

Abstract
Although estrogen and the enzymes responsible for its metabolism have been detected within the lung, the ability of this tissue to metabolize estrogen has not been demonstrated previously. The goal of this study was to characterize the profile of estrogen metabolites within the murine lung and to determine the effect of tobacco smoke exposure on metabolite levels. Use of liquid chromatography–tandem mass spectrometry led to the detection of three estrogens (E1, E2 and E3) and five estrogen metabolites (2-OHE1, 4-OHE1, 4-OHE2, 2-OMeE1 and 2-OMeE2) within the perfused lung, with 4-OHE1 being the most abundant species. Levels of 4-OHEs, carcinogenic derivatives produced primarily by cytochrome P450 1B1 (Cyp1b1), were 2-fold higher in females than males. Deletion of Cyp1b1 in females led to a dramatic reduction (21-fold) in 4-OHEs, whereas levels of 2-OHE1 and the putative protective estrogen metabolite 2-OMeE2 were increased (2.4- and 5.0-fold, respectively) (P = 0.01). Similar quantitative differences in estrogen metabolite levels were observed between Cyp1b1 null and wild-type males. Exposure of female mice to tobacco smoke for 8 weeks (2h per day, 5 days per week) increased the levels of 4-OHE1 (4-fold) and 2-OHE2 (2-fold) within the lung while reducing the total concentration of 2-OMeEs to 70% of those of unexposed controls. These data suggest that tobacco smoke accelerates the production of 4-OHEs within the lung; carcinogenic metabolites that could potentially contribute to lung tumor development. Thus, inhibition of CYP1B1 may represent a promising strategy for the prevention and treatment of lung cancer.
Introduction
Lung cancer is currently the leading cause of cancer-related death in the USA, with tobacco smoke exposure the primary risk factor for its development. Despite efforts to promote smoking cessation through public education and legislation, 20% of individuals >18 years of age in the USA continue to smoke (1). Understanding the mechanism of tobacco smoke-induced carcinogenesis, developing novel interventions that target pathways essential for tumor development and finding biomarkers to identify those individuals at highest risk of developing lung cancer remain promising strategies for the prevention of this dreadful disease.
Although the association between tobacco smoke exposure and lung tumor development remains very strong, emerging data suggest that other environmental factors may also be involved. Results from preclinical and clinical studies show a strong link between estrogen and lung cancer development. First, levels of 17β-estradiol (E2) and transcripts of aromatase, an enzyme that converts testosterone to E2, are elevated in human non-small cell lung carcinomas as compared with matched non-neoplastic tissue (2). Second, estrogen stimulates the growth of several lung cancer cell lines in culture (3,4) and promotes the development of lung adenocarcinomas in LSL-K-RasG12D/Tp53f/f mice that conditionally express an oncogenic mutant K-Ras with concurrent deletion of p53 (5). Third, results from the Women’s Health Initiative indicate that use of hormone therapy (estrogen plus progestin) significantly increases the risk of death from lung cancer in postmenopausal women (6). Fourth, use of the antiestrogen tamoxifen by breast cancer patients reduces mortality from lung cancer (7). Lastly, the presence of estrogen receptors (ERs, ERα and/or ERβ) on >90% of lung tumors suggests that ER-mediated mitogenic effects of estrogen promote lung cancer development (2,8). In addition to its ER-dependent activity, estrogen may stimulate tumor formation via the generation of genotoxic metabolites such as 4-hydroxyestrogens (4-OHEs) and 16α-hydroxyestrone; a mechanism that has not been investigated as extensively.
Expression profiling of genes within the normal human (9,10) and mouse (11) lung has led to the identification of cytochrome P450 1B1 (CYP1B1) as a gene consistently induced following exposure to tobacco smoke. This enzyme metabolizes both estrogen and procarcinogens present in tobacco smoke to carcinogenic derivatives (12). Furthermore, CYP1B1 is a central node of a high-scoring network of genes modulated by tobacco smoke, including those involved in xenobiotic metabolism and circadian rhythm (11). Its early induction (3 weeks) and its stable elevation throughout lung tumorigenesis suggest that CYP1B1 may play a critical role in tobacco smoke-induced lung tumorigenesis (9,10). Results from several studies indicate an association between polymorphisms in the coding region of the CYP1B1 gene and lung cancer risk (13–16).
CYP1B1 catalyzes the addition of a hydroxyl group to estrogen to generate predominantly 4-hydroxyestrogens (4-OHEs). 4-OHEs can either bind to the ERs to activate ER-mediated signaling pathways or be oxidized to semiquinones and quinones, resulting in DNA damage (17). In contrast, 2-hydroxyestrogens (2-OHEs) are produced primarily by CYP1A1, do not activate the ERs (18) and are less reactive with DNA when oxidized (19). Both 2-OHEs and 4-OHEs can be further conjugated to sulfate, glucuronide or methyl groups for excretion (17); methylation catalyzed by catechol-O-methyltransferase (COMT) is a ubiquitous source of conjugation in extrahepatic tissues (20). Preclinical studies indicate that 2-methoxyestrogens (2-OMeEs) are antineoplastic as they do not activate the ER, but instead inhibit angiogenesis and stimulate apoptosis (21). Hence, based on the varying genotoxic properties of the different metabolites, alterations in the profile of estrogen metabolites (EM) are expected to influence one’s risk of cancer.
Comprehensive and reproducible analysis of EM in tissue was made possible recently by the development of a liquid chromatography–tandem mass spectrometry (LC-MS2) assay that can concurrently measure 15 estrogens and EM. Application of this technology to an analysis of serum from breast cancer patients demonstrated more extensive 2-hydroxylation of estrogens in postmenopausal women who were at lower risk for breast cancer (22). EM levels have not been measured in organs other than the breast. Profiling EM within the lung will further enhance our understanding of the contribution of estrogen metabolism to lung carcinogenesis.
Our previous studies demonstrate that estrone (E1) and E2 are present within the normal murine lung and that exposure to tobacco smoke induces the expression of several genes involved in estrogen metabolism (11). The goal of this study was to extend these previous findings by determining the ability of the murine lung to metabolize estrogen through measurements obtained using an advanced LC-MS2 assay. The effect of tobacco smoke exposure on the homeostatic profile of EM was also examined. The contribution of CYP1B1 to the metabolism of estrogen within the lung was assessed in mice lacking the enzyme. The resulting data demonstrate for the first time that estrogen is metabolized extensively in the lung. The predominance of 4-OHEs within the lung provides additional support for the use of CYP1B1 as a molecular target for therapeutic intervention to prevent lung cancer.
Materials and methods
Animals
Heterozygous 129/SvJ Cyp1b1-KO mice (23) were purchased from the Mutant Mouse Regional Resource Center (MMRRC, supported by National Center for Research Resources–National Institutes of Health). Mice were bred to homozygosity in the Laboratory Animal Facility at Fox Chase Cancer Center and maintained on Teklad Global 18% Protein Rodent Diet 2018S (Harlan Laboratory, Indianapolis, IN). Smoke exposures were performed on female C57/B6 mice (1.5–2 years old) carrying a human APOE*4 transgene (24) that were part of an atherosclerosis study at Duke University and fed a high-fat diet TD.88051 (Harlan Laboratory). Animals had free access to food and water. All animal experimentation was approved by the Institutional Animal Care and Use Committees at Fox Chase Cancer Center and Duke University.
Genotyping
PCR-based genotyping of Cyp1b1 wild-type (Cyp1b1-WT) and knockout (Cyp1b1-KO) mice was performed using Choice Taq Blue DNA Polymerase Master mix (Denville Scientific, South Plainfield, NJ) according to the following protocol: Wild-type primers—AAATCAAAACAGATACCCGGATG versus TCCGGCCTCTCACTTGCA; KO/Neo primers—TGAATGAACTGC AGGACGAG versus ACGACTTGGGCTTAATGGTC; reaction conditions—95ºC for 5min, followed by 35 cycles at 95ºC for 30 s, 60ºC for 1min and 72ºC for 30 s.
Tobacco smoke exposure
Mainstream and sidestream cigarette smoke was pumped into sealed chambers (via sidestream exposure) containing APOE*4 transgenic mice using a custom-built microprocessor-controlled cigarette-smoking machine (Model TE-10z; Teague Enterprises, Davis, CA). This machine provided quantitative volumes of sidestream smoke from eight cigarettes [University of Kentucky reference cigarette (2R4F)] per cycle (8min). The animals were exposed to smoke for 2h per day, 5 days per week for 8 weeks. The total suspended particulate was 100–120mg/m3 and the carbon monoxide (CO) levels were 600–800 p.p.m. Animals remained unrestrained in their cages during smoke exposure, with full access to food and water.
Lung tissue collection
Following euthanasia, the lungs were perfused by intracardiac injection with 30ml phosphate buffered saline to flush out the blood. Perfused tissues were snap-frozen in liquid nitrogen and stored at –80ºC. The accessory lobe of the lung was reserved for RNA extraction, whereas the remaining lobes were processed for estrogen metabolite analyses.
Measurement of estrogen and its metabolites
Reagents and materials.
Estrogens and EM, including E1, E2, estriol (E3), 16-epiestriol (16-epiE3), 17-epiestriol (17-epiE3), 16-ketoestradiol (16-ketoE2), 16α-hydroxyestrone (16α-OHE1), 2-methoxyestrone (2-MeOE1), 4-methoxyestrone (4-MeOE1), 2-hydroxyestrone-3-methyl ether (3-MeOE1), 2-methoxyestradiol (2-MeOE2), 4-methoxyestradiol (4-MeOE2), 2-hydroxyestrone (2-OHE1), 4-hydroxyestrone (4-OHE1), 2-hydroxyestradiol (2-OHE2) and 4-hydroxyestradiol (4-OHE2), were obtained from Steraloids (Newport, RI). Stable isotope-labeled estrogens (SI-EM), including estradiol-13,14,15,16,17,18-13C6 (13C6-E2) and estrone-13,14,15,16,17,18-13C6 (13C6-E1), were purchased from Cambridge Isotope Laboratories (Andover, MA); estriol-2,4,17-d3 (d3-E3), 2-hydroxyestradiol-1,4,16,16,17-d5 (d5-2-OHE2) and 2-methoxyestradiol-1,4,16,16,17-d5 (d5-2-MeOE2) were obtained from C/D/N Isotopes (Pointe-Claire, Quebec, Canada). 16-Epiestriol-2,4,16-d3 (d3-16-epiE3) was purchased from Medical Isotopes (Pelham, NH). All steroid analytical standards have reported chemical and isotopic purity ≥98% and were used without further purification. Dichloromethane and methanol were obtained from EM Science (Gibbstown, NJ). Glacial acetic acid and sodium bicarbonate were purchased from J.T. Baker (Phillipsburg, NJ), and sodium hydroxide and sodium acetate were purchased from Fisher Scientific (Fair Lawn, NJ). Ethyl alcohol was obtained from Pharmco Products (Brookfield, CT). Formic acid, acetone, dansyl chloride and l-ascorbic acid were obtained from Sigma–Aldrich Chemical Co. (St Louis, MO). All chemicals and solvents used in this study were high-performance liquid chromatography or reagent grade unless otherwise noted.
Preparation of standard solutions.
Stock solutions containing 80 μg/ml of each estrogen and stable isotope-labeled estrogen were prepared in methanol containing 0.1% l-ascorbic acid. The stock solutions are stable for at least 2 months while stored at –20ºC. Working standard solutions of estrogens at 0.32 and 8ng/ml were prepared by diluting the stock solutions with methanol containing 0.1% l-ascorbic acid.
Sample preparation.
Lung tissue samples (0.1–0.2g per sample) from Cyp1b1-WT or Cyp1b1-KO mice (female and male, 12–14 weeks of age, n = 4–5 per group) were thawed at room temperature, minced with scissors and transferred into 1.5ml Eppendorf tubes. The tissue was snap-frozen in liquid nitrogen for 5min, pulverized and transferred into a clean screw-capped glass tube containing 1ml of ice-cold 12.5mM ammonium bicarbonate buffer. The tissue was homogenized on ice using a Tissue TearorTM (Cole-Parmer, Vernon Hills, IL) at low and high speeds in two consecutive 15 s increments for a total of 30 s, and further sonicated on ice (five cycles of 10 s pulses with 10 s breaks between pulses). Eight milliliters of ethanol:acetone and 50 μl each of stable isotope-labeled estrogen internal standards (0.32ng/ml working standard solutions) were added to each tissue homogenate. The mixture was incubated on a rotator at room temperature for 1h and centrifuged at 3000g for 30min. The ethanol:acetone tissue extract was transferred to a clean glass tube and dried under nitrogen gas at 60°C for 60min (Reacti-Vap IIITM, Pierce, Rockford, IL). The residue was redissolved in 4ml of methanol, vortexed for 1min, chilled at 80ºC for 1h, returned to room temperature and centrifuged at 3000g for 20min. The methanolic phase was transferred to a clean glass tube and dried under nitrogen gas. The residue was redissolved in 100 μl of ethanol and vortexed briefly. This step was followed by the addition of 1.5ml of 100mM sodium acetate buffer, pH 4.6, and 5ml of dichloromethane to the residue and incubation at room temperature on a rotator for 30min. The extract was chilled at –80ºC for 10min, returned to room temperature and centrifuged at 3000g for 20min. The dichloromethane phase was transferred to a clean tube and dried. To each dried sample, 40 μl of 0.1M sodium bicarbonate buffer, pH 9.0, and 40 μl of dansyl chloride solution (1mg/ml in acetone) were added. After vortexing for 10 s, samples were heated at 70ºC (Reacti-Therm IIITM Heating Module, Pierce, Rockford, IL) for 10min to form the EM and SI-EM dansyl derivatives. All samples were centrifuged at 3000g for 20min and analyzed using LC-MS2. The efficiency of extracting estrogen and its metabolites from the tissue cannot be measured accurately because a known amount of each metabolite cannot be placed in the tissue prior to extraction. Furthermore, the amount of estrogens/EM present at baseline is unknown. Use of this same extraction protocol to isolate EM from serum, another highly complex protein mixture, yielded extraction efficiencies ranging from 90 to 105% for the various metabolites (22).
Liquid chromatography–tandem mass spectrometry analysis.
LC-MS2 analysis was performed using a Shimadzu Prominence UFLC system (Shimadzu Scientific Instruments, Columbia, MD) coupled with a TSQ™ Quantum Ultra triple quadrupole mass spectrometer (Thermo Electron, San Jose, CA). The LC separation was carried out on a 50mm long × 2mm intradermally column packed with 2.5 μm Synergi Hydro-RP particles (Phenomenex, Torrance, CA) maintained at 40°C. A 20 μl aliquot of each sample was injected onto the column. The mobile phase, operating at a flow rate of 200 μl/min, consisted of methanol as solvent A and 0.1% (vol/vol) formic acid in water as solvent B. A linear gradient (increasing from 72 to 85% solvent A in 15min) was employed for the separation. The MS conditions were as follows: source, ESI; ion polarity, positive; spray voltage, 3500V; sheath and auxiliary gas, nitrogen; sheath gas pressure, 40 arbitrary units; ion transfer capillary temperature, 350ºC; scan type, selected reaction monitoring; collision gas, argon; collision gas pressure, 1.5 mTorr; scan width, 0.7 µm; scan time, 0.01 s; Q1 peak width, 0.70 µm full width at half maximum; Q3 peak width, 0.70 µm FWHM. The optimized selected reaction monitoring conditions for the protonated molecules [MH]+ of EM-dansyl and SI-EM-dansyl were similar to those described previously (25).
Quantitation of tissue estrogens.
Quantitation of lung tissue estrogens and EM was carried out using Xcalibur™ Quan Browser (Thermo Electron) as described previously (25). Briefly, calibration curves for each EM were constructed by plotting EM-dansyl/SI-EM-dansyl peak area ratios obtained from calibration standards versus amounts of the EM injected on column and fitting these data using linear regression with 1/X weighting. The amounts of EM in the tissue samples were then interpolated using this linear function. The lower limit of quantitation of the analytical method was 0.05 pg EM on column and the lower limit of detection was 5–10 times lower than the lower limit of quantitation.
Quantitative RT–PCR
Total RNA was extracted from frozen lung tissue using TRIzol® Reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Reverse transcription was carried out using 1 μg RNA and the High Capacity cDNA Kit (Applied Biosystems, Foster City, CA). Quantitative PCR reactions were performed on the ABI 7900 instrument using TaqMan® Universal Master Mix and gene-specific primer mixes (both from ABI): Cyp1a1 (Mm00487218_m1), Cyp1b1 (Mm00487229_m1), Comt (Mm01171183_m1) and Hprt (Mm00446968_m1). The Ct values for each gene were normalized to the housekeeping gene Hprt, and the fold change in the transcript level of samples from parallel groups (female versus male, smoke treated versus control) was computed using the comparative Ct method (∆∆Ct; Applied Biosystems Reference Manual, User Bulletin #2).
Statistical analyses
The two-sided Wilcoxon rank sum test was used to compare two groups. The difference was considered significant when the P value was ≤ 0.05.
Results
Detection of estrogen and its metabolites in murine lung tissue
Analysis of murine lung tissue has revealed the presence of eight biologically active estrogens/EM within the perfused lungs of male and female wild-type 129SvJ mice (Figure 1). In agreement with our previous LC-MS2 analyses of lung tissue from A/J mice (11), E1 and E2 were also detected within the lungs of 129SvJ mice. E3, the predominant estrogen produced during pregnancy, was also found in the lungs of both male and female mice, but at a concentration (~1 pg/g tissue) much less than that of E1 (≥3 pg/g) or E2 (≥6 pg/g) (Figure 2A). In addition to the three major forms of estrogen, five metabolites of estrogen (2-OHE1, 4-OHE1, 4-OHE2, 2-OMeE1 and 2-OMeE2) were detected in murine lung tissue. The levels of 4-OHE1 within the lungs of both females (7.26 pg/g) and males (3.26 pg/g) were much higher than those of the other metabolites (<1 pg/g). Interestingly, 4-OMeEs were not detected in the murine lung despite the abundance of its precursor 4-OHEs.
Fig. 1.

Representative chromatograms of estrogens and EM detected in the lungs of 129SvJ mice using LC-MS2.
Fig. 2.

Comparison of the levels of estrogen and its metabolites within the lungs of female and male 129SvJ mice (n = 5 per group) as determined by LC-MS2. (A) Absolute levels of each metabolite species. (B–D) Levels of 4-OH, 2-OH and 2-OMe metabolites expressed as a percentage of total estrogen (sum of all estrogens and EM). Values represent the mean ± SEM. Asterisks indicate the EM whose levels are significantly different in males and females. *P ≤ 0.05.
Gender differences in the metabolism of estrogen within the murine lung
Distinct differences were observed in the amount of EM within the lungs of male and female mice (Figure 2). As anticipated, the levels of most EM were higher in the female lung than in the male lung. Both 4-OHE1 and 4-OHE2 were 2-fold higher within the lungs of female mice as compared with male mice; the elevation was significant for the more abundant 4-OHE1 (P = 0.032) but not for the 4-OHE2 metabolite (P = 0.094) (Figure 2A). The concentrations of the putative protective estrogen species, 2-OMeE1 and 2-OMeE2, were also higher in female lungs (P = 0.008 and P = 0.032, respectively). In contrast, the level of 2-OHE1 was comparable in both genders (Figure 2A). Even after normalizing for the amount of total estrogen (sum of estrogen and its metabolites) within the lung, the level of 4-OHEs (4-OHE1 and 4-OHE2) was 60% higher in the female lung than in the male lung (P = 0.016; Figure 2B). The concentration of neither 2-OHE1 nor 2-OMeEs varied significantly between genders when expressed as a percentage of total estrogen (Figure 2C and D).
Impact of Cyp1b1 deletion on estrogen metabolism
Because 4-OHEs have been shown to be carcinogenic (26,27), the contribution of the major estrogen-metabolizing enzyme CYP1B1 to the production of 4-OHEs was investigated next by comparing the profile of EM within the lungs of Cyp1b1-WT and Cyp1b1-KO mice. Deletion of Cyp1b1 led to a dramatic decrease in 4-OHE1 levels in both males (14-fold) and females (21-fold) (7.4 and 4.7% of WT controls, respectively) (Figure 3A). The level of 4-OHE2 was reduced by ~50% in Cyp1b1-KO mice compared with Cyp1b1-WT controls (56% for males and 60% for females) (Figure 3A). When expressed as a percentage of total estrogens, 4-OHE levels dropped from 32 (WT) to 3% (KO) in females and from 23 (WT) to 3% (KO) in males (Figure 3A). These results confirm that 4-OHEs are produced primarily by CYP1B1 in the lung.
Fig. 3.

Impact of Cyp1b1 deletion on estrogen metabolism. (A) Comparison of estrogens and EM levels within the lungs of 129SvJ Cyp1b1-WT and Cyp1b1-KO mice as determined by LC-MS2. Asterisks indicate the metabolites levels that differ significantly in WT and KO mice. (B and C) Expression of Cyp1a1 and Comt in the lungs of Cyp1b1-WT and Cyp1b1-KO mice. The transcript level of each gene was determined by quantitative RT–PCR and normalized against that of the housekeeping gene Hprt. The fold difference was calculated using the Δ ΔCt method. The levels for WT female mice have been set arbitrarily to 1. Values represent the mean ± SEM (n = 5 females and 4 males). *P ≤ 0.05, **P ≤ 0.01.
In contrast with 4-OHEs, the level of 2-OHE1, the primary metabolite of CYP1A1, was elevated significantly in the lungs of both male and female Cyp1b1-KO mice as compared with WT controls (1.7-fold and 3-fold, respectively). These data suggest that estrogen metabolism is shifted toward 2-hydroxylation in the absence of Cyp1b1. Deletion of Cyp1b1 also increased pulmonary levels of 2-OMeE2, a product of the major conjugating enzyme COMT, in both males (3.5-fold) and females (5-fold) as compared with WT controls (Figure 3A). To determine if increases in the production of 2-OHEs and 2-MeOEs were accompanied by alterations in the expression of Cyp1a1 or Comt, transcript levels were measured in Cyp1b1-WT and Cyp1b1-KO mice by quantitative RT–PCR. The mean level of Cyp1a1 transcripts increased non-significantly in both females and males as a result of Cyp1b1 deletion (P = 0.056 and P = 0.28, respectively) (Figure 3B). However, Comt expression was ~2-fold higher within the lungs of Cyp1b1-KO mice compared with those of Cyp1b1-WT mice (P = 0.008 for females and P = 0.016 for males) (Figure 3C).
Significant differences in 4-OHE levels between WT males and females were ameliorated by deletion of Cyp1b1 (Figure 4A). Moreover, 4-OHE levels (percentage of total estrogen) were ~30% lower in female Cyp1b1-KO mice compared with males (P = 0.016) (Figure 4B). In contrast, 4-OHEs represented a larger percentage of total estrogens in female Cyp1b1-WT mice than in males. Consistent with the findings in Cyp1b1-WT mice, no significant difference was observed in 2-OHE1 or 2-OMeEs when expressed as a percentage of total estrogen in male and female Cyp1b1-KO mice (Figure 4C and D).
Fig. 4.

Comparison of estrogens and EM levels within the lungs of female versus male Cyp1b1-KO mice as described in Figure 2. (A) Absolute levels of each metabolite species. (B–D) Levels of 4-OH, 2-OH and 2-OMe metabolites expressed as a percentage of total estrogen (sum of all estrogens and EM). Values represent the mean ± SEM (n = 5 females and 4 males). Asterisks indicate the EM whose levels are significantly different in males and females. *P ≤ 0.05.
Tobacco smoke modulates pulmonary estrogen metabolism
To extend our previous microarray analysis of tobacco smoke-induced alterations in gene expression (11), the effect of smoke exposure on the transcript levels of the estrogen-metabolizing genes Cyp1a1, Cyp1b1 and Comt within the lung was examined. Exposure of female C57/B6-APOE*4 mice to tobacco smoke for 8 weeks led to a 2.3-fold increase in Cyp1b1 expression (P = 0.008; Figure 5B); a response consistent with our previous findings (11). Tobacco smoke also caused a 3.1-fold decrease in the level of Comt mRNA (P = 0.095; Figure 5C). However, no significant change in Cyp1a1 mRNA level was observed following tobacco smoke exposure (Figure 5A).
Fig. 5.
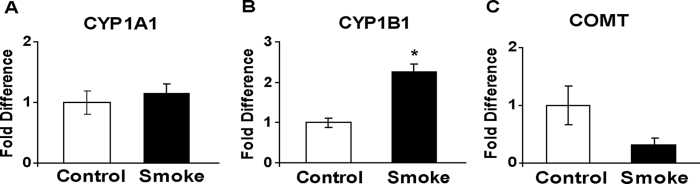
Effect of tobacco smoke on the expression of key estrogen-metabolizing genes. The transcript level of each gene was determined by quantitative RT–PCR and normalized against that of the housekeeping gene Hprt . The fold difference was calculated using the Δ ΔCt method. The levels for control mice have been set arbitrarily to 1. Values are expressed as the mean ± SEM (n = 5 per group). *P ≤ 0.05.
The profile of EM detected within the smoked lung was consistent with the changes in the expression of the estrogen-metabolizing genes that were observed following smoke exposure. Levels of both 4-OHE1 and 4-OHE2 were elevated (4- and 2-fold, respectively) in lung tissue from smoke-exposed mice as compared with those of lung tissue from control mice exposed in parallel to filtered air (Figure 6A). Furthermore, the 4-OHEs (4-OHE1 + 4-OHE2) represented a larger proportion of the total estrogen within the lung following smoke exposure (2-fold higher than that of lungs exposed to filtered air; Figure 6B). In contrast, 2-OHE1 levels, when expressed either as an absolute value or as a percentage of total estrogen, were not altered by smoke exposure (Figure 6C). Furthermore, levels of the putative protective EM 2-OMeE1 and 2-OMeE2 were decreased to 75 and 71% of control, respectively, in lungs exposed to tobacco smoke (Figure 6A). This reduction was also reflected in a decrease in 2-OMeEs as a percentage of total estrogen (49% of control; Figure 6D).
Fig. 6.

Comparison of estrogen metabolite levels within the lungs of mice exposed to filtered air (control) or tobacco smoke (smoked). (A) Absolute level of each metabolite species. (B–D) Levels of 4-OH, 2-OH and 2-OMe metabolites expressed as a percentage of total estrogen (sum of all estrogens and EM). Values represent the mean ± SEM (2 pools of 5 mice per group).
Discussion
Although this group and others have detected E1 and E2 in mouse and human lung previously (2,11), this study represents the first definitive demonstration of estrogen metabolism in lung tissue. Three estrogens and five EM were measured and quantified in murine lung tissue using a highly sensitive LC-MS2 assay. These findings are consistent with results from this group (unpublished data) and others indicating that several key estrogen-metabolizing genes, including CYP1A1 (28), CYP1B1 (23), COMT (20), SULT1A1 (29) and GSTP1 (30), are expressed at the RNA and/or protein level within the lung. In addition to E1 and E2, the detection of pulmonary E3 for the first time suggests the presence of 16-hydroxylation activity in murine lung tissue. Furthermore, successful measurement of 2-OHE and 4-OHE metabolites confirms the catalytic activity of CYP1A1 and CYP1B1, respectively, within the lung. It should be noted that this assay included an initial hydrolysis step to remove sulfate and glucuronide groups from conjugated estrogens and EM. The development and optimization of protocols for the detection of conjugated estrogens will lead to a more comprehensive assessment of the status of estrogen within the lung and enhance our understanding of the association of this profile with lung tumorigenesis.
Interestingly, 4-OHE1, produced primarily by CYP1B1, was found to be the most abundant EM within the normal murine lung. 4-OHE1 is the catechol estrogen most efficient in inducing DNA adducts and transforming cells in culture (31). Furthermore, preclinical studies indicate that 4-OHEs are carcinogenic (26,27). Daily subcutaneous injection of 4-OHE2 induces kidney tumors in Syrian hamsters (26) and uterine adenocarcinomas in CD-1 mice (27). In contrast, the ability of 4-OHEs to cause breast cancer remains controversial. The concentration of 4-OHEs were ~4-fold higher in breast biopsy tissues from women with breast carcinoma than in those from women without cancer (32). However, subcutaneous implants containing 4-OHE1 or 4-OHE2 did not induce mammary tumors in ACI rats, whereas those containing E2 did (33); a result that could be related to the instability of 4-OHEs in the implants.
The presence of higher levels of 4-OHE within the lungs of female mice, as compared with males, is consistent with reports of gender differences in tumor development in mouse models of lung cancer (5,34). In the lung adenocarcinoma Tp53R172H∆g/+/K-RasLA1/+ mouse model that carries oncogenic missense mutations in p53 and K-Ras, females had a significantly higher death rate from cancer than males (34). The higher multiplicity of lung tumors among female Tp53f/f/LSL-K-RasG12D mice has been attributed in part to estrogen, as tumor formation was diminished by ovariectomy, whereas E2 treatment significantly increased tumor multiplicity and volume in both males and ovariectomized females (5). The tumor promoting activity of estrogen is consistent with a higher level of 4-OHE in female mice, as observed in this study.
Results from this study suggest that CYP1B1 may be a potential target for preventing lung cancer: (i) Deletion of Cyp1b1 dramatically reduced levels of carcinogenic 4-OHEs. The small amounts of 4-OHEs that were still detectable within the lungs of Cyp1b1-KO mice (Figure 3A) are most likely produced by CYP1A1, as this enzyme is known to convert estrogens to 4-OHEs, albeit with a much lower efficiency than CYP1B1. (ii) Deletion of Cyp1b1 led to an inconsequential increase in Cyp1a1 transcripts and a significant increase in levels of 2-OHE1 (the only 2-hydroxyestrogen metabolite detected in the murine lung and the major product of CYP1A1). This represents a shift in estrogen metabolism toward the less toxic 2-hydroxylation pathway in the absence of CYP1B1. (iii) Deletion of Cyp1b1 increased the levels of Comt transcripts and 2-OMeE2 metabolites in the murine lung. 2-OMeE2 is not a ligand for the ERs and thus is not estrogenic. Although the antiangiogenic and proapoptotic properties of 2-OMeE2 suggest its potential activity against solid tumors (21), its utility in a clinical setting has been compromised by its low bioavailability (35). Thus, inhibition of CYP1B1 may represent a promising strategy by which to circumvent this barrier and increase 2-OMeE2 levels in humans. Lastly, because CYP1B1 sits at the hub of many signaling pathways (11), its inhibition may block multiple oncogenic events. Knockdown of CYP1B1 with siRNA led to a decrease in the motility of oral leukoplakia cells in vitro (36), suggesting that reducing CYP1B1 may impair tumor metastasis. Therefore, modulation of estrogen metabolism via inhibition of CYP1B1 may represent a novel strategy for the prevention of lung cancer. Indeed, a few selective CYP1B1 inhibitors, such as 2,2′,4,6′-tetramethoxystilbene (37) and homoeriodictyol (3′-methoxy-4′,5,7-trihydroxyflavanone) (38), have been identified. Although no toxicity was observed when tetramethoxystilbene was tested in animals at a low dose (39,40), the feasibility of using these inhibitors for the prevention of lung cancer in humans remains to be determined.
Previous work from this group (11) and others (9,41) demonstrates that tobacco smoke modulates the expression of genes involved in estrogen metabolism. However, this study is the first to report the ability of tobacco smoke to alter the profile of EM within the lung. Although the mice used in this experiment were older and on a high-fat diet, as required for an atherosclerosis study, the results generated are consistent with our previous finding that tobacco smoke significantly increased Cyp1b1 expression and 4-OHE levels. On the other hand, we also observed that the levels of the beneficial 2-OMEs were decreased following exposure to tobacco smoke. These data suggest that tobacco smoke not only promotes lung tumorigenesis by increasing the level of harmful EM but also by decreasing those metabolic species known to confer cellular protection.
Based on the putative protective and carcinogenic properties of certain EM (21,26,27), their profile in biospecimens may reflect one’s risk for cancer. Results from a recent study suggest that the level of EM in blood or urine serves as a surrogate biomarker of breast cancer risk (22,42). Higher ratios of 2-OHEs versus parental estrogens and 4-OMeEs versus 4-OHEs in the serum of postmenopausal women were associated with a lower risk of cancer (22). An inverse association was observed between levels of urinary E1, E2, 2-OHEs and 4-OHEs and breast cancer risk among premenopausal women, suggesting that enhanced excretion of harmful EM decreases one’s risk of cancer (42). Although the relationship between the levels of 2-OHEs and 4-OHEs within the lung and cancer risk remains to be established, it is unlikely that the EM profile of blood or urine will mimic the local synthesis and metabolism of estrogen within lung tissue. Based on the recent discovery of a strong similarity between the transcriptome of the aerodigestive (buccal or nasal) and bronchial epithelium (43,44), a comparison of the EM profile of these more accessible tissues of the upper airway with that of the lung is warranted. Such assays will require the optimization of the current LC-MS2 methodology for the analysis of much smaller samples of tissue.
In summary, the present demonstration of estrogen metabolism in the lung raises new and exciting questions about the potential contribution of EM to lung tumorigenesis. Although the predominance of CYP1B1-mediated 4-hydroxylation of estrogen within the normal lung is surprising, the ability of tobacco smoke to increase tissue levels of the carcinogenic 4-OHEs while decreasing the protective 2-OMeE species is consistent with its promotion of lung tumor development. The observed shift in estrogen metabolism toward the less harmful 2-hydroxylation pathway, with a dramatic decrease of 4-OHEs and an elevation of 2-OMeEs, in the absence of CYP1B1 suggests that CYP1B1 inhibition may represent a promising strategy for the prevention of lung cancer. The relevance of the present murine findings to a clinical setting will only be realized once the EM profile of human lung tissue has been characterized and the potential utility of EM as biomarkers of risk for lung cancer and other tobacco-related cancers (i.e. cervix, bladder, head and neck) has been established.
Funding
National Institutes of Health (CA006927 to Fox Chase Cancer Center and CA167721 to M.C.); Commonwealth of Pennsylvania; Fox Chase Cancer Center Keystone Program in Personalized Risk and Prevention (to M.C.); and generous support to the lab of M.C. from the Estate of Jane Villon; Jerome M. Spencer and Arnold Zaslow Family Fund; and the Kitty Jackson Fund.
Conflict of Interest Statement: None declared.
Acknowledgements
We thank Drs Denise Connolly, Edna Cukierman and Andres Klein-Szanto for their constructive comments, Dr Sibele Meireles for her insightful scientific discussions and Maureen Climaldi for her excellent assistance in preparing this article for publication. The following facilities were used: Biostatistics and Bioinformatics Facility and the Laboratory Animal Facility at Fox Chase Cancer Center; and the Laboratory Animal Facility at Duke University.
Glossary
Abbreviations:
- COMT
catechol-O-methyltransferase
- EM
estrogen metabolites
- ER
estrogen receptor
- KO
knockout
- LC-MS
liquid chromatography–tandem mass spectrometry
- OHEs
hydroxyestrogens
- OmeEs
methoxyestrogens
- WT
wild-type.
References
- 1. Centers for Disease Control and Prevention (2011) Vital signs: current cigarette smoking among adults aged ≥18 years—United States, 2005–2010. Morb. Mortal. Wkly. Rep., 60, 1207–1212 [PubMed] [Google Scholar]
- 2. Niikawa H, et al. (2008) Intratumoral estrogens and estrogen receptors in human non-small cell lung carcinoma. Clin. Cancer Res., 14, 4417–4426 [DOI] [PubMed] [Google Scholar]
- 3. Stabile L.P, et al. (2002) Human non-small cell lung tumors and cells derived from normal lung express both estrogen receptor α and β and show biological responses to estrogen. Cancer Res., 62, 2141–2150 [PubMed] [Google Scholar]
- 4. Dougherty S.M, et al. (2006) Gender difference in the activity but not expression of estrogen receptors α and β in human lung adenocarcinoma cells. Endocr. Relat. Cancer, 13, 113–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hammoud Z, et al. (2008) Estrogen promotes tumor progression in a genetically defined mouse model of lung adenocarcinoma. Endocr. Relat. Cancer, 15, 475–483 [DOI] [PubMed] [Google Scholar]
- 6. Chlebowski R.T, et al. (2009) Oestrogen plus progestin and lung cancer in postmenopausal women (Women’s Health Initiative trial): a post-hoc analysis of a randomised controlled trial. Lancet, 374, 1243–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouchardy C, et al. (2011) Lung cancer mortality risk among breast cancer patients treated with anti-estrogens. Cancer, 117, 1288–1295 [DOI] [PubMed] [Google Scholar]
- 8. Mollerup S, et al. (2002) Expression of estrogen receptors α and β in human lung tissue and cell lines. Lung Cancer, 37, 153–159 [DOI] [PubMed] [Google Scholar]
- 9. Port J.L, et al. (2004) Tobacco smoke induces CYP1B1 in the aerodigestive tract. Carcinogenesis, 25, 2275–2281 [DOI] [PubMed] [Google Scholar]
- 10. Spivack S.D, et al. (2003) Phase I and II carcinogen metabolism gene expression in human lung tissue and tumors. Clin. Cancer Res., 9(16 Pt 1)6002–6011 [PubMed] [Google Scholar]
- 11. Meireles S.I, et al. (2010) Early changes in gene expression induced by tobacco smoke: evidence for the importance of estrogen within lung tissue. Cancer Prev. Res. (Phila)., 3, 707–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murray G.I, et al. (2001) Regulation, function, and tissue-specific expression of cytochrome P450 CYP1B1. Annu. Rev. Pharmacol. Toxicol., 41, 297–316 [DOI] [PubMed] [Google Scholar]
- 13. Chen B, et al. (2010) The CYP1B1 Leu432Val polymorphism contributes to lung cancer risk: evidence from 6501 subjects. Lung Cancer, 70, 247–252 [DOI] [PubMed] [Google Scholar]
- 14. Cote M.L, et al. (2009) Tobacco and estrogen metabolic polymorphisms and risk of non-small cell lung cancer in women. Carcinogenesis, 30, 626–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rotunno M, et al. (2009) Phase I metabolic genes and risk of lung cancer: multiple polymorphisms and mRNA expression. PLoS ONE, 4, e5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu W, et al. (2012) Current evidence on the relationship between CYP1B1 polymorphisms and lung cancer risk: a meta-analysis. Mol. Biol. Rep., 39, 2821–2829 [DOI] [PubMed] [Google Scholar]
- 17. Cavalieri E.L, et al. (2004) A unifying mechanism in the initiation of cancer and other diseases by catechol quinones. Ann. N. Y. Acad. Sci., 1028, 247–257 [DOI] [PubMed] [Google Scholar]
- 18. Sepkovic D.W, et al. (2009) Estrogen hydroxylation: the good and the bad. Ann. N. Y. Acad. Sci., 1155, 57–67 [DOI] [PubMed] [Google Scholar]
- 19. Zahid M, et al. (2006) The greater reactivity of estradiol-3,4-quinone vs estradiol-2,3-quinone with DNA in the formation of depurinating adducts: implications for tumor-initiating activity. Chem. Res. Toxicol., 19, 164–172 [DOI] [PubMed] [Google Scholar]
- 20. Männistö P.T, et al. (1999) Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol. Rev., 51, 593–628 [PubMed] [Google Scholar]
- 21. Verenich S, et al. (2010) Therapeutic promises of 2-methoxyestradiol and its drug disposition challenges. Mol. Pharm., 7, 2030–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fuhrman B.J, et al. (2012) Estrogen metabolism and risk of breast cancer in postmenopausal women. J. Natl Cancer Inst., 104, 326–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buters J.T, et al. (1999) Cytochrome P450 CYP1B1 determines susceptibility to 7, 12-dimethylbenz[a]anthracene-induced lymphomas. Proc. Natl Acad. Sci. USA, 96, 1977–1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Knouff C, et al. (1999) Apo E structure determines VLDL clearance and atherosclerosis risk in mice. J. Clin. Invest., 103, 1579–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu X, et al. (2007) Quantitative measurement of endogenous estrogens and estrogen metabolites in human serum by liquid chromatography-tandem mass spectrometry. Anal. Chem., 79, 7813–7821 [DOI] [PubMed] [Google Scholar]
- 26. Liehr J.G, et al. (1986) Carcinogenicity of catechol estrogens in Syrian hamsters. J. Steroid Biochem., 24, 353–356 [DOI] [PubMed] [Google Scholar]
- 27. Newbold R.R, et al. (2000) Induction of uterine adenocarcinoma in CD-1 mice by catechol estrogens. Cancer Res., 60, 235–237 [PubMed] [Google Scholar]
- 28. Androutsopoulos V.P, et al. (2009) Cytochrome P450 CYP1A1: wider roles in cancer progression and prevention. BMC Cancer, 9, 187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Misra V, et al. (2007) Global expression profiles from C57BL/6J and DBA/2J mouse lungs to determine aging-related genes. Physiol. Genomics, 31, 429–440 [DOI] [PubMed] [Google Scholar]
- 30. Zhou J, et al. (2008) Glutathione transferase P1: an endogenous inhibitor of allergic responses in a mouse model of asthma. Am. J. Respir. Crit. Care Med., 178, 1202–1210 [DOI] [PubMed] [Google Scholar]
- 31. Yagi E, et al. (2001) The ability of four catechol estrogens of 17beta-estradiol and estrone to induce DNA adducts in Syrian hamster embryo fibroblasts. Carcinogenesis, 22, 1505–1510 [DOI] [PubMed] [Google Scholar]
- 32. Rogan E.G, et al. (2003) Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: potential biomarkers of susceptibility to cancer. Carcinogenesis, 24, 697–702 [DOI] [PubMed] [Google Scholar]
- 33. Turan V.K, et al. (2004) The effects of steroidal estrogens in ACI rat mammary carcinogenesis: 17β-estradiol, 2-hydroxyestradiol, 4-hydroxyestradiol, 16α-hydroxyestradiol, and 4-hydroxyestrone. J. Endocrinol., 183, 91–99 [DOI] [PubMed] [Google Scholar]
- 34. Zheng S, et al. (2007) A genetic mouse model for metastatic lung cancer with gender differences in survival. Oncogene, 26, 6896–6904 [DOI] [PubMed] [Google Scholar]
- 35. Dahut W.L, et al. (2006) Phase I clinical trial of oral 2-methoxyestradiol, an antiangiogenic and apoptotic agent, in patients with solid tumors. Cancer Biol. Ther., 5, 22–27 [DOI] [PubMed] [Google Scholar]
- 36. Shatalova E.G, et al. (2011) Estrogen and cytochrome P450 1B1 contribute to both early- and late-stage head and neck carcinogenesis. Cancer Prev. Res. (Phila)., 4, 107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim S, et al. (2002) Design, synthesis, and discovery of novel trans-stilbene analogues as potent and selective human cytochrome P450 1B1 inhibitors. J. Med. Chem., 45, 160–164 [DOI] [PubMed] [Google Scholar]
- 38. Doostdar H, et al. (2000) Bioflavonoids: selective substrates and inhibitors for cytochrome P450 CYP1A and CYP1B1. Toxicology, 144, 31–38 [DOI] [PubMed] [Google Scholar]
- 39. Kim T, et al. (2011) Tetra-methoxystilbene modulates ductal growth of the developing murine mammary gland. Breast Cancer Res. Treat., 126, 779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park H, et al. (2007) Effects of tetramethoxystilbene on hormone-resistant breast cancer cells: biological and biochemical mechanisms of action. Cancer Res., 67, 5717–5726 [DOI] [PubMed] [Google Scholar]
- 41. Tan X.L, et al. (2009) Smoking-related gene expression in laser capture-microdissected human lung. Clin. Cancer Res., 15, 7562–7570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eliassen A.H, et al. (2012) Urinary estrogens and estrogen metabolites and subsequent risk of breast cancer among premenopausal women. Cancer Res., 72, 696–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boyle J.O, et al. (2010) Effects of cigarette smoke on the human oral mucosal transcriptome. Cancer Prev. Res. (Phila)., 3, 266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sridhar S, et al. (2008) Smoking-induced gene expression changes in the bronchial airway are reflected in nasal and buccal epithelium. BMC Genomics, 9, 259 [DOI] [PMC free article] [PubMed] [Google Scholar]