


Abstract
Hepatitis C Virus (HCV) and other plus-strand RNA viruses typically require the generation of a small number of negative genomes (20–100× lower than the positive genomes) for replication, making the less-abundant antigenome an attractive target for RNA interference(RNAi)-based therapy. Because of the complementarity of duplex short hairpin RNA/small interfering RNA (shRNA/siRNAs) with both genomic and anti-genomic viral RNA strands, and the potential of both shRNA strands to become part of the targeting complexes, preclinical RNAi studies cannot distinguish which viral strand is actually targeted in infected cells. Here, we addressed the question whether the negative HCV genome was bioaccessible to RNAi. We first screened for the most active shRNA molecules against the most conserved regions in the HCV genome, which were then used to generate asymmetric anti-HCV shRNAs that produce biologically active RNAi specifically directed against the genomic or antigenomic HCV sequences. Using this simple but powerful and effective method to screen for shRNA strand selectivity, we demonstrate that the antigenomic strand of HCV is not a viable RNAi target during HCV replication. These findings provide new insights into HCV biology and have important implications for the design of more effective and safer antiviral RNAi strategies seeking to target HCV and other viruses with similar replicative strategies.
INTRODUCTION
HCV is an enveloped virus with a positive-sense single-stranded RNA genome belonging to the Flaviviridae family, which also includes viruses such as dengue virus, yellow fever virus and bovine viral diarrhea virus (1). HCV infection afflicts >150 million people worldwide, with infection rates >20% in some countries (2). The ∼9.6 kb HCV genome encodes for one long proteolyticaly processed polyprotein flanked by highly conserved essential non-translated regions (NTRs) at the 5′ and 3′ ends that encode essential regulatory signals for initiating antigenomic (3′NTR) and genomic (5′NTR) RNA synthesis. One of the most significant features of HCV RNA is its high degree of genomic variability (3). The low fidelity of virus-encoded RNA-dependent RNA polymerase, with mutation rates of 10−4–10−3 base substitution/nucleotide (4), is responsible for the high genetic diversity of HCV. This results in a classification of seven genotypes harbouring >90 subtypes and countless quasispecies present in patient populations (5–7).
The fact that HCV is an RNA virus with a single-stranded viral RNA genome of positive polarity, together with the fact that the viral genome does not enter the cell nucleus, and all stages of HCV replication occur in the cytoplasm, makes HCV an ideal target for RNAi-based therapy. The efficacy of RNAi against viral infections has been well documented [reviewed in (8,9)], and knockdown of replicating hepatitis viruses has been demonstrated (10,11). Furthermore, RNAi is well suited as a therapeutic strategy allowing for simultaneous targeting of multiple viral sequences (10) reducing the probability of the emergence of RNAi-resistant mutants (10,11).
It is now understood that there are constraints on the amount of RNAi that can be safely introduced into the target cells. There is a rate-limiting amount of RNAi tolerated in tissues (12–14), and at higher doses, combinatorial RNAi strategies can increase the risk of RNAi-related toxicity and can result in competition limiting their relative effectiveness (15). Thus, to increase the effectiveness of anti-HCV RNAi therapy, only the most active si/shRNA molecules targeting the most accessible and preserved HCV regions should be used. However, the exact HCV target for RNAi is not clear. Although HCV is thought to possess extensive internal base pairing throughout its genome (16), with the exception of very small regions of the genome for which experimental data exist, it is not known which regions are structured and which regions might be more accessible to RNAi targeting. Even less is known about the structure of the negative antigenome as well as the extent of its interactions with protein factors [reviewed in (17)]. Because the negative sense antigenome is present in 20–100 times lower quantities than the positive sense genome, the antigenome potentially represents a more efficacious and safer target, as in theory, a lower dose of duplex RNAs would be required to achieve clinically relevant levels of knockdown of this viral RNA species. However, it is not known whether the negative HCV genome is accessible to standard RNAi, with a number of reports demonstrating data supporting negative HCV strand targeting (8,18,19), whereas others present contradictory results where modifications aimed at favouring negative HCV strand targeting actually led to the overall decrease of anti-HCV RNAi (20). Owing to specific limitations in the experimental approaches used in the previous studies, the results were not able to answer the question of direct RNAi-mediated negative strand HCV genome targeting.
Recent advances in RNAi technology and better understanding of the mechanisms involved in si/shRNA processing allow the design of si/shRNA molecules that show preferential strand loading into an active RNA-induced silencing complex (RISC) (21). Should the accessibility of the positive and negative HCV genome to standard RNAi therapy be known, the design of si/shRNA molecules could be designed to optimize efficacy and minimize toxicity. If both strands of the viral genome were accessible to RNAi, it might be beneficial to use si/shRNA molecules that preferentially target the negative strand because of the lower amount of negative strand HCV genomes in infected cells. However, if the negative HCV genome were not accessible to RNAi therapy, it would be beneficial to use si/shRNA where the strand targeting the positive genome was preferentially loaded into RISC. Unnecessary loading of siRNAs against the negative strand genomes while being therapeutically irrelevant use the endogenous RNAi machinery and can lead to toxicity associated with RNAi pathway overload and/or off-target effects.
Studies of the negative HCV genome are technically challenging owing to the low abundance of this RNA species, making many of the standard techniques used in RNA analysis (like northern blotting) unreliable. Fluorescent in situ hybridization techniques have been effectively applied to study positive and negative genomes of other RNA viruses, like polio (22), but not HCV. Polymerase chain reaction (PCR)-based techniques are complicated by the co-existence of both RNA species. Even functional tests based on RNAi have proven difficult to interpret. As HCV replication occurs through a (−), antigenome, intermediate (23), and both strands of si/shRNA can be incorporated into active RISC complexes (24,25), both strands can possibly participate in RNAi-mediated HCV degradation. Thus, standard RNAi screens cannot distinguish between positive and/or negative strand targeting. Furthermore, shRNAs with preferential RISC loading properties based on thermodynamic stability are not absolute, and even a small amount of ‘unwanted’ active RISC can lead to powerful knockdown of the target RNAs, making it difficult to definitively establish HCV negative strand accessibility to RNAi.
To overcome these limitations and provide proof of positive versus negative HCV strand targeting, in the present study, we screened and selected efficient HCV RNAi targets, and then designed ‘asymmetric’ shRNAs directed against individual polarities of HCV genome. By quantifying the ability of each shRNA to functionally reduce HCV replication, we were able to address the accessibility of HCV genomes to standard shRNA-based RNAi.
MATERIALS AND METHODS
rAAV–shRNA cloning and vector production
All newly designed shRNAs have a 24 nt stem and 5′-GAAGCTTG-3′ loop B (13). Each shRNA was ordered from Integrated DNA Technologies (IDTDNA.com) as two complementary oligos, 5′-CACC-G(+1)-sense-GAAGCTTG-antisense-3′ and 3′-C-antisense′-CTTCGAAG-sense′-AAAA-5′. G was introduced at position +1, unless natural G existed at +1 position. Oligos were annealed and ligated downstream from the H1 promoter in a BbsI digested scAAV-RSV-GFP-H1 plasmid. The scAAV–shRNA vectors were produced by triple transfection of 293 cells in 225 cm2 flasks as previously described (13). Bulges in sense or antisense strands of shRNA were introduced by changing the 9th, 10th and 11th nucleotides to their complements and doubling the 10th nucleotide.
psiCheck reporter cloning, Huh7.5 cell transfection and dual luciferase assay
To generate psiCheck reporter constructs, appropriate short template were cloned into the Renilla luciferase (hRLuc) gene 3′UTR in psiCheck-2 construct (Promega). Templates were ordered from Integrated DNA Technologies as two complementary oligos, which on annealing generated double-stranded templates with appropriate overhangs for cloning into XhoI-NotI-digested psiCheck construct. For large HCV templates, PCR oligos were designed to amplify the 5′NTR-CORE (from position 1–547 in recombinant HCV J6 (5′UTR-NS2)/JFH1 GenBank: JF343782.1) or 3′ region of NS5B-3′NTR (from position 9185–9679 in J6 (5′UTR-NS2)/JFH1 GenBank: JF343782.1) regions of HCV genome 2 a. Oligos contained XhoI or NotI cloning sites. PCR products were cloned into Zero Blunt® TOPO® PCR Cloning Kit (Invitrogen) and sequenced to ensure that mutations were not introduced during the PCR step. TOPO plasmids containing appropriate HCV templates were subsequently digested with XhoI-NotI, and released HCV templates were cloned into XhoI-NotI-digested psiCheck construct. Each HCV target (both short and long) was cloned in sense and antisense orientations with respect to the hRLuc gene, to mimic positive and negative HCV genomes on a RNA level when expressed as hRLuc–HCV fusions.
Each psiCheck construct containing HCV template was checked for Renilla:Firefly signal ratio to ensure that addition of shRNA targets did not significantly affect the expression level of Renilla luciferase.
The psiCheck reporters were transfected into Huh7.5 cells using Lipofectamine 2000 reagent (Invitrogen) as per the manufacturer’s instructions. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) media supplemented with 2 mM L-glutamine and 0.1 mM MEM non-essential amino but without Pen/Strep. Eight hours after transfection, cells were washed once with DMEM and an appropriate volume of DMEM media with supplements and with Pen/Strep was added. Cells were harvested 64 h later and used for luciferase expression analysis using Dual-Luciferase® Reporter Assay System (Promega) using Tecan M-1000 luminometer.
Cells and media
Huh7.5 cells were propagated in complete media DMEM media (Mediatech, VA, USA), supplemented with 2 mM L-glutamine (Life Technologies, NY), 100 U/ml of penicillin (Life Technologies, NY), 100 ug/ml of streptomycin (Life Technologies, NY), 0.1 mM MEM non-essential amino acids (Life Technologies, NY) and 10% fetal bovine serum (HyClone). The cells were grown in a 37°C incubator adjusted to 5% CO2.
RNA transcription and transfection
HCV RNA (J6/JFH chimeric genome or FL-J6/JFH-5′C19Rluc2AUbi reporter genome) was transcribed in vitro as described (26). Five micrograms of in vitro transcribed RNA were mixed with 4 × 106 Huh7.5 cells in RNase-free phosphate buffered saline (Biowhittaker) and transferred into a 2-mm diameter gap cuvette (BTX). The cells were pulsed using a BTX model 830 electroporator (0.68 kV and five periods of 99 µs at 500-ms intervals) and then left to recover for 15 min at room temperature followed by dilution in 10 ml of pre-warmed complete medium. For transient replication experiments, the cells were counted and plated at a density of 1.5 × 104 cells per well in a 96-well plate. The cells were incubated for 3–4 h at 37°C and then transduced with rAAV as described later in the text. For stable replication, the cells were plated in a 10-cm dish and grown at 37°C. Forty-eight hours post electroporation, the cells were trypsinized and plated at a density of 1.5 × 104 cells per well in a 96-well plate and 24 h later were infected with AAV as described later in the text. For virus production, the cells were plated in 75 cm2 flasks and maintained as described later in the text.
Cell viability and Luciferase assay
Cell viability in each well was assessed using the Alamar Blue assay (Invitrogen) according to the manufacturer’s protocol. Following viability measurement, the cells were washed twice with ice-cold phosphate buffered saline and 30 µl of Renilla luciferase assay buffer (Promega, WI, USA) was added to lyse the cells. Renilla luciferase activity was measured and integrated for 10 s using a Tecan M-1000 luminometer. Cell viability was used for normalization purposes.
Virus particle production
Huh7.5 cells were electroporated with RNA of the J6/JFH HCV chimeric genome as described earlier in the text. The cells were maintained in complete media at low density (maximum of 60% confluence), and the media was collected for a period of 2 weeks and stored at −80°C. The collected media was pooled and concentrated 100-fold using a Stirred Cell (Millipore) equipped with a 100-Kd filter. The resulting concentrated media was filtered through a 0.2 µm syringe filter (Millipore), aliquoted and kept at −80°C until use. The viral titer was determined following infecting naïve Huh7.5 cells with increasing dilutions of the virus for 72 h. The cells were immunostained, and core positive foci were counted to determine the inoculum’s infectivity in focus-forming unit (FFU)/ml.
HCV infection and rAAV transduction
Naïve Huh7.5 cells were plated at high density a day before the experiment in a 96-well plate (1.5 × 104 cells per well). The virus was diluted in complete media to enable infection at a multiplicity of infection of 0.1 when applied onto the cells in a volume of 50 µl and incubated at 37°C for 2 h. Two hours post infection, the virus was removed from the cells, and the wells were washed four times with 100 µl of complete media to completely remove leftover virus. Following the last wash, 50 µl of media was added to the cells and supplemented with 50 µl scAAV-RSV-GFP-H1-shRNA. All rAAV vectors were previously titrated on Huh7.5 cells and diluted so that 50 ul of rAAV resulted in 80–90% transduction based on GFP expression.
Real-time PCR
RNA was extracted from infected cells using RNeasy 96 kit (Qiagen) according to the manufacturer’s protocol. Real-time PCR was performed with 5 µl of extracted RNA using AgPath ID RT-PCR Master Mix (Applied Biosystems, Foster City, CA), primers AGAGCCATAGTGGTCT and CCAAATCTCCAGGCATTGAGC, and probe 6-carboxyfluorescein-CACCGGAATTGCCAGGACGACCGG-6-carboxytetramethylrhodamine. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) RNA was quantified using GAPDH control reagents (Applied Biosystems) according to the manufacturer’s instructions. The relative amount of HCV RNA was adjusted to GAPDH RNA and normalized to scrambled shRNA-treated sample.
Delta-G determinations
Delta-G (dG) values were determined by the widely used programme ‘RNAstructure’ that enabled to predict secondary RNA structure and their dG values (27) and dG value for the most favourable structure is reported.
RESULTS
To investigate whether the negative HCV genome is bioaccessible to standard RNAi therapy, we wanted to take advantage of asymmetric shRNAs, where introduction of a bulge at the cleavage site eliminates the shRNA passenger strand from RNAi-mediated cleavage of its target even if it is loaded into RISC (Figure 1A). This allows one to distinguish the strand-specific activity of an sh/siRNA when there is a potential to target complementary RNA strands as in the case of HCV infection/replication (23). We started by performing an extensive screen to identify which regions of the HCV genome can be most effectively targeted by rAAV-encoded shRNA. To do so, we examined the HCV genome and identified regions of homology between genotypes 1b and 2a and then designed 24 nt overlapping shRNAs staggered every 4 bp targeting the homologous regions in 5′NTR, CORE, NS5B and 3′NTR (Figure 1B). All shRNAs were selected without taking into consideration the thermodynamic stability of the hairpin and thus without any bias that might affect strand loading into active RISC. We also included a few shRNA sequences previously reported to effectively knockdown HCV (20,28). Eighty-six unique shRNAs (Supplementary Table S1) were designed and cloned into recombinant self-complementary AAV (scAAV) vectors (scAAV-RSV-GFP-H1, Figure 1C) under the H1 promoter, which allows for safer shRNA expression in vivo than the U6 promoter (13). All shRNAs were tested for their activity against replication of a chimeric J6/JFH HCV genome with a luciferase reporter gene (FL-J6/JFH-5′C19Rluc2AUbi, here referred to as FL-J6/JFH-Luc) (29) in both transient- (shRNA treatment of cells shortly after HCV RNA transfection, Figure 2A) and stable- (shRNA treatment 72 h post establishment of HCV replication, Figure 2B)-replication models, as well as against fully infectious virus-HCVcc (26) (Figure 2C). As shown in Figure 2A–C, a number of highly active shRNAs capable of specifically knocking down HCV were identified in the 5′NTR, CORE and NS5B regions, whereas, as reported previously (30,31), even the most active shRNA against the 3′NTR led only to 55% inhibition of HCV replication. Any specific activity against the 3′NTR was observed only in the transient-replication model and against HCVcc (Figure 2A and C), suggesting that the 3′NTR-specific shRNAs were only active against incoming and not replicating HCV RNAs. Similar results were also obtained when a number of different shRNAs were tested against HCV 1b in a transient-replication model (data not shown).
Figure 1.

(A) Graphic representation of asymmetric shRNAs and the effect of introduced bulges on shRNA function against (−) HCV and (+) HCV genomes. HCV templates cloned into psiCheck reporter system in the sense orientation mimic the (+) HCV genome, whereas HCV templates cloned in antisense orientation mimic (−) HCV genome. (B) Anti-HCV-shRNA coverage plot. The y-axis represents number of shRNA sequences covering each nucleotide at a given position along the HCV genome (x-axis). (C) Graphic representation of scAAV-RSV-GFP-H1 vectors used in the study. ITR, AAV inverted terminal repeat; RSV, Rous sarcoma virus promoter; eGFP, enhanced green fluorescent protein.
Figure 2.

All shRNAs were tested against replicating FL-J6/JFH-Luc HCV in a transient-replication model (A), a stable-replication model (B), as well as against HCVcc infection (C) in Huh7.5 cells. Upper portions of panels A–C show graphic representation of each experimental design. The number corresponding to each shRNA represents the position of the first nucleotide in the recombinant HCV J6(5′UTR-NS2)/JFH1 (GenBank: JF343782.1). The shRNA-SCR was used as negative control and was assigned a value of 100. In (A) and (B), FL-J6/JFH-Luc luciferase levels were normalized to AAV-shRNA encoded eGFP to account for transduction efficiency, whereas in (C), GAPDH quantitative PCR levels and AAV-shRNA eGFP levels were used to normalize the HCV RNA levels measured by quantitative PCR. Error bars represent SD from n = 3–6.
To gain insight into which strand of the shRNA is incorporated into an active RISC complex, we took advantage of the psiCheck reporter system. Based on the results obtained with FL-J6/JFH-Luc and HCVcc infection (Figure 2A–C), 26 active and inactive shRNAs were selected for further analysis. All selected shRNAs were tested using the psiCheck reporter system harbouring specific targets for each shRNA cloned into the 3′UTR of the RLuc reporter gene in the sense (psiCheck-short-sense) or antisense (psiCheck-short-antisense) orientation (Supplementary Figure S1A) so as to mimic positive and negative HCV genomes targets, respectively.
The data obtained with the psiCheck system did not always correspond to what was observed when targeting full-length HCV genomes (compare Figure 2A–C and Supplementary Figure S2A). All but three of the shRNAs that were effective at knocking down FL-J6/JFH-Luc and the HCVcc replication effectively knocked down luciferase expression in this simplified system. Three shRNA constructs that were highly active in FL-J6/JFH-Luc replication and HCVcc infection screens (14, 302 and 353) showed specific knockdown activity only against the psiCheck-sense sequences. However, the converse was not always true (Supplementary Figure S2A). Of the inactive control shRNAs, some exhibited good activity against psiCheck reporter both in forward and reverse orientation (clones 52, 9589, 9601 and 9633, Supplementary Figure S2), and two (shRNA 74 and 9625) showed significant activity only against the reporter in forward orientation.
These results are in agreement with previously published work (24,25) showing that both of the shRNA strands (5p and 3p) can be loaded into an active RISC complex and thus potentially target either viral strand during viral replication. Thus, the use of conventional shRNAs will not allow one to distinguish positive from negative HCV genome targeting. To be able to distinguish which HCV genome undergoes shRNA-mediated targeting, we needed to uncouple positive genome targeting from negative genome targeting. Thus, we generated asymmetric shRNAs capable of targeting only the positive or the negative genome of HCV. To do so, a bulge was introduced between positions 9–11 in the sense or antisense strand (sense bulge and antisense bulge, respectively) of each shRNA (Figure 1A and Supplementary Table S1), and all asymmetric shRNAs were tested for their specific activity using the psiCheck reporter system (Supplementary Figure S2B and C).
Introduction of bulges into the shRNA at the predicted cleavage site led to a dramatic change in the activity of many of the shRNAs (Supplementary Figure S2), an observation in agreement with our previously published data (32). When a bulge was introduced into the sense strand of shRNA, which should only affect function against the psiCheck with target sequences in the antisense orientation (psiCheck-short-antisense), a few shRNAs (e.g. 6, 74, 353 and 9255) demonstrated decrease in activity against the psiCheck-short-sense reporter. Similar observations were made for a few shRNAs with bulges in the antisense strand, where the presence of the bulge affected function against the reporter that is not targeted by antisense strand (psiCheck-short-sense) (e.g. shRNA 134, 276, 318, 9589, 9601 and 9633). In one case (shRNA 14), the activity against psiCheck-short-antisense was actually improved on introduction of a bulge in the antisense strand. These data clearly show that the effect of a bulge on shRNA-gene knockdown is impossible to predict, and each shRNA has to be tested in psiCheck or a similar reporter system to confirm the activity. Similar effects were also seen when shRNA with bulges were tested against FL-J6/JFH-Luc replication and HCVcc infection (Supplementary Figure S3), where introduction of a sense bulge affected shRNA function by lowering the specific activity when compared with an shRNA without bulges. The efficacy and sensitivity of the asymmetric shRNA was further confirmed in a rescue experiment, in which psiCheck-HCV templates were mutated to match the bulges in corresponding shRNA (Supplementary Figures S4 and S5).
Based on the results presented in Figure 2A–C and Supplementary Figure S2A–C, we selected six shRNAs against various regions of the HCV genome that despite the presence of a sense or an antisense bulge retained the proper processing and strand-specific activity when tested using the psiCheck system (Figure 3). As observed previously, shRNAs without bulges effectively targeted HCV sequences cloned in both the sense and antisense orientation in the psiCheck reporter system (Figure 3A). When a bulge was introduced in the sense strand of each shRNA, leaving only the antisense strand of shRNA available to direct HCV-specific cleavage, as predicted (Figure 1A), only the psiCheck reporter constructs harbouring target sequences cloned in the sense orientation underwent specific knockdown (Figure 3B). The converse was observed when shRNAs with bulges in the antisense strand were tested against psiCheck reporter constructs harbouring HCV target sequences in sense or antisense orientation (Figure 3C).
Figure 3.
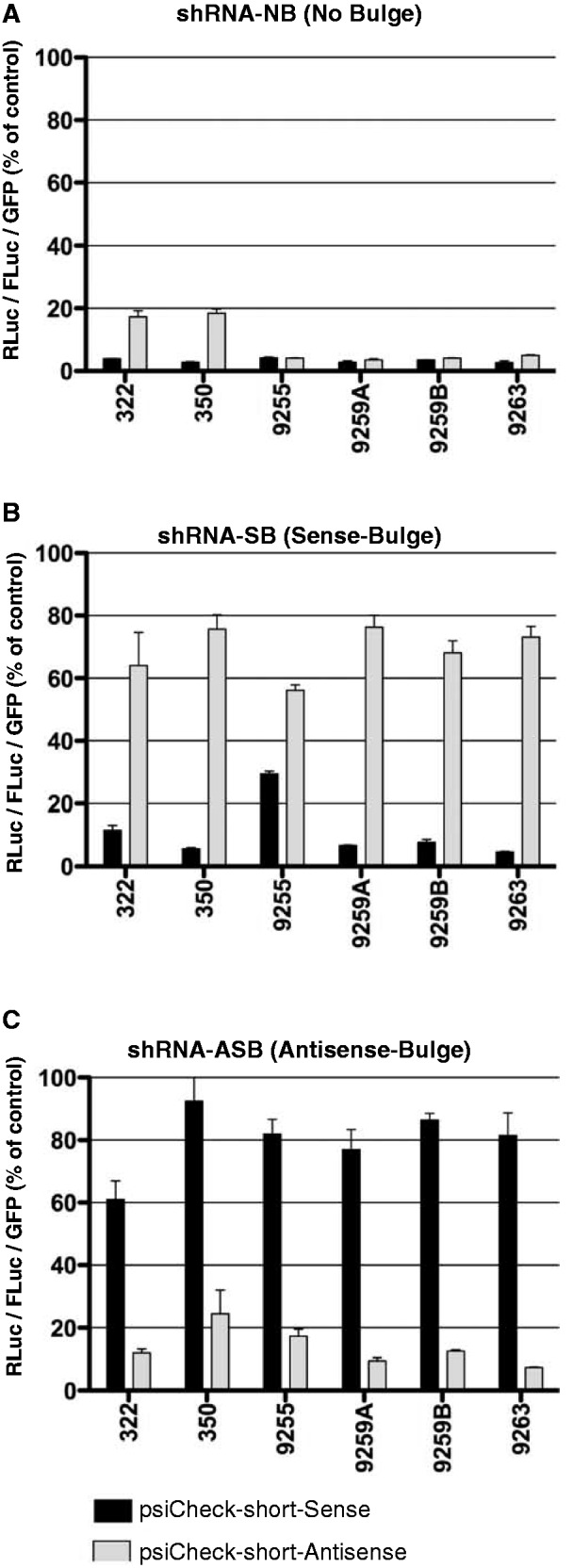
Asymmetric shRNA validation experiment. Selected shRNAs without bulges (A), or with bulges in the sense strand (B) or antisense strand (C) were tested using psiCheck reporter system containing specific short targets in the 3′UTR of RLuc cloned in sense (black bars) or antisense (gray bars) orientations. Both strands of shRNA without bulges can be incorporated into active RISC leading to specific cleavage and degradation of template cloned in sense and antisense orientation (A). Introduction of a bulge into sense shRNA strand (B) makes it inactive against template cloned in reverse orientation into psiCheck and thus mimicking (−) HCV genome target, leading to rescue of RLuc expression (gray bars). The opposite effect is observed with shRNA containing bulges in the antisense shRNA strand (C), which interferes with targeting of the specific templates cloned in sense orientation (black bars). Each psiCheck reporter was also treated with shRNA-SCR, which was assigned a value of 100 (not shown in the graphs). Signal from RLuc containing HCV templates in the 3′UTR were normalized to levels of FLuc encoded on the same plasmid, whereas AAV-shRNA vector encoded eGFP was used to normalize for transduction efficiency (shRNA levels). Error bars represent SD from n = 3.
Now that the shRNA-bulge system was fully validated and we were able to select a number of shRNAs that were active only against sense (+HCV genome) or antisense (−HCV genome) templates, we were able to directly establish whether the negative genome of HCV could be selectively targeted with RNAi (Figure 1A).
When FL-J6/JFH-Luc HCV was targeted with an shRNA without a bulge (shRNA-NB) or with a bulge in the sense strand (shRNA-SB), HCV replication was specifically reduced as compared with scrambled (SCR) shRNA control (Figure 4A white and gray bars, respectively). However, when an shRNA containing a bulge in the antisense strand (shRNA-ASB) was used (black bars in Figure 4A), only allowing for cleavage of the (−)HCV genome, no significant knockdown of HCV was detected for any of the selected shRNAs targeting different regions of the HCV genome. This clearly demonstrated that the negative genome of replicating FL-J6/JFH-Luc HCV was not targetable by standard vector encoded shRNA.
Figure 4.
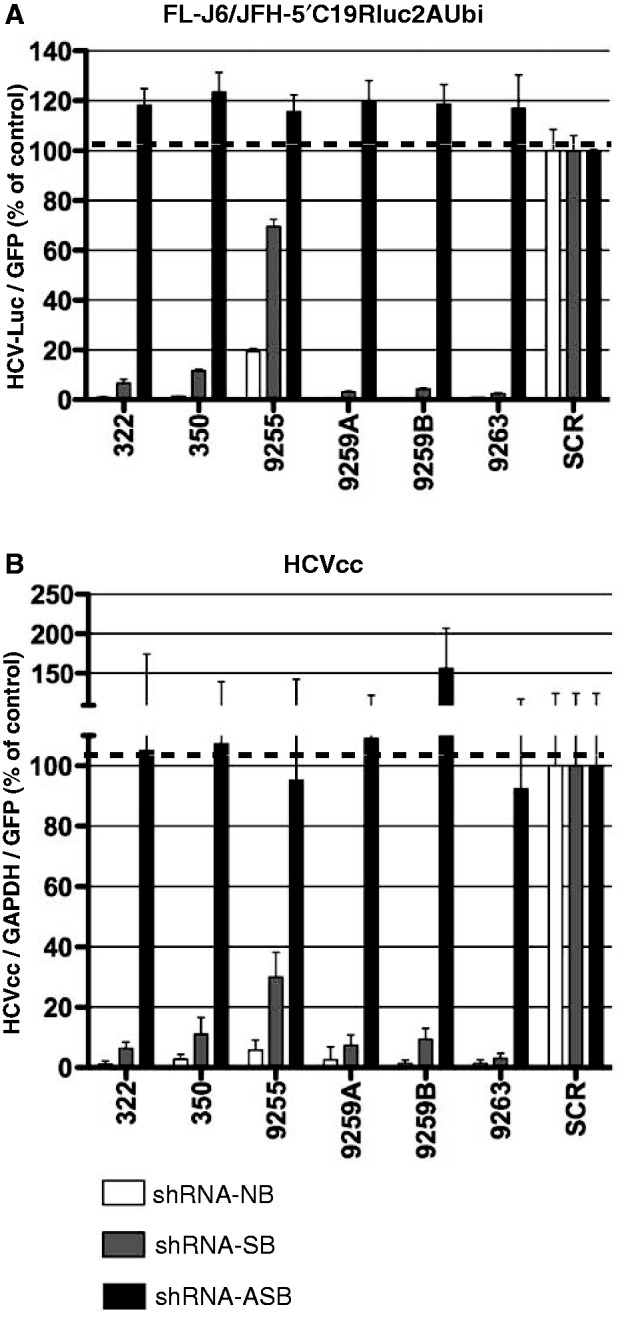
shRNA without bulges (NB-no bulge, white bars) and asymmetric shRNA with bulges in sense strand (SB-sense bulge, gray bars) or antisense strand (ASB-antisense bulge, black bars) were tested against replicating FL-J6/JFH-Luc HCV (A) and HCVcc infection (B) in Huh7.5 cells. SCR shRNA was used as the control and assigned a value of 100. In (A), the luciferase signal was normalized to GFP expressed from the AAV-shRNA vectors to standardize for the transduction efficiency. In (B), quantitative PCR was used to detect HCV levels, and values were normalized to GAPDH quantitative PCR levels as well as AAV-shRNA vector encoded eGFP. Error bars represent SD from n = 3–6.
In concordance, the same observations were made when the same shRNAs were tested in HCVcc-infected cells (Figure 4B). As in the case of replicating FL-J6/JFH-Luc, the use of shRNA without bulges (shRNA-NB) and thus capable of targeting both genomes of HCV (white bars) or with bulge in the sense-strand of shRNA (shRNA-SB), and thus capable of targeting only the positive genome (gray bars) led to significant knockdown of HCV infection. In contrast, all of the shRNAs containing a bulge in the antisense-strand (shRNA-ASB, black bars in Figure 4B) only capable of cleaving the (−)HCV genome were not effective at knocking down HCVcc infection. These results clearly support the conclusion that the negative genome of HCV is not targetable by standard shRNA therapy.
DISCUSSION
The fact that HCV is an RNA virus with a single-stranded genome of positive polarity, together with the fact that all stages of HCV replication occur in the cytoplasm, makes HCV an ideal target for RNAi-based therapy. The high rate of HCV mutations (4) leading to resistant quasispecies lowers the efficiency of RNAi therapy (33). To lower the chances of accumulation of inactivating mutations, it would be beneficial to target the most conserved viral sequences and to use multiple si/shRNAs simultaneously. Wilson and Richardson (33) demonstrated using an HCV replicon system that repeated treatments with the same siRNA can lead to accumulation of inhibiting mutations. Use of two distinct siRNAs significantly lowered the ability of the virus to accumulate mutations required to escape the treatment (33). Based on these observations, multiple groups have now examined the possibility of co-expressing multiple shRNAs to improve the efficiency of anti-HCV RNAi (10,34,35). Biological data are supported by mathematical modeling, which predicts that in the case of suboptimal siRNAs, simultaneous use of at least four siRNAs targeting different regions of the HCV genome would be required to efficiently knockdown HCV infection and avoid viral escape (36). According to the same model, the more effective the individual siRNA used, the fewer siRNAs needs to be co-expressed to achieve total viral clearance.
Our study was designed to select effective shRNA candidates directed against conserved regions of HCV genotypes 1b and 2a. Most activity was observed against the 3′ region of 5′NTR, the 5′ region of the core gene and a 32 nt homology region in the NS5B gene. The results obtained were concordant between transient- and stable-replicating FL-J6/JFH-Luc and HCVcc infection. In contrast, we observed minor differences in the ability to target the 3′ NTR in transient- versus stable-replication or acute infection with HCVcc. This may reflect a better accessibility of the 3′ NTR to shRNA targeting early after introduction of the HCV RNA into cells. Overall, in contrast to data reported by Korf et al. (31), and in agreement with data obtained from an siRNA screen by Smith et al. (20), the 3′NTR invariant X-tail region (37) proved not to be a good target for RNAi in the context of established FL-J6/JFH-Luc replication and HCVcc infection. Although shRNA 9621 used in our study targets the same region as the shRNA used by Korf et al., the observed differences could be caused by the different length of the shRNA stem and/or 1 nt base difference between the replicons used in both studies.
Our results are furthermore supported by results obtained by Chandra et al. (35) who tested 13 siRNA sequences targeting the 5′NTR IRES region of HCV. The authors identified three siRNAs (si279, si321 and si359), which effectively inhibited HCV replication. The si279 overlaps with shRNA274, 276 and 268 from our shRNA panel, whereas si321 overlaps with shRNA318, all of which demonstrated very strong anti-HCV effects in our study. Interestingly, si359 overlaps with our shRNA354, which showed no effect in our system; however, shRNA353, which overlaps with si359 perfectly with the exception of an additional A at the 3′end, demonstrated very strong anti-HCV effect in our study.
Overall, we were able to identify six shRNA molecules that retained their predicted specific functions on addition of the bulge in the sense or antisense strand. Using those verified asymmetric shRNAs, we were able to show in the context of the HCV replicon and HCVcc that the negative HCV genome is not accessible to the standard shRNA-based RNAi. Our data are supported by previous reports from other RNA viruses (38–40), which demonstrated resistance of viral genomic RNA to RNAi, as well as is in agreement with observations made by Smith et al. (20) in HCV system. Smith et al. (20) performed extensive analysis of multiple siRNAs, which, based on their thermodynamic stability, were predicted to exhibit biased RISC loading and thus preferential positive or negative HCV genome targeting. Although the authors included additional alterations to the siRNA sequence (Z and Y modifications) in order to further shift the balance between positive and negative genome targeting, strand specific targeting was not detected in the psiCheck reporter system. In the context of replicon system, however, the authors did observe a decrease in the overall HCV knockdown when siRNAs predicted to preferentially target the negative genome were used. Although the observed effects were small, the authors concluded that the negative genome seems to be more resistant to RNAi than the positive genome (20). It is important to mention that their experimental design could not accurately establish the degree of si/shRNA strand loading bias and, even if such bias existed, it was not an all-or-none effect. Thus, all studies reported by Smith et al. (20) suffered from the co-existence of two active RISC complexes capable of targeting the positive and negative genomes, making the final interpretation of their results difficult. Our system overcomes this limitation, as only the strand without a bulge is complementary to the HCV target and can function in anti-HCV RNAi (as observed in the validation experiments).
To date, the reason for the lack of negative strand targeting is not clear. The structure and sub-cellular localization of the negative HCV genome is not fully known. On HCV infection, the host cell undergoes dramatic internal rearrangements driven by viral protein factors, which convert the cell into a virus-making factory. Multiple studies demonstrate that most of the non-structural HCV proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B), when expressed in the context of full-length HCV genome or as individual proteins, associate with endoplasmic reticulum and other cellular membranes (41–45). Like most positive strand RNA viruses, HCV replicate its genome in close association with modified cellular membranes (46). Egger et al. (47) demonstrated that expression of NS4B alone or in context of the entire HCV polyprotein caused dramatic alterations of the endogenous membranes giving rise to the so-called membranous web, a structure with which all HCV proteins were associated. Recently, NS5A was implicated in generation of double membrane vesicles, the appearance of which correlated with the kinetics of viral replication (48). It has been proposed that the membranous web is the site of viral RNA synthesis (47,48) and that these vesicular membrane structures protects HCV’s replication machinery and double-stranded HCV RNA from host cell proteases and nucleases (potentially including RNAi machinery) and from detection by the innate immune system (49). In addition, the double-stranded nature of the negative genome (50,51) and/or its high occupancy by replication machinery may also block access to the RISC machinery. Of note, similar observations were made for other RNA viruses, e.g. HIV-1, where despite viral RNA involvement at many steps during the viral replication cycle, only a small fraction of viral RNA is accessible to RNAi (52,53).
In terms of screening for active anti-HCV RNAi molecules, the results obtained with the replicon system and HCVcc were not always concordant with the simplified reporter systems commonly used in HCV RNA studies (20,54–56). We believe that observed differences between bona fide replicating HCV and psiCheck reporter system harbouring short specific target sequences may have been related to the virus having different (i) subcellular localization of the genomes; (ii) interactions with protein factors; or (iii) target RNA structure. Our results obtained with psiCheck reporters harbouring long HCV target sequences (Supplementary Figure S6 and S7), which presumably would be able to acquire a structure more closely resembling those of the full-length HCV genome, failed to imply that the HCV RNA structure at least in the absence of HCV proteins as the main determinant of the efficiency of RNAi-mediated knockdown in the context of the more simplified reporter system. Furthermore, dG values calculated for all the short and long (5′NTR-CORE and NS5B-3′NTR) targets in both orientations failed to allow for the generation of simple predictive algorithms to identify a potential suppressive RNA template as an explanation of observed differences in targeting tested sense and antisense targets (Supplementary Table S2). This further supports the conclusion that an appropriate and reliable RNAi screen should be performed in the context of replicating HCV.
When considering clinical implementation, the use of multiple RNAi species must be balanced against two additional concerns—the chance of generating more off-targeting species (32) and limits to the total level of si/shRNA(s)—both of which can lower the relative effectiveness of RNAi and lead to toxicity (12,13,15). Ultimately, the goal is to limit RISC loading to those si/shRNAs that are most effective at reducing viral replication. Our study establishing that the negative strand is not RNAi accessible further suggests that an anti-HCV RNAi therapeutic strategy should avoid the use of siRNA/shRNAs that result in the incorporation of RNAi into active RISC that targets the HCV anti-genome. This would potentially leave more flexibility for using additional positive strand targets, lessening the chance of generating escape quasispecies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2, Supplementary Figures 1–8, Supplementary Results and Discussion, Supplementary Figure Legends and Supplementary Reference [57].
FUNDING
National Institutes of Health (NIH) [NIAI071068 to M.A.K., RO1AI087917 to J.S.G.]; Berry Foundation Fellowship (in part to L.L.). Funding for open access charge: NIH [NIAI071068].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr. Shuo Gu for discussions on shRNA designs.
REFERENCES
- 1.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 2.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005;5:558–567. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 3.Martell M, Esteban JI, Quer J, Genesca J, Weiner A, Esteban R, Guardia J, Gomez J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J. Virol. 1992;66:3225–3229. doi: 10.1128/jvi.66.5.3225-3229.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartenschlager R, Lohmann V. Replication of hepatitis C virus. J. Gen. Virol. 2000;81:1631–1648. doi: 10.1099/0022-1317-81-7-1631. [DOI] [PubMed] [Google Scholar]
- 5.Nakano T, Lau GM, Sugiyama M, Mizokami M. An updated analysis of hepatitis C virus genotypes and subtypes based on the complete coding region. Liver Int. 2012;32:339–345. doi: 10.1111/j.1478-3231.2011.02684.x. [DOI] [PubMed] [Google Scholar]
- 6.Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Halfon P, Inchauspe G, Kuiken C, Maertens G, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 7.Bukh J, Miller RH, Purcell RH. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin. Liver Dis. 1995;15:41–63. doi: 10.1055/s-2007-1007262. [DOI] [PubMed] [Google Scholar]
- 8.Randall G, Rice CM. Interfering with hepatitis C virus RNA replication. Virus Res. 2004;102:19–25. doi: 10.1016/j.virusres.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 9.Caplen NJ. RNAi as a gene therapy approach. Expert Opin. Biol. Ther. 2003;3:575–586. doi: 10.1517/14712598.3.4.575. [DOI] [PubMed] [Google Scholar]
- 10.Yang X, Haurigot V, Zhou S, Luo G, Couto LB. Inhibition of hepatitis C virus replication using adeno-associated virus vector delivery of an exogenous anti-hepatitis C virus microRNA cluster. Hepatology. 2010;52:1877–1887. doi: 10.1002/hep.23908. [DOI] [PubMed] [Google Scholar]
- 11.McCaffrey AP, Nakai H, Pandey K, Huang Z, Salazar FH, Xu H, Wieland SF, Marion PL, Kay MA. Inhibition of hepatitis B virus in mice by RNA interference. Nat. Biotechnol. 2003;21:639–644. doi: 10.1038/nbt824. [DOI] [PubMed] [Google Scholar]
- 12.Grimm D, Wang L, Lee JS, Schurmann N, Gu S, Borner K, Storm TA, Kay MA. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J. Clin. Invest. 2010;120:3106–3119. doi: 10.1172/JCI43565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 14.Gu S, Jin L, Zhang Y, Huang Y, Zhang F, Valdmanis PN, Kay MA. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell. 2012;151:900–911. doi: 10.1016/j.cell.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castanotto D, Sakurai K, Lingeman R, Li H, Shively L, Aagaard L, Soifer H, Gatignol A, Riggs A, Rossi JJ. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 2007;35:5154–5164. doi: 10.1093/nar/gkm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simmonds P, Tuplin A, Evans DJ. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: Implications for virus evolution and host persistence. RNA. 2004;10:1337–1351. doi: 10.1261/rna.7640104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008;6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korf M, Meyer A, Jarczak D, Beger C, Manns MP, Kruger M. Inhibition of HCV subgenomic replicons by siRNAs derived from plasmids with opposing U6 and H1 promoters. J. Viral Hepat. 2007;14:122–132. doi: 10.1111/j.1365-2893.2006.00793.x. [DOI] [PubMed] [Google Scholar]
- 19.Seo MY, Abrignani S, Houghton M, Han JH. Small interfering RNA-mediated inhibition of hepatitis C virus replication in the human hepatoma cell line Huh-7. J. Virol. 2003;77:810–812. doi: 10.1128/JVI.77.1.810-812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith RM, Smolic R, Volarevic M, Wu GY. Positional effects and strand preference of RNA interference against hepatitis C virus target sequences. J. Viral Hepat. 2007;14:194–212. doi: 10.1111/j.1365-2893.2006.00794.x. [DOI] [PubMed] [Google Scholar]
- 21.Gu S, Jin L, Zhang F, Huang Y, Grimm D, Rossi JJ, Kay MA. Thermodynamic stability of small hairpin RNAs highly influences the loading process of different mammalian Argonautes. Proc. Natl Acad. Sci. USA. 2011;108:9208–9213. doi: 10.1073/pnas.1018023108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolten R, Egger D, Gosert R, Schaub G, Landmann L, Bienz K. Intracellular localization of poliovirus plus- and minus-strand RNA visualized by strand-specific fluorescent In situ hybridization. J. Virol. 1998;72:8578–8585. doi: 10.1128/jvi.72.11.8578-8585.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartenschlager R, Frese M, Pietschmann T. Novel insights into hepatitis C virus replication and persistence. Adv. Virus Res. 2004;63:71–180. doi: 10.1016/S0065-3527(04)63002-8. [DOI] [PubMed] [Google Scholar]
- 24.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 25.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 26.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 27.Mathews DH. RNA secondary structure analysis using RNAstructure. Curr. Protoc. Bioinformatics. 2006 doi: 10.1002/0471250953.bi1206s13. Chapter 12, Unit 12.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanda T, Steele R, Ray R, Ray RB. Small interfering RNA targeted to hepatitis C virus 5′ nontranslated region exerts potent antiviral effect. J. Virol. 2007;81:669–676. doi: 10.1128/JVI.01496-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 2006;80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarczak D, Korf M, Beger C, Manns MP, Kruger M. Hairpin ribozymes in combination with siRNAs against highly conserved hepatitis C virus sequence inhibit RNA replication and protein translation from hepatitis C virus subgenomic replicons. FEBS J. 2005;272:5910–5922. doi: 10.1111/j.1742-4658.2005.04986.x. [DOI] [PubMed] [Google Scholar]
- 31.Korf M, Jarczak D, Beger C, Manns MP, Kruger M. Inhibition of hepatitis C virus translation and subgenomic replication by siRNAs directed against highly conserved HCV sequence and cellular HCV cofactors. J. Hepatol. 2005;43:225–234. doi: 10.1016/j.jhep.2005.02.046. [DOI] [PubMed] [Google Scholar]
- 32.Gu S, Jin L, Zhang Y, Huang Y, Zhang F, Valdmanis PN, Kay MA. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell. 2012;151:900–911. doi: 10.1016/j.cell.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson JA, Richardson CD. Hepatitis C virus replicons escape RNA interference induced by a short interfering RNA directed against the NS5b coding region. J. Virol. 2005;79:7050–7058. doi: 10.1128/JVI.79.11.7050-7058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henry SD, van der Wegen P, Metselaar HJ, Tilanus HW, Scholte BJ, van der Laan LJ. Simultaneous targeting of HCV replication and viral binding with a single lentiviral vector containing multiple RNA interference expression cassettes. Mol. Ther. 2006;14:485–493. doi: 10.1016/j.ymthe.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 35.Chandra PK, Kundu AK, Hazari S, Chandra S, Bao L, Ooms T, Morris GF, Wu T, Mandal TK, Dash S. Inhibition of hepatitis C virus replication by intracellular delivery of multiple siRNAs by nanosomes. Mol. Ther. 2012;20:1724–1736. doi: 10.1038/mt.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leonard JN, Schaffer DV. Computational design of antiviral RNA interference strategies that resist human immunodeficiency virus escape. J. Virol. 2005;79:1645–1654. doi: 10.1128/JVI.79.3.1645-1654.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka T, Kato N, Cho MJ, Shimotohno K. A novel sequence found at the 3′ terminus of hepatitis C virus genome. Biochem. Biophys. Res. Commun. 1995;215:744–749. doi: 10.1006/bbrc.1995.2526. [DOI] [PubMed] [Google Scholar]
- 38.Barik S. Control of nonsegmented negative-strand RNA virus replication by siRNA. Virus Res. 2004;102:27–35. doi: 10.1016/j.virusres.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 39.Ge Q, McManus MT, Nguyen T, Shen CH, Sharp PA, Eisen HN, Chen J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl Acad. Sci. USA. 2003;100:2718–2723. doi: 10.1073/pnas.0437841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silvestri LS, Taraporewala ZF, Patton JT. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. J. Virol. 2004;78:7763–7774. doi: 10.1128/JVI.78.14.7763-7774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tu H, Gao L, Shi ST, Taylor DR, Yang T, Mircheff AK, Wen Y, Gorbalenya AE, Hwang SB, Lai MM. Hepatitis C virus RNA polymerase and NS5A complex with a SNARE-like protein. Virology. 1999;263:30–41. doi: 10.1006/viro.1999.9893. [DOI] [PubMed] [Google Scholar]
- 42.Mottola G, Cardinali G, Ceccacci A, Trozzi C, Bartholomew L, Torrisi MR, Pedrazzini E, Bonatti S, Migliaccio G. Hepatitis C virus nonstructural proteins are localized in a modified endoplasmic reticulum of cells expressing viral subgenomic replicons. Virology. 2002;293:31–43. doi: 10.1006/viro.2001.1229. [DOI] [PubMed] [Google Scholar]
- 43.Hwang SB, Park KJ, Kim YS, Sung YC, Lai MM. Hepatitis C virus NS5B protein is a membrane-associated phosphoprotein with a predominantly perinuclear localization. Virology. 1997;227:439–446. doi: 10.1006/viro.1996.8357. [DOI] [PubMed] [Google Scholar]
- 44.Hijikata M, Mizushima H, Tanji Y, Komoda Y, Hirowatari Y, Akagi T, Kato N, Kimura K, Shimotohno K. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc. Natl Acad. Sci. USA. 1993;90:10773–10777. doi: 10.1073/pnas.90.22.10773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003;77:5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rice CM. Flaviviridae: the viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. 3rd edn. Philadelphia, PA: Lippincott-Raven Publications; 1996. pp. 931–959. [Google Scholar]
- 47.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002;76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012;8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quinkert D, Bartenschlager R, Lohmann V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005;79:13594–13605. doi: 10.1128/JVI.79.21.13594-13605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Targett-Adams P, Boulant S, McLauchlan J. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 2008;82:2182–2195. doi: 10.1128/JVI.01565-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iacovacci S, Manzin A, Barca S, Sargiacomo M, Serafino A, Valli MB, Macioce G, Hassan HJ, Ponzetto A, Clementi M, et al. Molecular characterization and dynamics of hepatitis C virus replication in human fetal hepatocytes infected in vitro. Hepatology. 1997;26:1328–1337. doi: 10.1002/hep.510260535. [DOI] [PubMed] [Google Scholar]
- 52.Gao Y, Lobritz MA, Roth J, Abreha M, Nelson KN, Nankya I, Moore-Dudley DM, Abraha A, Gerson SL, Arts EJ. Targets of small interfering RNA restriction during human immunodeficiency virus type 1 replication. J. Virol. 2008;82:2938–2951. doi: 10.1128/JVI.02126-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Westerhout EM, ter Brake O, Berkhout B. The virion-associated incoming HIV-1 RNA genome is not targeted by RNA interference. Retrovirology. 2006;3:57. doi: 10.1186/1742-4690-3-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rose SD, Kim DH, Amarzguioui M, Heidel JD, Collingwood MA, Davis ME, Rossi JJ, Behlke MA. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33:4140–4156. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leaman D, Chen PY, Fak J, Yalcin A, Pearce M, Unnerstall U, Marks DS, Sander C, Tuschl T, Gaul U. Antisense-mediated depletion reveals essential and specific functions of microRNAs in Drosophila development. Cell. 2005;121:1097–1108. doi: 10.1016/j.cell.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 56.Solda G, Robusto M, Primignani P, Castorina P, Benzoni E, Cesarani A, Ambrosetti U, Asselta R, Duga S. A novel mutation within the MIR96 gene causes non-syndromic inherited hearing loss in an Italian family by altering pre-miRNA processing. Hum. Mol. Genet. 2012;21:577–585. doi: 10.1093/hmg/ddr493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.