


Abstract
Transgenic animals are extensively used to model human disease. Typically, the transgene copy number is estimated, but the exact integration site and configuration of the foreign DNA remains uncharacterized. When transgenes have been closely examined, some unexpected configurations have been found. Here, we describe a method to recover transgene insertion sites and assess structural rearrangements of host and transgene DNA using microarray hybridization and targeted sequence capture. We used information about the transgene insertion site to develop a polymerase chain reaction genotyping assay to distinguish heterozygous from homozygous transgenic animals. Although we worked with a bacterial artificial chromosome transgenic mouse line, this method can be used to analyse the integration site and configuration of any foreign DNA in a sequenced genome.
INTRODUCTION
Stable transgenic animal lines are a valuable resource for studies of gene function and disease. Bacterial artificial chromosomes (BACs) and yeast artificial chromosomes are often used as transgenes because of their size and their capacity to preserve the local regulatory elements around the gene of interest. Unfortunately, their large size and potential to undergo breakage and rearrangement present a challenge when it is necessary to define the outcome of integration. For practical reasons, transgenic lines are often only assessed by Southern blot to estimate copy number. The site of integration, the possibility of rearrangements of the transgene and potential deletions of native DNA at the site of integration remain unknown for most transgenic lines. The exact mechanism of foreign DNA integration is also unknown in the specific case, although general evidence suggests the role of non-homologous end-joining (NHEJ) as well as non-NHEJ DNA repair systems (1).
For studies assessing dosage effects, there is a compelling need to determine the transgene integration site to design a facile assay that can distinguish heterozygotes from homozygotes. Although some investigators have reported success in genotyping using quantitative real-time polymerase chain reaction (PCR) (2), we were unable to distinguish between one and two copies of the transgene reliably in our transgenic model. Fluorescence in situ hybridization (FISH) can be used to make the distinction, but this technique is slow, expensive and inappropriate for high-throughput applications. For optimum speed and reliability, there is currently no effective substitute for rescuing sequence from the integration site and using this to develop a PCR assay that can accurately distinguish genotypes.
Identifying the location of random foreign DNA integration would also be useful when it is important to assess the possibility of gene disruption at the integration site. There are many examples in the literature for ‘insertional mutations’ that result from transgene integration disrupting or deleting an endogenous gene (3). To readily pinpoint the location of transgene insertion and identify the gene or genes that are disrupted or deleted would be of considerable use.
We have previously developed a transgenic mouse model of Hutchinson–Gilford progeria syndrome (HGPS) by injecting a circular BAC carrying the human lamin A gene (LMNA). The 164.4-kb insert had been recombineered to carry the G608G mutation in LMNA that is found in majority of the children with this rare disorder (4). We have observed an interesting dose effect of the transgene, where homozygotes have a substantially more severe phenotype than heterozygotes (unpublished data), but the ability to distinguish genotypes reliably between the two groups was presenting significant challenges and project delays. The BAC’s length and circular configuration at the time of injection made it impossible to determine the integration site with established methods that are designed to rescue the ends of the transgene. Instead, we devised a different approach by creating a custom microarray using DNA probes tiled across the BAC transgene, with the aim of capturing adjacent BAC and mouse genomic sequences. We then used next-generation sequencing to recover enriched DNA sequences and aligned them to mouse and BAC reference sequence. With this process, we were able to identify the BAC integration site and uncover the complex sequence anatomy of the transgene. This information allowed us to develop a three-primer PCR genotyping assay that readily distinguishes wild-type, heterozygous transgenic and homozygous transgenic mice.
MATERIALS AND METHODS
Transgenic animals
The sites of integration and rearrangements were assessed for the previously reported G608G H line. BAC clone RP11-702H12 (RPCI-11 Human BAC Library, BACPAC Resource Center at Children’s Hospital Oakland Research Institute, Oakland, CA, USA) was recombineered to contain the most common mutation that causes HGPS, a C > T transition at base 1824 in LMNA exon 11, also denoted as G608G for the amino acid sequence which remains unchanged. The circular recombineered BAC (total size with vector, 173.2 kb) was microinjected to create the G608G H transgenic mouse line (4). By FISH, it was determined that the BAC was integrated into mouse chromosome 4 (data not shown).
Microarray
See Figure 1 for an overview of the workflow. A custom NimbleGen Sequence Capture Developer 385 K Array (Roche) was designed with probes tiled across BAC RP11-702H12. Probes were screened for uniqueness against the mm9 mouse build, the human hg19 build (the portion contained in RP11-702H12) and the vector portion of the BAC (pBACe3.6).
Figure 1.

Overview of microarray workflow. The microarray was designed with probes complementary to the entire BAC sequence. An Illumina next-generation DNA sequencing library was created from homozygous transgenic mouse DNA. Adapters (in red) were ligated to the ends of the DNA fragments. BAC DNA is shown in blue (both in the library and the BAC specific probes on the microarray), and mouse DNA is shown in black. The library was heat denatured to generate single-stranded DNA, then applied to the microarray. Non-hybridized DNA was washed off, and the hybridized DNA was eluted and collected. The eluted DNA was PCR amplified using primers specific to the adapters, sequenced and the paired-end reads were analysed to determine the location of the BAC/mouse or non-contiguous BAC/BAC junctions, an example of which is simplified here for illustrative purposes.
The protocols for library preparation and hybridization were modified from Teer et al. (5). Briefly, 5 µg of genomic DNA from a G608G (line H) homozygous transgenic mouse was used to prepare a non-indexed DNA library. Adapter ligated fragments were selected for a size of 300 bp with the Pippin Prep DNA size selection system (Sage Science) and then purified on a MinElute PCR purification column (QIAgen) (for future applications of this protocol, we would recommend selecting for a size of 400–500 bp to achieve a higher likelihood that any given fragment crosses a break point without being too large to sequence on the Illumina platform). The library was LM–PCR (ligation-mediated PCR) amplified with adaptor specific primers and purified using Agencourt AMPure XP beads (Beckman Coulter Inc.).
In all, 2.5 µg of the amplified library was hybridized to the custom microarray, washed and eluted with sodium hydroxide per manufacturer’s instructions. Mouse Cot-I DNA at 100X molar excess (Life Technologies) and blocking oligos were used to prevent non-specific hybridization. The hybridized DNA was eluted, purified and PCR amplified with adaptor specific primers (sequences available on request) and, finally, purified using AMPure XP beads. An aliquot of this amplified DNA was evaluated for enrichment of microarray probe targeted regions before submitting DNA for sequencing.
Enrichment of the regions targeted by the microarray was evaluated by qPCR with QuantiTect SYBR Green (QIAgen) on a 7900 HT Fast Real-Time PCR system (ABI) under standard conditions in triplicate. To assess the level of enrichment of the BAC target, qPCR primers were designed to amplify eight regions within the BAC. Similarly, qPCR primers were designed to amplify three non-targeted mouse regions as negative controls. Primer sequences are available on request. The negative control region sequences were depleted in the post-microarray DNA sample compared with the pre-microarray DNA sample (data not shown). The eight regions in the BAC were highly enriched in the post-microarray sample (∼3000- to 26 000-fold more) compared with the pre-microarray DNA (data not shown).
Sequencing, assembly and identification of junctions
The enriched libraries were sequenced on a single lane of an Illumina HiSeq2000, which generated 101-bp paired-end reads. Reads were aligned to a hybrid reference consisting of the BAC (the pBACe3.6 vector and the EcoRI partial 164.4-kb human insert) and the mouse mm9 (NCBI37) reference genome using Novoalign (Novocraft). A total of 197 million reads were mapped to the reference genomes with average nucleotide coverage of 82 186× in the enriched area. Regions with break points were identified by SAMtools (6) and refined by manual inspection of individual reads spanning two genomes or non-contiguous genomic regions. Around the four BAC/mouse junctions that were identified, there were 8118 reads with one paired-end matching BAC sequence and the other paired-end matching mouse sequence in chromosome 4. All of the identified junctions were confirmed by PCR followed by capillary sequencing.
Genotyping PCR
Genomic DNA was PCIA (phenol:chloroform:isoamyl alcohol 25:24:1 v/v) extracted, ethanol precipitated and resuspended in tris-ethylenediaminetetraacetic acid buffer. PCR was performed under standard conditions using QIAgen HotStarTaq DNA polymerase. Primers used were GenoCh4-F1, 5′-CAAACAAGTACATATCATAGGC-3′; GenoCh4-R1, 5′-ATGATAGTGACAGGTATACGG-3′; and GenoBAC-R2, 5′-ATTCTAGTGGAGGGAGACAG-3′. GenoCh4-F1 and GenoCh4-R1 amplify an endogenous sequence of 493 bp that is not observed in the presence of the transgene. GenoCh4-F1 and GenoBAC-R2 amplify a hybrid BAC/mouse sequence of 233 bp.
RESULTS AND DISCUSSION
Our sequencing efforts unexpectedly revealed not two, but four junctions between mouse chromosome 4 and the BAC, and three additional unexpected junctions between non-contiguous regions of the BAC. We were unable to recover sequences from the transgenic mouse genome in two regions of the original BAC, despite the fact that probes for these two regions were present on the hybridization microarray. One of the deleted regions is 2.1 kb (from 67 871 through 69 972), and the other is 31.7 kb (14 391 through 46 042). We also observed a 90-bp deletion and a 1090-bp deletion at the sites of integration in the mouse genome (Ch4: 81 461 498-81 461 588 and Ch4: 81 461 734-81 462 824, respectively). These observations are consistent with previous reports of deletions occurring within large BAC transgenes (7,8) and deletions at the integration site (9). With this collection of junctions and deletions, several configurations are possible, one of which is shown here (Figure 2). Although it is clear that not all copies of the LMNA gene are likely to be functional, there is at least one intact copy that faithfully expresses the mutated human LMNA gene at roughly the same levels as the endogenous mouse locus (4,10). In all of the possible configurations of the transgenic locus, there are sections of the BAC represented multiple times, suggesting that fragments of more than one BAC were incorporated. We suspect that the high-molecular weight BAC DNA was partially degraded and underwent concatamerization before integration. Given that a fragment of mouse sequence is also interspersed in the transgene, some rearrangement must have happened at the locus itself.
Figure 2.

The original BAC, and one possible configuration of the transgenic locus. The original 173.2-kb BAC was recombineered to carry a mutation in LMNA that causes HGPS, then microinjected in circular form (A) to create the G608G H transgenic mouse. A possible configuration of the transgenic locus is shown with grey boxes representing mouse DNA and all other boxes representing foreign DNA (B). The black bars between the boxes indicate junctions, and the base number and direction of each section of DNA are labelled. BAC coordinates are in blue and numbered based on the circular BAC, starting at one end of the human insert (i.e. BAC coordinate 1 corresponds to Chr1: 155 981 195 of the human hg19 build). Mouse coordinates are shown in black. All copies and partial copies of LMNA are shown in yellow boxes, and the BAC vector (pBACe3.6) is shown in a dark blue box. The junction between two SINE elements is indicated by the green arrow. Although only one option is shown here, there are several possible configurations with the same junctions, same deleted mouse and BAC sequence and at least one intact copy of LMNA. Not to scale.
At six of the junctions, we found microhomology of three bases or less, with only one base of homology at two of the three BAC/BAC junctions. At the remaining BAC/BAC junction, we observed a much larger region of homology between two short interspersed nuclear elements (SINEs), specifically Alu elements. SINEs are hot spots of homologous recombination, and mouse SINEs can greatly enhance recombination when placed at the ends of targeting vectors because of homologous recombination with endogenous mouse SINEs (11,12). Looking primarily at balanced germline rearrangements (which undergo double-strand break repair as is also the case for transgenics), Chiang et al. found that long interspersed nuclear elements (LINEs), not SINEs, were slightly enriched at break points. This is discordant with previous studies reporting SINEs as prime locations for homologous recombination. Chiang et al. also found that NHEJ was the most common process involved in generating rearrangements. These results indicate that both homologous recombination (which created this particular BAC/BAC junction) and NHEJ can occur to yield a complex transgenic allele.
Complex rearrangements of the transgene and the host genome have previously been observed in multiple transgenic lines. Reported rearrangements include deletions, duplications, inversions and translocations (7–9,13,14) reviewed earlier by Wrutele et al. (15). These integrations and rearrangements of genomic structure are of particular interest to the fields of cancer and gene therapy.
We identified several BAC/mouse junctions in our alignment, and used the sequence information to develop a genotyping PCR across one of the sites of integration. Any of the recovered mouse/BAC junctions could be selected for this purpose, although a junction without any repetitive elements is preferable. In this three-primer PCR assay, the forward primer anneals to the mouse DNA near a mouse/BAC junction, and the reverse primers anneal either within the BAC transgene or to the endogenous mouse sequence (Figure 3). The product sizes indicate whether the mouse is wild-type, heterozygous or homozygous for the transgene. This assay has proven to be rapid and robust in distinguishing wild-type, heterozygous and homozygous BAC transgenic mice.
Figure 3.
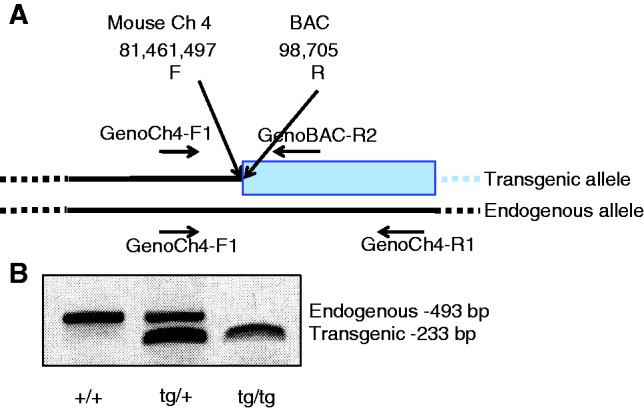
Genotyping by PCR using one of the BAC/mouse junctions identified by the microarray sequencing protocol. The primers GenoCh4-F1 and GenoBAC-R2 amplify across one of the BAC/mouse junctions on the transgenic allele, whereas the primers GenoCh4-F1 and GenoCh4-R1 amplify a region in the endogenous allele (A). A PCR reaction with these three primers amplifies only the endogenous allele from wild-type DNA (493 bp), both endogenous and transgenic alleles from heterozygous transgenic DNA (493 bp and 233 bp, respectively) and only transgenic alleles from homozygous transgenic DNA (233 bp) (B).
In summary, we present a method to define the complex anatomy of a transgene, which allows rapid development of a genotyping assay that can distinguish heterozygous and homozygous transgenic animals. This method can also be used in any situation requiring the recovery of the location of randomly integrated foreign DNA in a sequenced genome, including transgene integrations on an isogenic background. In the case of transgene integration on an isogenic background, mismatched paired ends (with one end within the transgene and the other located elsewhere in the mouse genome) could readily be used to discover the transgene/mouse and transgene/transgene junctions, despite the presence of additional reads deriving from the endogenous locus. Designing a custom microarray for targeted sequence capture followed by next-generation sequencing involves a significant cost (currently ∼$4000 US dollars), but it should be noted that the same microarray design could be used to assess multiple different independent lines for the same transgene. Furthermore, genotyping transgenic animals by FISH or qPCR is an expensive and labour-intensive process, whereas the costs of microarray design, microarray production and next-generation sequencing will likely continue to decline over time. Many transgenic lines such as the one described here are ultimately used for a prolonged programme of experiments, often in multiple laboratories; therefore, defining the anatomy of the transgene and the integration site may be well worth the investment.
FUNDING
Funding for open access charge: NIH Division of Intramural Research/NHGRI Project Number Z01-HG200305.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Casey Matthews at Roche Applied Science for technical advice. They also thank Michael Erdos for ongoing discussions.
REFERENCES
- 1.Iiizumi S, Kurosawa A, So S, Ishii Y, Chikaraishi Y, Ishii A, Koyama H, Adachi N. Impact of non-homologous end-joining deficiency on random and targeted DNA integration: implications for gene targeting. Nucleic Acids Res. 2008;36:6333–6342. doi: 10.1093/nar/gkn649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tesson L, Remy S, Menoret S, Usal C, Anegon I. Analysis by quantitative PCR of zygosity in genetically modified organisms. Methods Mol. Biol. 2010;597:277–285. doi: 10.1007/978-1-60327-389-3_19. [DOI] [PubMed] [Google Scholar]
- 3.Woychik RP, Alagramam K. Insertional mutagenesis in transgenic mice generated by the pronuclear microinjection procedure. Int. J. Dev. Biol. 1998;42:1009–1017. [PubMed] [Google Scholar]
- 4.Varga R, Eriksson M, Erdos MR, Olive M, Harten I, Kolodgie F, Capell BC, Cheng J, Faddah D, Perkins S, et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl Acad. Sci. USA. 2006;103:3250–3255. doi: 10.1073/pnas.0600012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teer JK, Bonnycastle LL, Chines PS, Hansen NF, Aoyama N, Swift AJ, Abaan HO, Albert TJ, Margulies EH, Green ED, et al. Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–1431. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandler KJ, Chandler RL, Broeckelmann EM, Hou Y, Southard-Smith EM, Mortlock DP. Relevance of BAC transgene copy number in mice: transgene copy number variation across multiple transgenic lines and correlations with transgene integrity and expression. Mamm. Genome. 2007;18:693–708. doi: 10.1007/s00335-007-9056-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Saux A, Houdebine LM, Jolivet G. Chromosome integration of BAC (bacterial artificial chromosome): evidence of multiple rearrangements. Transgenic Res. 2010;19:923–931. doi: 10.1007/s11248-010-9368-7. [DOI] [PubMed] [Google Scholar]
- 9.Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR, et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat. Genet. 2012;44:390–397, S391. doi: 10.1038/ng.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capell BC, Olive M, Erdos MR, Cao K, Faddah DA, Tavarez UL, Conneely KN, Qu X, San H, Ganesh SK, et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc. Natl Acad. Sci. USA. 2008;105:15902–15907. doi: 10.1073/pnas.0807840105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang YK, Park JS, Lee CS, Yeom YI, Chung AS, Lee KK. Efficient integration of short interspersed element-flanked foreign DNA via homologous recombination. J. Biol. Chem. 1999;274:36585–36591. doi: 10.1074/jbc.274.51.36585. [DOI] [PubMed] [Google Scholar]
- 12.Kang YK, Park JS, Lee CS, Yeom YI, Han YM, Chung AS, Lee KK. Effect of short interspersed element sequences on the integration and expression of a reporter gene in the preimplantation-stage mouse embryos. Mol. Reprod. Dev. 2000;56:366–371. doi: 10.1002/1098-2795(200007)56:3<366::AID-MRD6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 13.Kasai F, Yoshihara M, Matsukuma S, O’Brien P, Ferguson-Smith MA. Emergence of complex rearrangements at translocation breakpoints in a transgenic mouse; implications for mechanisms involved in the formation of chromosome rearrangements. Cytogenet. Genome Res. 2007;119:83–90. doi: 10.1159/000109623. [DOI] [PubMed] [Google Scholar]
- 14.Abrahams BS, Chong AC, Nisha M, Milette D, Brewster DA, Berry ML, Muratkhodjaev F, Mai S, Rajcan-Separovic E, Simpson EM. Metaphase FISHing of transgenic mice recommended: FISH and SKY define BAC-mediated balanced translocation. Genesis. 2003;36:134–141. doi: 10.1002/gene.10205. [DOI] [PubMed] [Google Scholar]
- 15.Wurtele H, Little KC, Chartrand P. Illegitimate DNA integration in mammalian cells. Gene Ther. 2003;10:1791–1799. doi: 10.1038/sj.gt.3302074. [DOI] [PubMed] [Google Scholar]