91. doi: 10.1016/j.cell.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freeman M. Annu. Rev. Genet. 2008;42:191–210. doi: 10.1146/annurev.genet.42.110807.091628. [DOI] [PubMed] [Google Scholar]
- 13.Loureiro J, et al. Nature. 2006;441:894–897. doi: 10.1038/nature04830. [DOI] [PubMed] [Google Scholar]
- 14.Fleig L, et al. Mol. Cell. 2012;47:558–569. doi: 10.1016/j.molcel.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 15.McIlwain DR, et al. Science. 2012;335:229–232. doi: 10.1126/science.1214448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adrain C, Zettl M, Christova Y, Taylor N, Freeman M. Science. 2012;335:225–228. doi: 10.1126/science.1214400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hessa T, et al. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- 18.Bondar AN, del Val C, White SH. Structure. 2009;17:395–405. doi: 10.1016/j.str.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallio M, Sturgill G, Rather P, Kylsten P. Proc. Natl. Acad. Sci. USA. 2002;99:12208–12213. doi: 10.1073/pnas.192138799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urban S, Freeman M. Mol. Cell. 2003;11:1425–1434. doi: 10.1016/s1097-2765(03)00181-3. [DOI] [PubMed] [Google Scholar]
- 21.Erez E, Bibi E. Biochemistry. 2009;48:12314–12322. doi: 10.1021/bi901648g. [DOI] [PubMed] [Google Scholar]
- 22.Urban S. Biochem. J. 2010;425:501–512. doi: 10.1042/BJ20090861. [DOI] [PMC free article] [PubMed] [Google Scholar]